
A Simple Zinc-Mediated Method for Selenium Addition to Michael Acceptors

 ,
,  ,
, 
Abstract
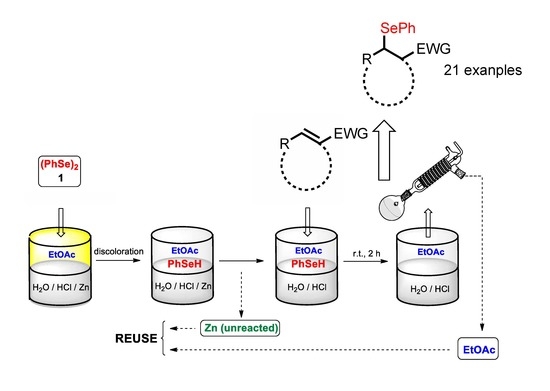
:
1. Introduction
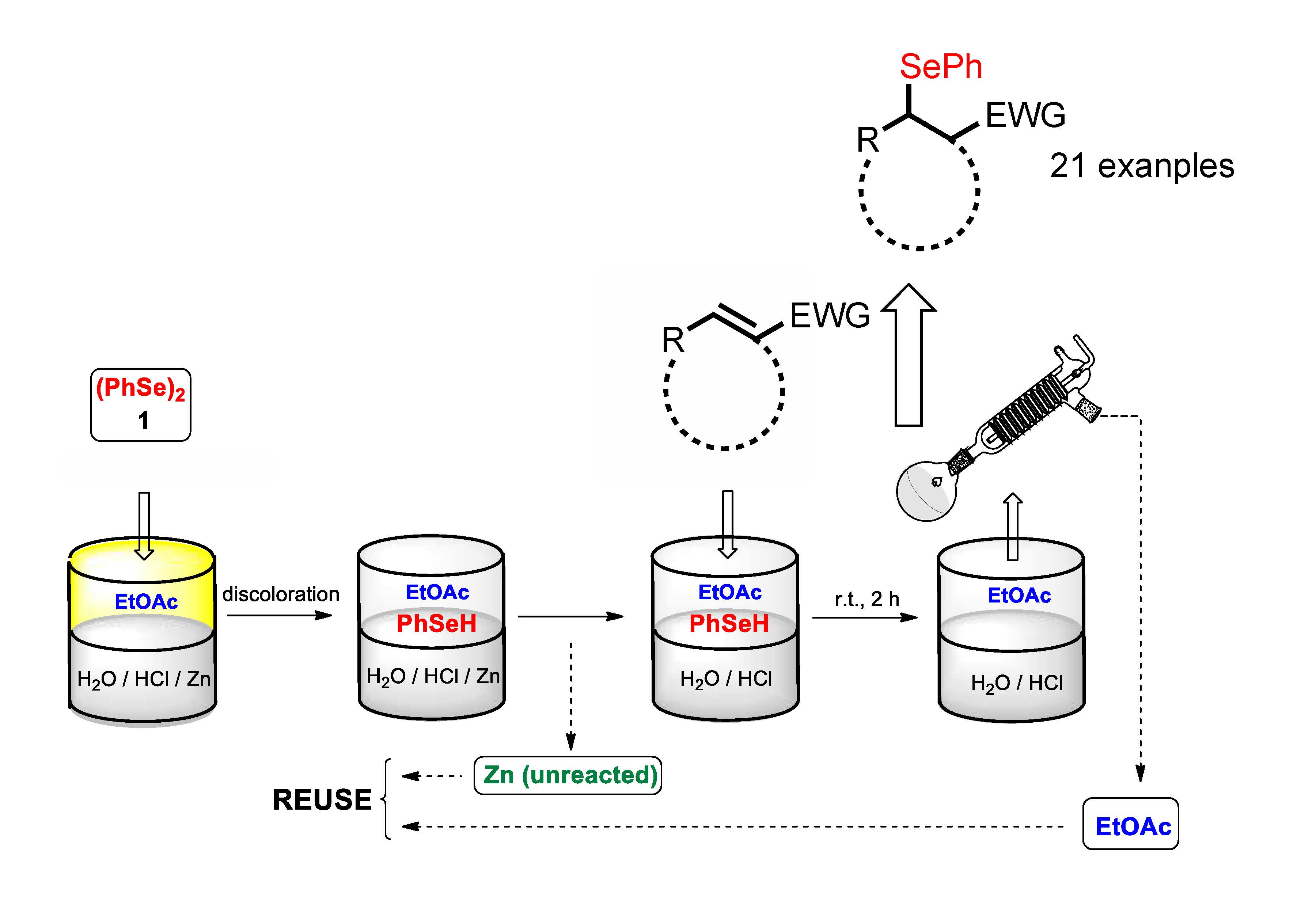
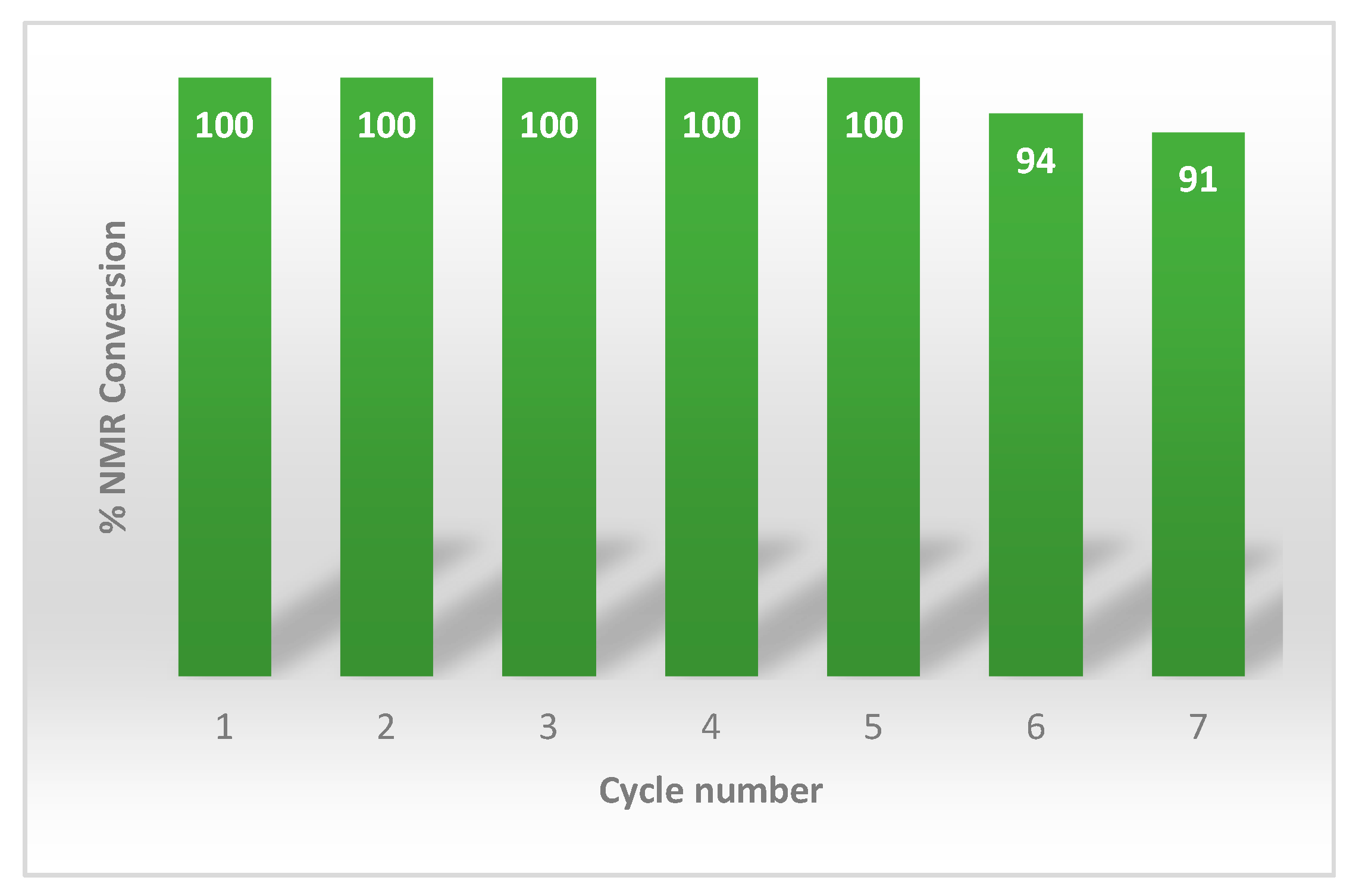
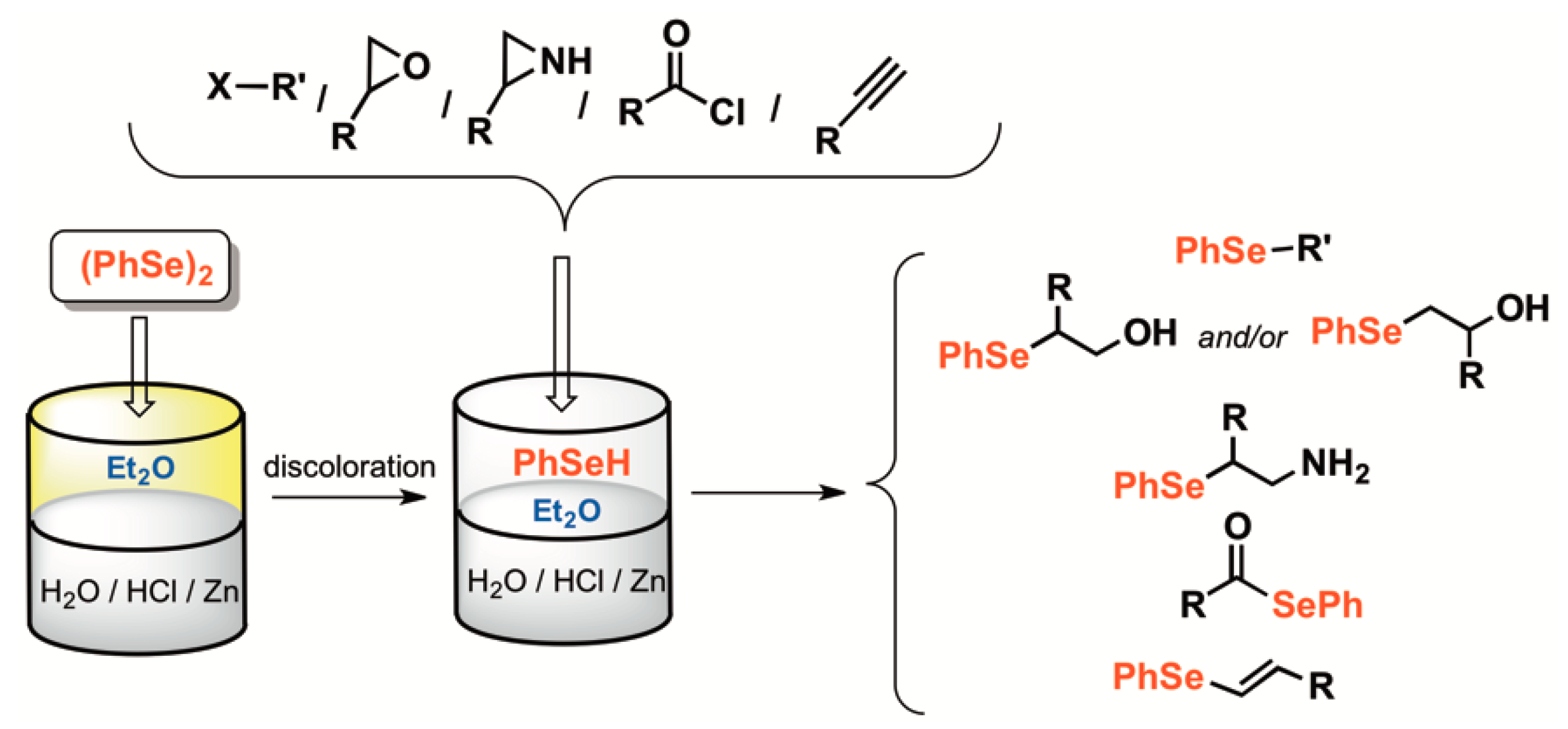
2. Results and Discussion
3. Materials and Methods
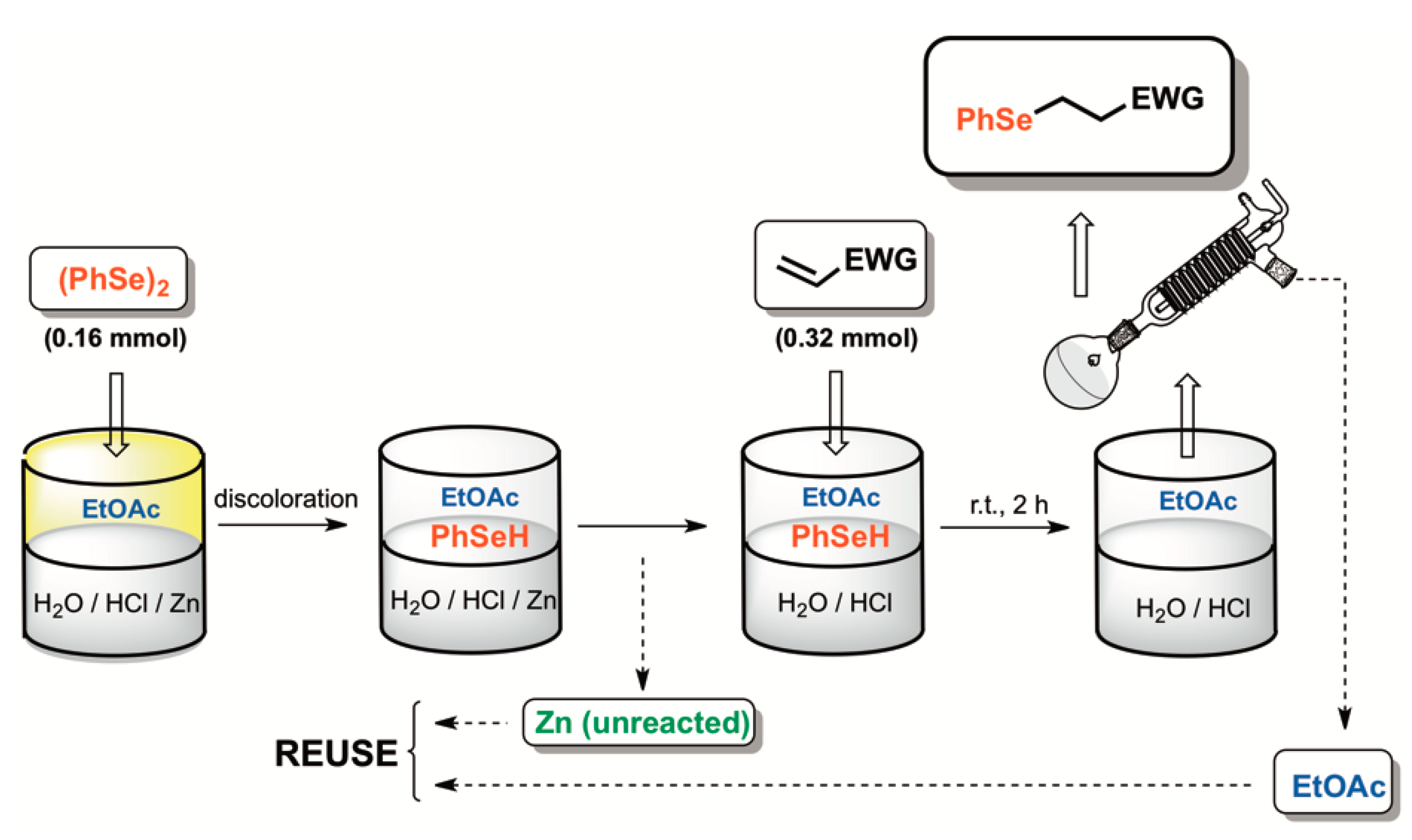
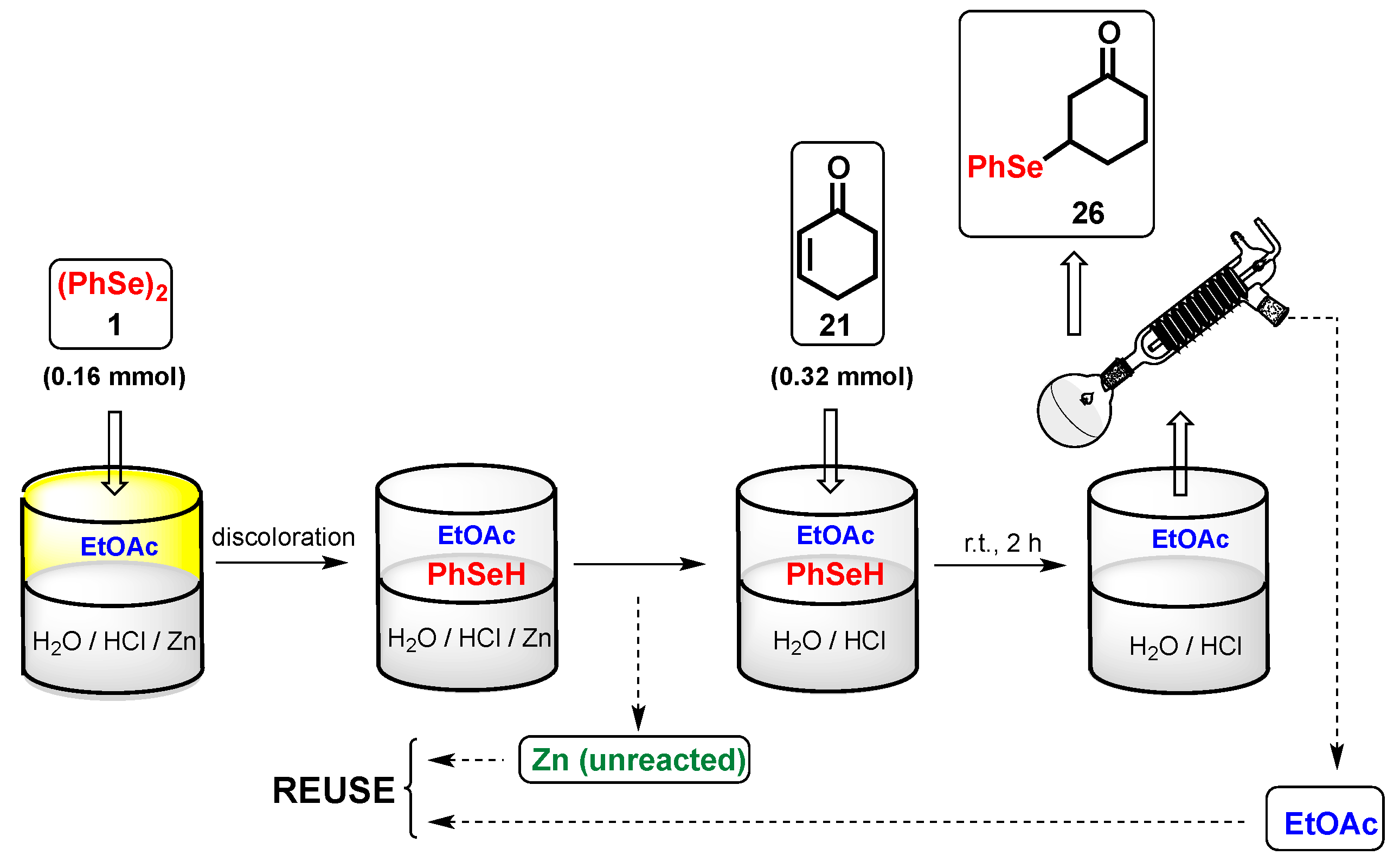
General Procedure for the Se-Michael-Type Addition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nicolaou, K.C.; Petasis, N.A. Selenium in Natural Products Synthesis; CIS: Philadelphia, PA, USA, 1984. [Google Scholar]
- Paulmier, C. Selenium Reagents and Intermediates in Organic Synthesis; Pergamon: Oxford, UK, 1986. [Google Scholar]
- Patai, S.; Rappoport, Z. The Chemistry of Organic Selenium and Tellurium Compounds; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Liotta, D. Organoselenium Chemistry; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Back, T.G. Organoselenium Chemistry: A Practical Approach; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Reich, H.J. Functional Group Manipulation Using Organoselenium Reagents. Acc. Chem. Res. 1979, 12, 22–30. [Google Scholar] [CrossRef]
- Liotta, D. New Organoselenium Methodology. Acc. Chem. Res. 1984, 17, 28–34. [Google Scholar] [CrossRef]
- Wirth, T. Organoselenium Chemistry–Modern Developments in Organic Synthesis, Topics in Current Chemistry; Spring: Heidelberg, Germany, 2000. [Google Scholar] [CrossRef]
- Mugesh, G.; Singh, H.B. Heteroatom-Directed Aromatic Lithiation: A Versatile Route to the Synthesis of Organochalcogen (Se, Te) Compounds. Acc. Chem. Res. 2002, 35, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Rigby, J.H.; Maharoof, U.S.M.; Mateo, M.E. Studies on the Narciclasine Alkaloids: Total Synthesis of (+)-Narciclasine and (+)-Pancratistatin. J. Am. Chem. Soc. 2000, 122, 6624–6628. [Google Scholar] [CrossRef]
- Knapp, S.; Zhao, D. Synthesis of the Sialidase Inhibitor Siastatin B. Org. Lett. 2000, 25, 4037–4040. [Google Scholar] [CrossRef]
- Treadwell, E.M.; Neighbors, J.D.; Wiemer, D.F. A Cascade Cyclization Approach to Schweinfurthin B. Org. Lett. 2002, 21, 3639–3642. [Google Scholar] [CrossRef]
- Hossain, S.U.; Sharma, A.K.; Ghosh, S.; Bhattacharya, S. Synthesis and Biological Evaluation of Novel Spiro 6-methoxytetralin-1,3′-pyrrolidine Based Organoselenocyanates Against Cadmium-induced Oxidative and Hepatic Damage in Mice. Eur. J. Med. Chem. 2010, 45, 3265–3273. [Google Scholar] [CrossRef]
- Jeong, L.S.; Choi, Y.N.; Tosh, D.K.; Choi, W.J.; Kim, H.O.; Choi, J.J. Design and Synthesis of Novel 2′,3′-dideoxy-4′-selenonucleosides as Potential Antiviral Agents. Bioorg. Med. Chem. 2008, 16, 9891–9897. [Google Scholar] [CrossRef]
- De Souza, D.; Mariano, D.O.; Nedel, F.; Schultze, E.; Campos, V.F.; Seixas, F.; da Silva, R.S.; Munchen, T.S.; Ilha, V.; Dornelles, L.; et al. New Organochalcogen Multitarget Drug: Synthesis and Antioxidant and Antitumoral Activities of Chalcogenozidovudine Derivatives. J. Med. Chem. 2015, 58, 3329–3339. [Google Scholar] [CrossRef]
- Pietrella, D. Antimicrobial activity of Organoselenium Compounds. In Organoselenium Chemistry: Between Synthesis and Biochemistry; Santi, C., Ed.; Bentham eBooks: Sharjah, UAE, 2014; pp. 328–344. [Google Scholar] [CrossRef] [Green Version]
- Legnaioli, S.; Piroddi, M.; Tidei, C.; Santi, C.; Galli, F. Targeting the GSTP-dependent Control of Cell Kinases and Apoptosis with PhSeZnCl: A New Seleno-organic Drug. Free Radical. Biol. Med. 2012, 53, S112–S113. [Google Scholar] [CrossRef]
- Sancineto, L.; Mariotti, A.; Bagnoli, L.; Marini, F.; Desantis, J.; Iraci, N.; Santi, C.; Pannecouque, C.; Tabarrini, O. Design and Synthesis of DiselenoBisBenzamides (DISeBAs) as Nucleocapsid Protein 7 (NCp7) Inhibitors with anti-HIV Activity. J. Med. Chem. 2015, 58, 9601–9614. [Google Scholar] [CrossRef]
- Krief, A.; Havesi, L. Organoselenium Chemistry; Springer: Berlin/Heidelberg, Germany, 1988. [Google Scholar]
- Krief, A. Alkylations of Sulfur- and Selenium-containing Carbanions. In Comprehensive Organometallic Chemistry; Trost, B.M., Fleming, I., Pattenden, G., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; pp. 85–191. [Google Scholar]
- Wirth, T. Selenium. In Comprehensive Organometallic Chemistry III; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 457–500. [Google Scholar]
- Zhang, Y.; Bao, W.; Zheng, Y. Synthesis of Allyl-Type Selenides and α-Selenoesters in Aqueous Media Promoted by Cadmium. Synth. Commun. 2000, 30, 1731–1736. [Google Scholar] [CrossRef]
- Sonoda, N.; Okada, M.; Kuroki, T.; Watanabe, T.; Nishiyama, Y.; Nishino, T. One-Pot Synthetic Method of Unsymmetrical Diorganyl Selenides: Reaction of Diphenyl Diselenide with Alkyl Halides in the Presence of Lanthanum Metal. J. Org. Chem. 2002, 67, 8696–8698. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Y.; Zhou, J.; Bao, W. A Novel Synthesis of Allyl and Propargyl Selenides in Aqueous Media Promoted by Indium. Tetrahedron Lett. 1996, 37, 9333–9334. [Google Scholar] [CrossRef]
- Mandal, T.; Samanta, S.; Ranu, B.C. Indium(I) Iodide-Mediated Cleavage of Diphenyl Diselenide. An Efficient One-Pot Procedure for the Synthesis of Unsymmetrical Diorganyl Selenides. Org. Lett. 2003, 5, 1439–1441. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Y.; Guo, H. Convenient One-Pot Synthesis of α-Selenonitriles, α-Selenoesters and Asymmetrical Selenides by a Sm/ZnCl2 system in DMF-H2O. J. Chem. Res. 2001, 160–161. [Google Scholar]
- Guo, H.; Zhang, Y.; Zheng, Y. Synthesis of Allyl Selenides Promoted by an Sm/ZnCl2 Bimetal System in the Presence of Water. Chin. J. Chem. 2001, 19, 530–532. [Google Scholar] [CrossRef]
- Fujinami, T.; Sakai, S.; Fukuzawa, S. Samarium(II) Di-iodide Induced Synthesis of Allylic Phenyl Selenides from Allylic Acetates and Diphenyl Diselenide in the Presence of Palladium Catalyst. Chem. Lett. 1990, 927–930. [Google Scholar] [CrossRef]
- Bao, W.; Zhang, Y.; Liao, P. A Novel Synthesis of Allyl and Prop-2-ynyl Selenides Promoted by Tin in the Presence of Water. J. Chem. Res. (S) 1998, 150–151. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, Y.; Li, Y.; Su, W. Facile Formation of Ytterbium Diiodide and Its Use in the Synthesis of Allyl Selenides. Chin. J. Chem. 2002, 20, 174–177. [Google Scholar] [CrossRef]
- Roy, S.; Kundu, A. Copper(II)/Tin(II) Reagent for Allylation, Propargylation, Alkynylation, and Benzylation of Diselenides: A Novel Bimetallic Reactivity. Organometallics 2000, 19, 105–107. [Google Scholar] [CrossRef]
- Reddy, K.M.; Mugesh, G. Application of Dehydroalanine as a Building Block for the Synthesis of Selenocysteine-containing Peptides. RSC Adv. 2019, 9, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Krief, A.; Cravador, A. Nouvelles Methods de Synthèse des Méthylsélénoacétals et Cétals. C. R. Acad. Sci. Paris 1979, 289c, 267–269. [Google Scholar]
- Katsumata, K.; Watanabe, Y.; Toru, T.; Veno, Y.; Sakakakibara, M. A Convenient Procedure for the Preparation of Organic Selenides. Synthesis 1992, 377–379. [Google Scholar] [CrossRef]
- Miyazaki, T.; Ando, M.; Matsumura, Y.; Sakane, S.; Hattori, K.; Yamamoto, H.; Maruoka, K. Organoaluminum-promoted Beckmann Rearrangement of Oxime Sulfonates. J. Am. Chem. Soc. 1983, 105, 2831–2843. [Google Scholar] [CrossRef]
- Berkowitz, D.B.; Pedersen, M.L. Formal α-vinylation of Amino Acids. Use of a New Benzeneselenolate Equivalent. J. Org. Chem. 1993, 58, 6966–6975. [Google Scholar] [CrossRef]
- Block, H.D.; Schmidt, M. Notiz zur Vereinfachten Darstellung von Alkanselenolen. Chem. Ber. 1970, 103, 3348–3349. [Google Scholar] [CrossRef]
- Mlochowsky, J.; Syper, L. The Convenient Syntheses of Organoselenium Reagents. Synthesis 1984, 439–442. [Google Scholar] [CrossRef]
- Dan, W.; Deng, H.; Chen, J.; Liu, M.; Ding, M.; Wu, H. A New Odorless One-pot Synthesis of Thioesters and Selenoesters Promoted by Rongalite. Tetrahedron 2010, 66, 7384–7388. [Google Scholar] [CrossRef]
- Lakouraj, M.M.; Movassagh, B.; Fadaei, Z. Convenient Synthesis of Thiol Esters from Acyl Chlorides and Disulfides Using Zn/AlCl3. Monatsh. Chem. 2002, 133, 1085–1088. [Google Scholar] [CrossRef]
- Movassagh, B.; Lakouraj, M.M.; Fadaei, Z. A Convenient One-pot Synthesis of Thiol Esters from Disulfides Using a Zn/AlCl3 System. J. Chem. Res. (S) 2001, 22–23. [Google Scholar] [CrossRef]
- Tian, F.S.; Zhu, Y.M.; Zhang, S.L.; Wang, Y.L. A Novel Method for the Synthesis of Unsymmetrical Sulfides, Thioesters and β-thioesters. J. Chem. Res. (S) 2002, 11, 582–583. [Google Scholar] [CrossRef]
- Movassagh, B.; Tatar, A. Zn/RuCl3-Promoted Cleavage of Diselenides: An Efficient Michael Addition of Zinc Selenolates to Conjugated Alkenes in Aqueous Media. Synlett 2007, 12, 1954–1956. [Google Scholar] [CrossRef]
- Zhou, L.-H.; Zhang, Y.-M. An Effective Synthesis of β- Selenium and β-Tellurium Carbonyl Compounds Via Reaction of Diaryldiselenides Or Diarylditellurides with α,β-Unsaturated Carbonyl Compounds Induced by Low-Valent Titanium. Synth. Commun. 1999, 29, 533–540. [Google Scholar] [CrossRef]
- Nunes, V.L.; de Oliveira, I.C.; do Rego Barros, O.S. Organylzinc Chalcogenolate Promoted Michael-Type Addition of α,β-Unsaturated Carbonyl Compounds. Eur. J. Org. Chem. 2014, 1525–1530. [Google Scholar] [CrossRef]
- Sancineto, L.; Vargas, J.P.; Monti, B.; Arca, M.; Lippolis, V.; Perin, G.; Lenardão, E.J.; Santi, C. Atom Efficient Preparation of Zinc Selenates for the Synthesis of Selenol Esters under “On Water” Conditions. Molecules 2017, 22, 953. [Google Scholar] [CrossRef] [Green Version]
- Santi, C.; Santoro, S.; Battistelli, B.; Testaferri, L.; Tiecco, M. Preparation of the First Bench-Stable Phenyl Selenolate: An Interesting “On Water” Nucleophilic Reagent. Eur. J. Org. Chem. 2008, 5387–5390. [Google Scholar] [CrossRef]
- Santoro, S.; Battistelli, B.; Testaferri, L.; Tiecco, M.; Santi, C. Vinylic Substitutions Promoted by PhSeZnCl: Synthetic and Theoretical Investigations. Eur. J. Org. Chem. 2009, 4921–4925. [Google Scholar] [CrossRef]
- Santi, C.; Battistelli, B.; Testaferri, L.; Tiecco, M. On Water Preparation of Phenylselenoesters. Green Chem. 2012, 14, 1277–1280. [Google Scholar] [CrossRef]
- Battistelli, B.; Testaferri, L.; Tiecco, M.; Santi, C. “On-Water” Michael-Type Addition Reactions Promoted by PhSeZnCl. Eur. J. Org. Chem. 2011, 1848–1851. [Google Scholar] [CrossRef]
- Salman, S.; Schwab, R.; Alberto, E.E.; Vargas, J.; Dornelles, L.; Rodrigues, O.E.D.; Braga, A.L. Efficient Ring Opening of Protected and Unprotected Aziridines Promoted by Stable Zinc Selenolate in Ionic Liquid. Synlett 2011, 69–72. [Google Scholar] [CrossRef]
- Nagasawa, T.; Shimada, N.; Torihata, M.; Kuwahara, S. Enantioselective Total Synthesis of Idesolide via NaHCO3-promoted Dimerization. Tetrahedron 2010, 66, 4965–4969. [Google Scholar] [CrossRef]
- Jiang, H.; Pan, X.; Li, N.; Zhang, Z.; Zhu, J.; Zhu, X. Selenide-containing High Refractive Index Polymer Material with Adjustable Refractive Index and Abbe’s Number. React. Funct. Polym. 2017, 111, 1–6. [Google Scholar] [CrossRef]
- Kim, Y.; Mulay, S.V.; Choi, M.; Yu, S.B.; Jon, S.; Churchill, D.G. Exceptional Time Response, Stability and Selectivity in Doubly-activated Phenyl Selenium-based Glutathione-selective Platform. Chem. Sci. 2015, 6, 5435–5439. [Google Scholar] [CrossRef] [Green Version]
- Jardim, G.A.M.; Bozzi, I.A.O.; Oliveira, W.X.C.; Mesquita-Rodrigues, C.; Menna-Barreto, R.F.S.; Kumar, R.A.; Gravel, E.; Doris, E.; Braga, A.L.; da Silva Júnior, E.N. Copper Complexes and Carbon nanotube–copper Ferrite-catalyzed Benzenoid A-ring Selenation of Quinones: An Efficient Method for the Synthesis of Trypanocidal agents. New J. Chem. 2019, 43, 13751–13763. [Google Scholar] [CrossRef]
- Santi, C.; Santoro, S.; Testaferri, L.; Tiecco, M. A Simple Zinc-Mediated Preparation of Selenols. Synlett 2008, 1471–1474. [Google Scholar] [CrossRef]
- Braga, A.L.; Schwab, R.S.; Alberto, E.E.; Salman, S.M.; Vargas, J.; Azeredo, J.B. Ring Opening of Unprotected Aziridines by Zinc Selenolates in a Biphasic System. Tetrahedron Lett. 2009, 50, 2309–2311. [Google Scholar] [CrossRef]
- Bellino, G.; Scisciani, M.; Vargas, J.P.; Sancineto, L.; Bagnoli, L.; Marini, L.; Lüdtke, D.S.; Lenardão, E.J.; Santi, C. Reaction of Acyl Chlorides with In Situ Formed Zinc Selenolates: Synthesis of Selenoesters versus Ring-Opening Reaction of Tetrahydrofuran. J. Chem. 2016, 1–8. [Google Scholar] [CrossRef]
- Flemer, S., Jr. A Comprehensive One-Pot Synthesis of Protected Cysteine and Selenocysteine SPPS Derivatives. Prot. Pept. Lett. 2014, 21, 1257–1264. [Google Scholar]
- Flemer, S., Jr. Fmoc-Sec(Xan)-OH: Synthesis and Utility of Fmoc Selenocysteine SPPS Derivatives with Acid-Labile Sidechain Protection. J. Pept. Sci. 2015, 21, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Tidei, C.; Sancineto, L.; Bagnoli, L.; Battistelli, B.; Marini, F.; Santi, C. A Recyclable Biphasic System for Stereoselective and Easily Handled Hydrochalcogenations. Eur. J. Org. Chem. 2014, 5968–5975. [Google Scholar] [CrossRef]
- Santi, C.; Jacob, R.G.; Monti, B.; Bagnoli, L.; Sancineto, L.; Lenardão, E.J. Water and Aqueous Mixtures as Convenient Alternative Media for Organoselenium Chemistry. Molecules 2016, 21, 1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, S.-C.; Chen, F.; Hou, D.; Kim-Meade, A.; Bernard, C.; Liu, J.; Levy, S.; Wu, G.G. A Novel Enantioselective Alkylation and Its Application to the Synthesis of an Anticancer Agent. J. Org. Chem. 2013, 68, 4984–4987. [Google Scholar] [CrossRef] [PubMed]
- Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, C.R.; Abou-Shehada, S.; Dunn, P.J. CHEM21 Selection Guide of Classical- and Less Classical-solvents. Green Chem. 2016, 18, 288–296. [Google Scholar] [CrossRef] [Green Version]
- Bickley, J.F.; Evans, P.; Meek, A.; Morgan, B.S.; Roberts, S.M. Novel Preparation of (−)-4-Hydroxycyclohex-2-enone: Reaction of 4-hydroxycyclohex-2-enone and 4-hydroxycyclopent-2-enone with some Thiols. Tetrahedron: Asymmetry 2006, 17, 355–362. [Google Scholar] [CrossRef]
- O’Byrne, A.; Murray, C.; Keegan, D.; Palacio, C.; Evans, P.; Morgan, B.S. The Thio-adduct Facilitated, Enzymatic Kinetic Resolution of 4-hydroxycyclopentenone and 4-hydroxycyclohexenone. Org. Biomol. Chem. 2010, 8, 539–545. [Google Scholar] [CrossRef]
- Johnson, C.R.; Braun, M.P. A Two-Step, Three-Component Synthesis of PGE1: Utilization of α-Iodoenones in Pd(0)-Catalyzed Cross-Couplings of Organoboranes. J. Am. Chem. Soc. 1993, 115, 11014–11015. [Google Scholar] [CrossRef]
- Dauvergne, J.; Happe, A.M.; Jadhav, V.; Justice, D.; Matos, M.-C.; McCormack, P.J.; Pitts, M.R.; Roberts, S.M.; Singh, S.K.; Snape, T.J.; et al. Synthesis of 4-azacyclopent-2-enones and 5,5-diakyl-4-azacyclopent-2-enones. Tetrahedron 2004, 60, 2559–2567. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, Z.; Zeng, F.; Chen, J.; Wu, X.; Wu, S.; Xiao, W.-J.; Zheng, Z. Highly Efficient Route to Diselenides from the Reactions of Imines and Selenium in the Presence of Carbon Monoxide and Water. Adv. Synth. Catal. 2005, 347, 877–882. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, F.; Cheng, R.; Li, S.; Rozovsky, S.; Wang, Q.; Wang, L. Site-Specific Incorporation of Selenocysteine Using an Expanded Genetic Code and Palladium-Mediated Chemical Deprotection. J. Am. Chem. Soc. 2018, 140, 8807–8816. [Google Scholar] [CrossRef]
- Chu, C.-M.; Gao, S.; Sastry, M.N.V.; Kuo, C.-W.; Lu, C.; Liu, J.-T.; Yao, C.-F. Ceric Ammonium Nitrate (CAN) as a Green and Highly Efficient Promoter for the 1,4-addition of Thiols and Benzeneselenol to α,β-Unsaturated Ketones. Tetrahedron 2007, 63, 1863–1871. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andrés, C.; Duque-Soladana, J.P.; Rosón, C.D. Regio- and Stereoselective 6-exo-trig Radical Cyclisations onto Chiral Perhydro-1,3-benzoxazines: Synthesis of Enantiopure 3-alkylpiperidines. Tetrahedron 2000, 11, 2809–2821. [Google Scholar] [CrossRef]
- Sanz, X.; Vogels, C.M.; Decken, A.; Bo, C.; Westcott, T.A.; Fernandez, E. Face to Face Activation of a Phenylselenium Borane with α,β-Unsaturated Carbonyl Substrates: Facile Synthesis of C–Se Bonds. Chem. Comm. 2014, 50, 8420–8423. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Asano, T.; Kishimoto, Y.; Itoh, K.; Ishii, Y. Selective Synthesis of 1-Alkoxy-3-phenylseleno-1-alkenes and 3-Phenylselenoalkanals by the Reaction of Diisobutylaluminum Phenylselenolate with α,β-Unsaturated Acetals. Tetrahedron Lett. 1998, 39, 8685–8686. [Google Scholar] [CrossRef]
- Bhalla, A.; Sharma, S.; Bhasin, K.K.; Bari, S.S. Convenient Preparation of Benzylseleno- and Phenylselenoalkanoic Acids: Reagents for Synthesis of Organoselenium Compounds. Synth. Commun. 2007, 37, 783–793. [Google Scholar] [CrossRef]
- Miyashita, M.; Toshikoshi, A. Facile and Highly Efficient Conjugate Addition of Benzeneselenol to α,β-Unsaturated Carbonyl Compounds. Synthesis 1980, 8, 664–666. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Substrate a | Se-Adduct | Yield b |
| 1 |  2 |  11 | 91% |
| 2 |  3 |  12 | 44% |
| 3 |  4 |  13 | 70% |
| 4 |  5 |  14 | 60% |
| 5 |  6 |  15 | 22% |
| 6 |  7 |  16 | 79% |
| 7 |  8 |  17 | 60% |
| 8 |  9 |  18 | 25% |
| 9 |  10 |  19 | 95% c (62:38) |
 | |||
|---|---|---|---|
| Entry | Substrate | Se-Adduct | Yield a |
| 1 |  20 |  25 | 93% |
| 2 |  21 |  26 | 88% |
| 3 |  22 |  27 | 95% |
| 4 |  23 |  28 | 90% b (75:25) |
| 5 |  24 |  29 | 29% c |
 | |||
|---|---|---|---|
| Entry | Substrate | Se-Adduct | Yield a |
| 1 |  21 |  33 | 65% |
| 2 |  32 |  34 | 90% b (89:11) |
| 3 |  2 |  35 | 30% |
| 4 |  21 |  36 | 53% c (66:34) |
| 5 |  23 |  37 | 30% c (55:45) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nacca, F.G.; Monti, B.; Lenardão, E.J.; Evans, P.; Santi, C. A Simple Zinc-Mediated Method for Selenium Addition to Michael Acceptors. Molecules 2020, 25, 2018. https://doi.org/10.3390/molecules25092018
Nacca FG, Monti B, Lenardão EJ, Evans P, Santi C. A Simple Zinc-Mediated Method for Selenium Addition to Michael Acceptors. Molecules. 2020; 25(9):2018. https://doi.org/10.3390/molecules25092018
Chicago/Turabian StyleNacca, Francesca Giulia, Bonifacio Monti, Eder João Lenardão, Paul Evans, and Claudio Santi. 2020. "A Simple Zinc-Mediated Method for Selenium Addition to Michael Acceptors" Molecules 25, no. 9: 2018. https://doi.org/10.3390/molecules25092018
APA StyleNacca, F. G., Monti, B., Lenardão, E. J., Evans, P., & Santi, C. (2020). A Simple Zinc-Mediated Method for Selenium Addition to Michael Acceptors. Molecules, 25(9), 2018. https://doi.org/10.3390/molecules25092018