
Excited State Lifetimes of Sulfur-Substituted DNA and RNA Monomers Probed Using the Femtosecond Fluorescence Up-Conversion Technique


Abstract
:1. Introduction
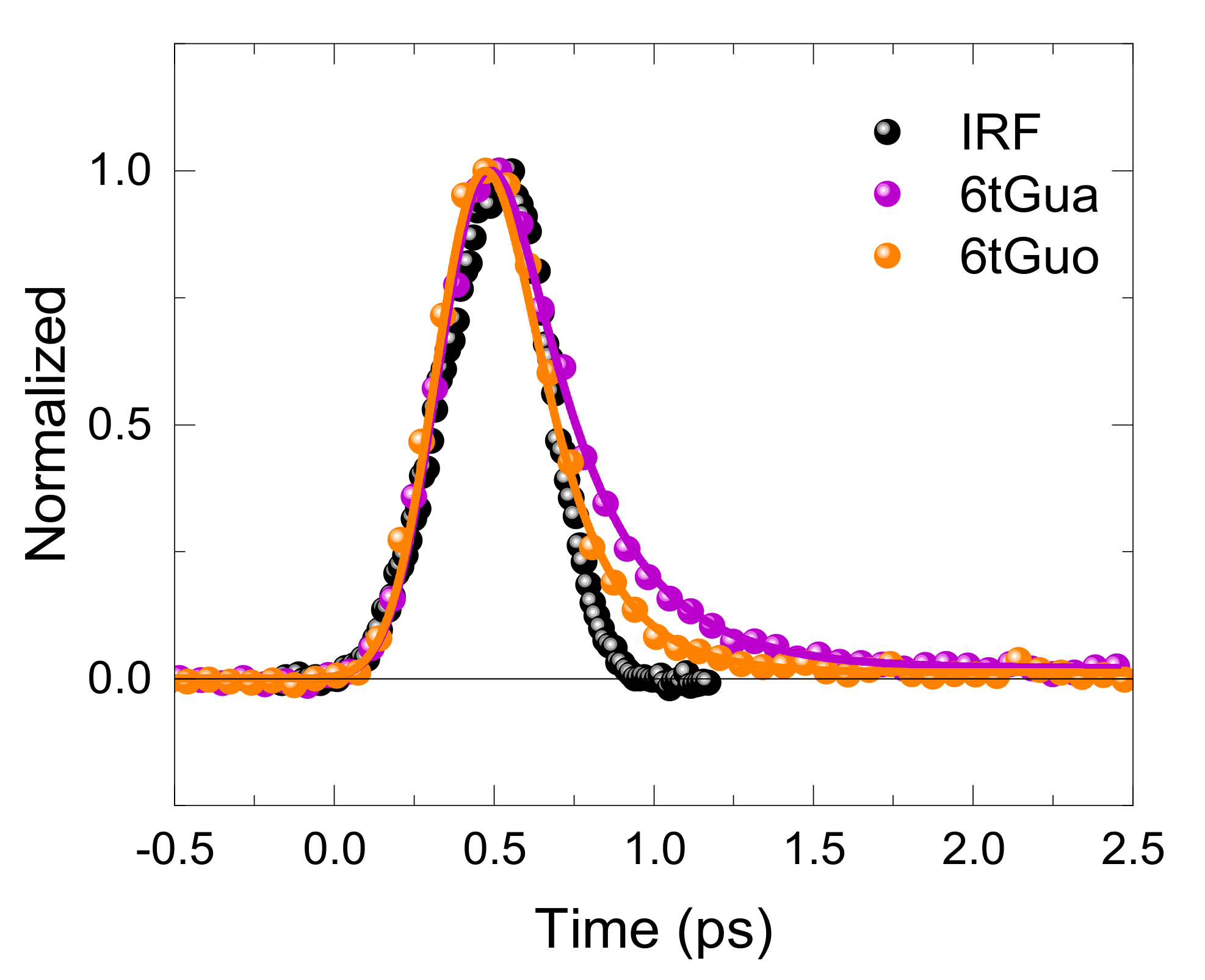
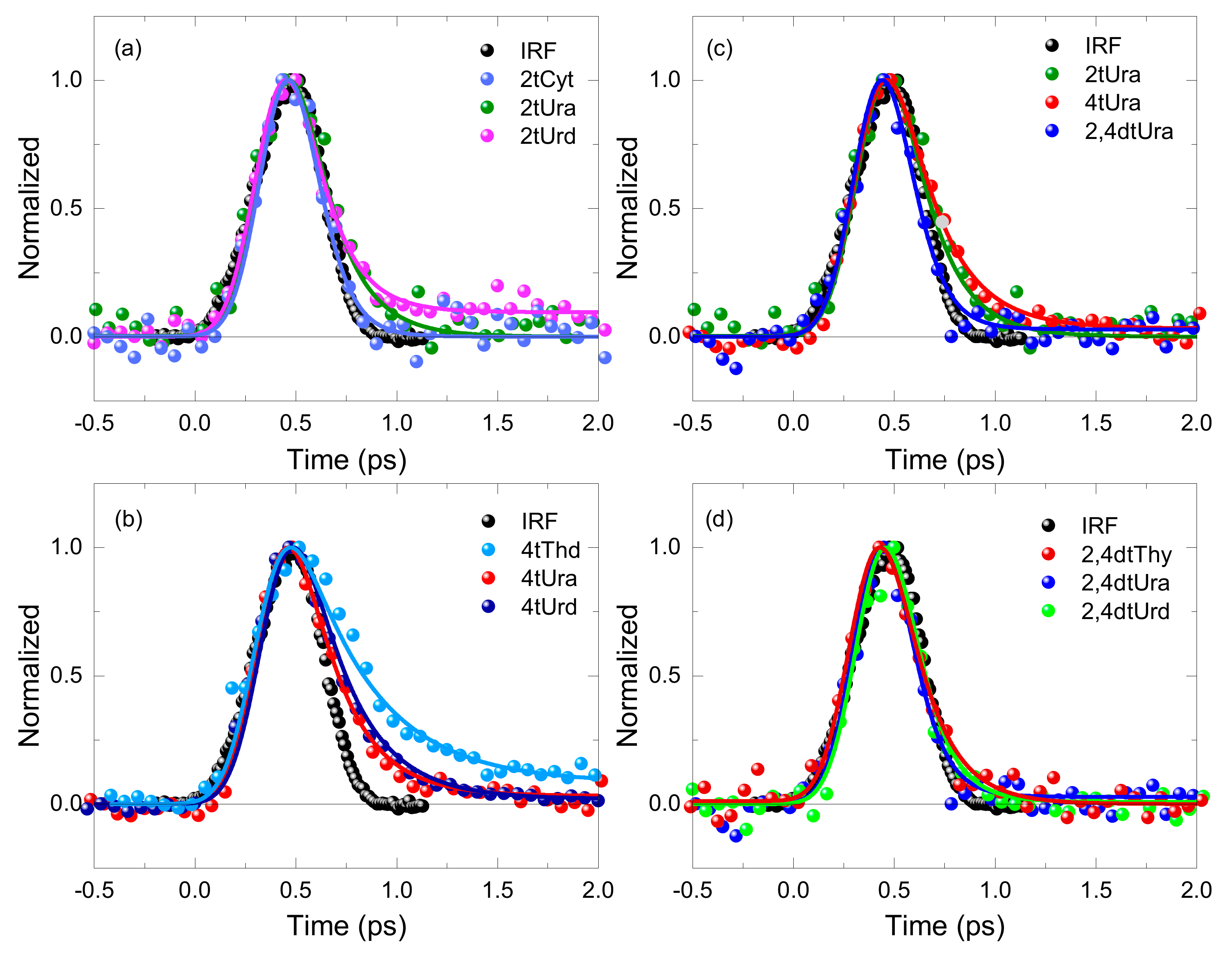
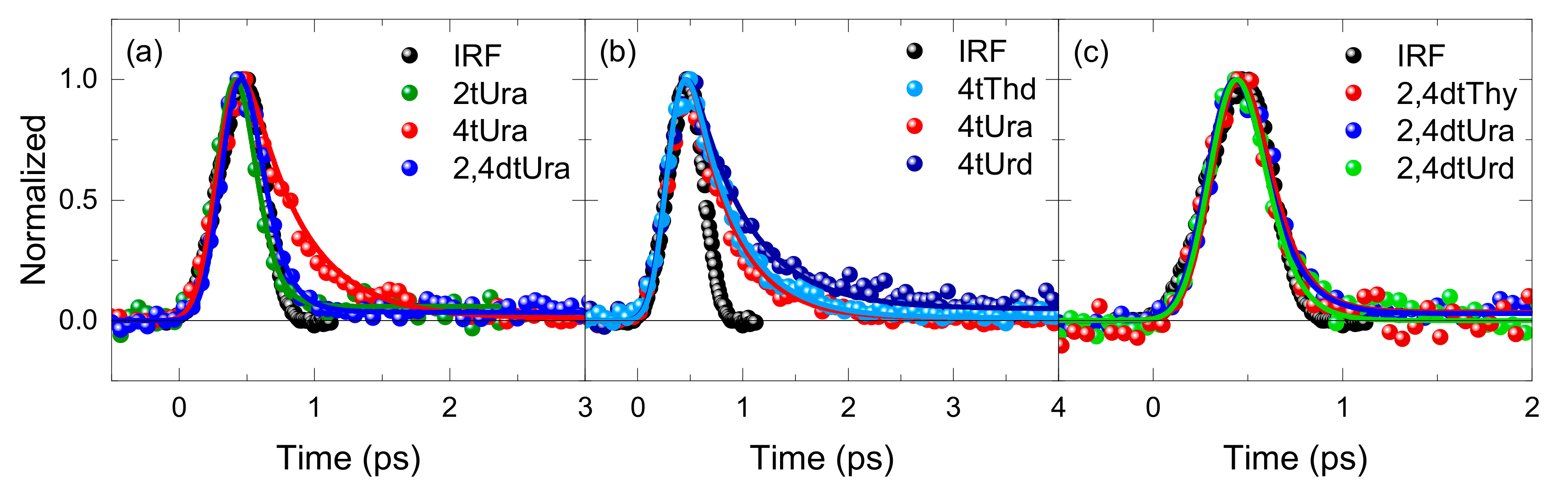
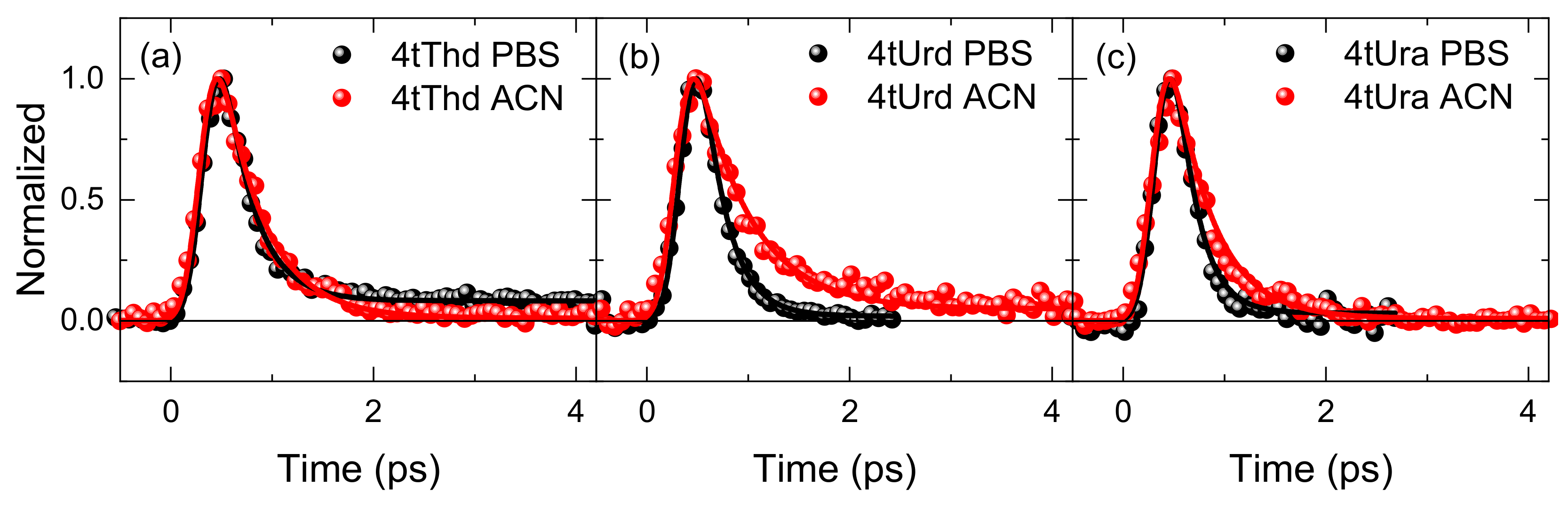
2. Results
3. Discussion
3.1. Effect of the Excitation Wavelength on the Fluorescence Lifetimes
3.2. Effects of N9-Glycosylation on the Fluorescence Lifetime of 6tGua
3.3. Fluorescence Decay Lifetimes of the Thiopyrimidine Monomers
3.4. Solvent Effects on the Fluorescence Decay Lifetimes
3.5. Comparison with DNA and RNA Canonical Nucleobases
4. Materials and Methods
4.1. Chemicals
4.2. Steady-State Absorption and Emission
4.3. Femtosecond Fluorescence Spectroscopy
4.4. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Attard, N.R.; Karran, P. UVA photosensitization of thiopurines and skin cancer in organ transplant recipients. Photochem. Photobio. Sci. 2012, 11, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kaba, S.E.; Kyritsis, A.P. Recognition and management of gliomas. Drugs 1997, 53, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.; Xu, Y.-Z.; Karran, P. Ambiguous coding is required for the lethal interaction between methylated DNA bases and DNA mismatch repair. DNA Repair 2002, 1, 275–286. [Google Scholar] [CrossRef]
- Ren, X.; Li, F.; Jeffs, G.; Zhang, X.; Xu, Y.-Z.; Karran, P. Guanine sulphinate is a major stable product of photochemical oxidation of DNA 6-thioguanine by UVA irradiation. Nucleic Acids Res. 2010, 38, 1832–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Xu, Y.-Z.; Swann, P.F. Photochemical cross-linking of λ-Cro repressor to operator DNA containing 4-thiothymine or 6-thioguanine. Nucleos. Nucleot. 1997, 16, 1799–1803. [Google Scholar] [CrossRef]
- Wang, Z.; Rana, T.M. Efficient DNA interstrand crosslinking by 6-thioguanine and UVA radiation. Biochemistry 1998, 37, 4235–4243. [Google Scholar] [CrossRef]
- Favre, A.; Saintomé, C.; Fourrey, J.-L.; Clivio, P.; Laugâa, P. Thionucleobases as intrinsic photoaffinity probes of nucleic acid structure and nucleic acid-protein interactions. J. Photochem. Photobiol. B 1998, 42, 109–124. [Google Scholar] [CrossRef]
- Pollum, M.; Ashwood, B.; Jockusch, S.; Lam, M.; Crespo-Hernández, C.E. Unintended consequences of expanding the genetic alphabet. J. Am. Chem. Soc. 2016, 138, 11457–11460. [Google Scholar] [CrossRef]
- Pollum, M.; Guan, L.; Ahsanuddin, S.; Baron, E.; Lam, M.; Crespo-Hernández, C. Photoactivation of sulfur-modified DNA and RNA analogs induces cytotoxicity in epidermoid carcinoma cells. J. Invest. Derm. 2016, 136, S105. [Google Scholar] [CrossRef]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. 2, 4-Dithiothymine as a potent UVA chemotherapeutic agent. J. Am. Chem. Soc. 2014, 136, 17930–17933. [Google Scholar] [CrossRef]
- Pollum, M.; Minh, L.; Jockusch, S.; Crespo-Hernández, C.E. Dithionated nucleobases as effective photodynamic agent against human epidermoid carcinoma cells. ChemMedChem 2018, 13, 1044–1050. [Google Scholar] [CrossRef]
- Reelfs, O.; Karran, P.; Young, A.R. 4-Thiothymidine sensitization of DNA to UVA offers potential for a novel photochemotherapy. Photochem. Photobio. Sci. 2012, 11, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.; Xu, Y.-Z.; Karran, P. Photoactivation of DNA thiobases as a potential novel therapeutic option. Curr. Biol. 2001, 11, 1142–1146. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Jeffs, G.; Ren, X.; O’Donovan, P.; Montaner, B.; Perrett, C.M.; Karran, P.; Xu, Y.-Z. Novel DNA lesions generated by the interaction between therapeutic thiopurines and UVA light. Dna Repair 2007, 6, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Prodgeon, S.W.; Heer, R.; Taylor, G.A.; Newell, D.R.; O’Toole, K.; Robinson, M.; Xu, Y.-Z.; Karran, P.; Boddy, A.V. Thiothymidine combined with UVA as a potential novel therapy for bladder cancer. Br. J. Cancer 2011, 104, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Brem, R.; Karran, P. Multiple forms of DNA damage caused by UVA photoactivation of DNA 6-thioguanine. Photochem. Photobiol. 2012, 88, 5–13. [Google Scholar] [CrossRef]
- Penn, I. The problem of cancer in transplant patients: An overview. Transpl. Sci. 1994, 4, 23–32. [Google Scholar]
- Perrett, C.M.; Wlker, S.L.; O’Donovan, P.; Warwick, J.; Harwood, C.A.; Karran, P.; McGregor, J.M. Azathioprine treatment sensitizes human skin to ultraviolet A radiation. Br. J. Derm. 2008, 159, 198–204. [Google Scholar] [CrossRef]
- Karran, P.; Attard, N. Thiopurines in current medical practice: Molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer 2008, 8, 24–36. [Google Scholar] [CrossRef]
- Swann, P.F.; Waters, T.R.; Moulton, D.C.; Xu, Y.-Z. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 1996, 273, 1109–1111. [Google Scholar] [CrossRef]
- Karran, P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br. Med. Bull. 2006, 79, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Weinshilboum, R. Thiopurine pharmacogenetics: Clinical and molecular studies of thiopurine methyltransferase. Drug Metab. Dispos. 2001, 29, 601–605. [Google Scholar] [PubMed]
- Cooke, M.S.; Duarte, T.L.; Cooper, D.; Chen, J.; Nandagopal, S.; Evans, M.D. Combination of azathioprine and UVA irradiation is a major source of cellular 8-oxo-7, 8-dihydro-2′-deoxyguanosine. Dna Repair (Amst.) 2008, 7, 1982–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favre, A. 4-thiouridine as an intrinsic photoaffinity probe of nucleic acid structure and interactions. In Bioorganic Photochemistry, Photochemistry and the Nucleic Acids; Morrison, H., Ed.; Wiley: New Jersey, USA, 1990; Volume 1, pp. 379–425. [Google Scholar]
- Pollum, M.; Martínez-Fernández, L.; Crespo-Hernández, C.E. Photochemistry of nucleic acid bases and their thio-and aza-analogues in solution. In Photoinduced Phenomena in Nucleic Acids I; Barbatti, M.B., Borin, A.C., Susanne, U., Eds.; Springer: Cham, Switzerland, 2015; Volume 355, pp. 245–327. [Google Scholar]
- Ashwood, A.; Pollum, M.; Crespo-Hernández, C.E. Photochemical and photodynamical properties of sulfur-substituted nucleic acid bases. Photochem. Photobiol. 2019, 95, 33–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslancan, S.; Martínez-Fernández, L.; Corral, I. Photophysics and photochemistry of canonical nucleobases’ thioanalogs: From quantum mechanical studies to time resolved experiments. Molecules 2017, 22, 998. [Google Scholar] [CrossRef]
- Teles-Ferreira, D.; Conti, I.; Borrego-Varillas, R.; Nenov, A.; van Stokkum, I.H.M.; Ganzer, L.; Manzoni, C.; de Paula, A.M.; Cerullo, G.; Garavelli, M. A unified experimental/theoretical description of the ultrafast photophysics of single and double thionated uracils. Chem. Eur. J. 2020, 26, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Pollum, M.; Crespo-Hernández, C.E. Communication: The dark singlet state as a doorway state in the ultrafast and efficient intersystem crossing dynamics in 2-thiothymine and 2-thiouracil. J. Chem. Phys. 2014, 140, 071101. [Google Scholar] [CrossRef] [Green Version]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. Increase in the photoreactivity of uracil derivatives by doubling thionation. Phys. Chem. Chem. Phys. 2015, 17, 27851–27861. [Google Scholar] [CrossRef]
- Pollum, M.; Ortiz-Rodríguez, L.A.; Jockusch, S.; Crespo-Hernández, C.E. The triplet state of 6-thio-2′-deoxyguanosine: Intrinsic properties and reactivity toward molecular oxygen. Photochem. Photobiol. 2016, 92, 286–292. [Google Scholar] [CrossRef]
- Reichardt, C.; Crespo-Hernández, C.E. Ultrafast spin crossover in 4-thiothymidine in an ionic liquid. Chem. Commun. 2010, 46, 5963–5965. [Google Scholar] [CrossRef]
- Reichardt, C.; Crespo-Hernández, C.E. Room-temperature phosphorescence of the DNA monomer analogue 4-thiothymidine in aqueous solutions after UVA excitation. J. Phys. Chem. Lett. 2010, 1, 2239–2243. [Google Scholar] [CrossRef]
- Reichardt, C.; Guo, C.; Crespo-Hernández, C.E. Excited-state dynamics in 6-thioguanosine from the femtosecond to microsecond time scale. J. Phys. Chem. B. 2011, 115, 3263–3270. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Pollum, M.; Martínez-Fernández, L.; Dunn, N.; Marquetand, P.; Corral, I.; Crespo-Hernández, C.E.; González, L. The origin of efficient triplet state population in sulfur-substituted nucleobases. Nat. Commun. 2016, 7, 13077. [Google Scholar] [CrossRef]
- Ashwood, B.; Pollum, M.; Crespo-Hernández, C.E. The effects of sugar substitution on the excited-state dynamics of thiopyrimidines. Unpublished work. 2020. [Google Scholar]
- Ashwood, B.; Jockusch, S.; Crespo-Hernández, C.E. Excited-state dynamics of the thiopurine prodrug 6-thioguanine: Can N9-glycosylation affect its phototoxic activity? Molecules 2017, 22, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrego-Varillas, R.; Teles-Ferreira, D.C.; Nenov, A.; Conti, I.; Ganzer, L.; Manzoni, C.; Garavelli, M.; de Paula, A.M.; Cerullo, G. Observation of the sub-100 femtosecond population of a dark state in a thiobase mediating intersystem crossing. J. Am. Chem. Soc. 2018, 140, 16087–16093. [Google Scholar] [CrossRef]
- Martínez-Fernández, L.; Granucci, G.; Pollum, M.; Crespo-Hernández, C.; Persico, M.; Corral, I. Decoding the molecular basis for the population mechanism of the triplet phototoxic precursors in UVA light-activated pyrimidine anticancer drugs. Chem. Eur. J. 2017, 23, 2619–2627. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, J.A.; Mohamadzade, A.; Mai, S.; Ashwood, B.; Pollum, M.; Marquetand, P.; González, L.; Crespo-Hernández, C.; Ullrich, S. 2-Thiouracil intersystem crossing photodynamics studied by wavelength-dependent photoelectron and transient absorption spectroscopies. Phys. Chem. Chem. Phys. 2017, 19, 19756–19766. [Google Scholar] [CrossRef] [Green Version]
- Crespo-Hernández, C.E.; Cohen, B.; Kohler, B. Complexity of excited-state dynamics in DNA. Nature 2006, 441, E8. [Google Scholar] [CrossRef]
- Markovitsi, D.; Talbot, F.; Gustavsson, T.; Onidas, D.; Lazzarotto, E.; Marguet, S. Molecular spectroscopy: Complexity of excited-state dynamics in DNA. Nature 2006, 441, E7. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Fernández, L.; Corral, I.; Granucci, G.; Persico, M. Competing ultrafast intersystem crossing and internal conversion: A time resolved picture for the deactivation of 6-thioguanine. Chem. Sci. 2014, 5, 1336–1347. [Google Scholar] [CrossRef]
- Mai, S.; Marquetand, P.; González, L. Intersystem crossing pathways in the noncanonical nucleobase 2-thiouracil: A time-dependent picture. J. Phys. Chem. Lett. 2016, 7, 1978–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Fernández, L.; González, L.; Corral, I. An ab initio mechanism for efficient population of triplet states in cytotoxic sulfur substituted DNA bases: The case of 6-thioguanine. Chem. Commun. 2012, 48, 2134–2136. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, J.; Mazzone, G.; Russo, N.; Bertini, L. Photophysical properties of S, Se, and Te-substituted deoxyguanosines: Insight into their ability to act as chemotherapeutic agents. J. Chem. Inf. Model. 2017, 57, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Okabe, C.; Kobayashi, T.; Suzuki, T.; Ichimura, T.; Nishi, N.; Xu, Y.-Z. Ultrafast intersystem crossing of 4-thiothymidine in aqueous solution. J. Phys. Chem. Lett. 2010, 1, 480–484. [Google Scholar] [CrossRef]
- Mai, S.; Ashwood, B.; Marquetand, P.; Crespo-Hernández, C.E.; González, L. Solvatochromic effects on the absorption spectrum of 2-thiocytosine. J. Phys. Chem. B 2017, 121, 5187–5196. [Google Scholar] [CrossRef]
- Koyama, D.; Milner, M.J.; Orr-Ewing, A.J. Evidence for a double well in the first triplet excited state of 2-thiouracil. J. Phys. Chem. B 2017, 121, 9274–9280. [Google Scholar] [CrossRef]
- Zhao, G.J.; Han, K.L. Hydrogen bonding in the electronic excited state. Acc. Chem. Res. 2012, 45, 404–413. [Google Scholar] [CrossRef]
- Improta, R.; Barone, V. Excited States Behavior of Nucleobases in Solution: Insights from Computational Studies. Top. Curr. Chem. 2015, 355, 329–357. [Google Scholar]
- Improta, R.; Barone, V.; Lami, A.; Santoro, F. Quantum dynamics of the ultrafast pp*/np* population transfer in uracil and 5-fluoro-uracil in water and acetonitrile. J. Phys. Chem. B 2009, 113, 14491–14503. [Google Scholar] [CrossRef]
- Gustavsson, T.; Banyasz, A.; Lazzarotto, E.; Markovitsi, D.; Scalmani, G.; Frisch, M.J.; Barone, V.; Improta, R. Singlet excited-state behavior of uracil and thymine in aqueous solution: A combined experimental and computational study of 11 uracil derivatives. J. Am. Chem. Soc. 2006, 128, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, T.; Sarkar, N.; Bányász, A.; Markovitsi, D.; Improta, R. Solvent effects on the steady-state absorption and emission spectra of the three pyrimidine bases uracil, thymine and 5-fluorouracil. Photochem. Photobiol. 2007, 83, 595–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustavsson, T.; Sarkar, N.; Vayá, I.; Jiménez, M.C.; Markovitsi, D.; Improta, R. A joint experimental/theoretical study of the ultrafast excited state deactivation of deoxyadenosine and 9-methyladenine in water and acetonitrile. Photochem. Photobiol. Sci. 2013, 12, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C.; Wen, C.; Vogt, R.A.; Crespo-Hernández, C.E. Role of intersystem crossing in the fluorescence quenching of 2-aminopurine 2’-deoxyriboside in solution. Photochem. Photobiol. Sci. 2013, 12, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Taras-Goślińska, K.; Burdziński, G.; Wenska, G. Relaxation of the T1 excited state of 2-thiothymine, its riboside and deoxyriboside-enhanced nonradiative decay rate induced by sugar substituent. J. Photochem. Photobiol. A 2014, 275, 89–95. [Google Scholar] [CrossRef]
- Gustavsson, T.; Improta, R.; Markovitsi, D. DNA/RNA: Building blocks of life under UV irradiation. J. Phys. Chem. Lett. 2010, 1, 2025–2030. [Google Scholar] [CrossRef] [Green Version]
- Crespo-Hernández, C.E.; Cohen, B.; Hare, P.M.; Kohler, B. Ultrafast excited-state dynamics in nucleic acids. Chem. Rev. 2004, 104, 1977–2019. [Google Scholar] [CrossRef]
- Middleton, C.T.; de La Harpe, K.; Su, C.; Law, Y.K.; Crespo-Hernández, C.E.; Kohler, B. DNA excited-state dynamics: From single bases to the double helix. Annu. Rev. Phys. Chem. 2009, 60, 217–239. [Google Scholar] [CrossRef] [Green Version]
- Hare, P.M.; Crespo-Hernández, C.E.; Kohler, B. Internal conversion to the electronic ground state occurs via two distinct pathways for pyrimidine bases in aqueous solution. Proc. Natl. Acad. Sci. USA 2007, 104, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Hare, P.M.; Crespo-Hernández, C.E.; Kohler, B. Solvent-dependent photophysics of 1-cyclohexyluracil: Ultrafast branching in the initial bright state leads nonradiatively to the electronic ground state and a long-lived 1nπ* state. J. Phys. Chem. B 2006, 110, 18641–18650. [Google Scholar] [CrossRef]
- Hare, P.M.; Middleton, C.T.; Mertel, K.I.; Herbert, J.M.; Kohler, B. Time-resolved infrared spectroscopy of the lowest triplet state of thymine and thymidine. Chem. Phys. 2008, 347, 383–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brister, M.M.; Crespo-Hernández, C.E. Direct observation of triplet-state population dynamics in the RNA uracil derivative 1-cyclohexyluracil. J. Phys. Chem. Lett. 2015, 6, 4404–4409. [Google Scholar] [CrossRef] [PubMed]
- Brister, M.M.; Crespo-Hernández, C.E. Excited-state dynamics in the RNA nucleotide uridine 5’-Monophosphate investigated using femtosecond broadband transient absorption spectroscopy. J. Phys. Chem. Lett. 2019, 10, 2156–2161. [Google Scholar] [CrossRef] [PubMed]
- Pilles, B.M.; Maerz, B.; Chen, J.; Bucher, D.B.; Gilch, P.; Kohler, B.; Zinth, W.; Fingerhut, B.P.; Schreier, W.J. Decay pathways of thymine revisited. J. Phys. Chem. A 2018, 122, 4819–4828. [Google Scholar] [CrossRef]
- Gustavsson, T.; Sharonov, A.; Markovitsi, D. Thymine, thymidine and thymidine 5’-monophosphate studied by femtosecond fluorescence upconversion spectroscopy. Chem. Phys. Lett. 2002, 351, 195–200. [Google Scholar] [CrossRef]
- O’Connor, D.V.; Phillips, D. Time-correlated Single Photon Counting; Academic Press: London, UK, 1984. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thiobase | Fl Lifetime (ps) a λexc = 267 nm | TA Lifetime (ps) b λexc = 270 nm | Fl Lifetime (ps) c λexc = 362 nm | TA Lifetime (ps) d λexc = UVB/UVA |
|---|---|---|---|---|
| 2tCyt | 0.11 ± 0.02 | 0.21 ± 0.05 [35] | ||
| 2tCyd | 0.19 ± 0.07 (0.74) e 1.9 ± 0.7 (0.26) e | 0.20 ± 0.05 [36] | ||
| 6tGua | 0.27 ± 0.02 | 0.56 ± 0.06 [37] | ||
| 6tGuo | 0.19 ± 0.01 | 0.31 ± 0.05 [34] | ||
| 2tUra | 0.18 ± 0.02 | 0.167 [28] | 0.35 ± 0.06 [29] | |
| 2tUrd | 0.14 ± 0.02 (0.94) 4 ± 3 (0.06) e | 0.25 ± 0.04 [36] | ||
| 4tUra | 0.24 ± 0.02 | 0.24 ± 0.02 [30] 0.076 ± 0.016 [38] | ||
| 4tUrd | 0.26 ± 0.02 | |||
| 4tThd | 0.28 ± 0.03 (0.94) 12 ± 7 (0.06) e | 0.21 ± 0.04 [39] | 0.24 ± 0.08 (0.98) 2.6 ± 0.5 (0.02) e | 0.24 ± 0.02 [33] 0.25 ± 0.05 [39] |
| 2,4dtThy | 0.16 ± 0.02 | 0.16 ± 0.02 | 0.18 ± 0.04 [10] | |
| 2,4dtUra | 0.12 ± 0.02 | 0.136 [28], 0.217 [28] | 0.11 ± 0.01 | 0.22 ± 0.04 [30] |
| 2,4dtUrd | 0.14 ± 0.03 | 0.09 ± 0.01 |
| Thiobase | Fl Lifetime (ps) a λexc = 267 nm | TA Lifetime (ps) b λexc = 268/270 nm | Fl Lifetime (ps) c λexc = 362 nm | TA Lifetime (ps) d λexc = 316/340 nm |
|---|---|---|---|---|
| 2tUra | 0.14 ± 0.03 | 0.44 ± 0.03 [40] | 0.34 ± 0.09 [29] | |
| 4tUra | 0.41 ± 0.03 | 0.84 ± 0.03 | ||
| 4tUrd | 0.66 ± 0.03 | 0.72 ± 0.02 | ||
| 4tThd | 0.45 ± 0.02 | 0.19 ± 0.04 [39] 0.49 ± 0.09 [39] | 0.50 ± 0.03 | 0.54 ± 0.01 [32] |
| 2,4dtThy | 0.11 ± 0.02 | 0.15 ± 0.03 | ||
| 2,4dtUra | 0.16 ± 0.02 | 0.23 ± 0.03 | ||
| 2,4dtUrd | 0.17 ± 0.02 | 0.18 ± 0.03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brister, M.M.; Gustavsson, T.; Crespo-Hernández, C.E. Excited State Lifetimes of Sulfur-Substituted DNA and RNA Monomers Probed Using the Femtosecond Fluorescence Up-Conversion Technique. Molecules 2020, 25, 584. https://doi.org/10.3390/molecules25030584
Brister MM, Gustavsson T, Crespo-Hernández CE. Excited State Lifetimes of Sulfur-Substituted DNA and RNA Monomers Probed Using the Femtosecond Fluorescence Up-Conversion Technique. Molecules. 2020; 25(3):584. https://doi.org/10.3390/molecules25030584
Chicago/Turabian StyleBrister, Matthew M., Thomas Gustavsson, and Carlos E. Crespo-Hernández. 2020. "Excited State Lifetimes of Sulfur-Substituted DNA and RNA Monomers Probed Using the Femtosecond Fluorescence Up-Conversion Technique" Molecules 25, no. 3: 584. https://doi.org/10.3390/molecules25030584
APA StyleBrister, M. M., Gustavsson, T., & Crespo-Hernández, C. E. (2020). Excited State Lifetimes of Sulfur-Substituted DNA and RNA Monomers Probed Using the Femtosecond Fluorescence Up-Conversion Technique. Molecules, 25(3), 584. https://doi.org/10.3390/molecules25030584