
Synthesis, Biological Evaluation and In Silico Studies of Certain Oxindole–Indole Conjugates as Anticancer CDK Inhibitors

 ,
,  , and
, and
Abstract
:
1. Introduction
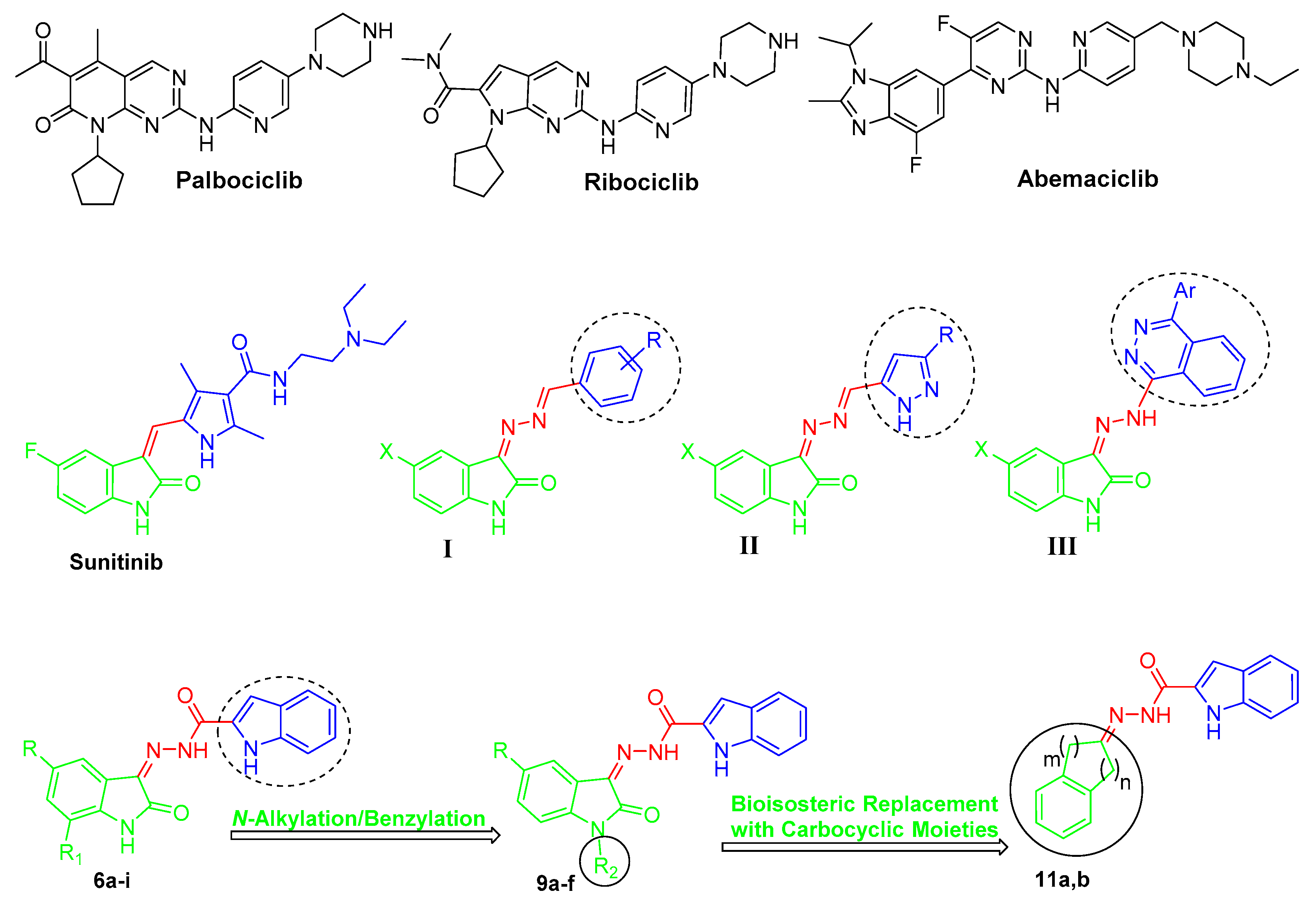
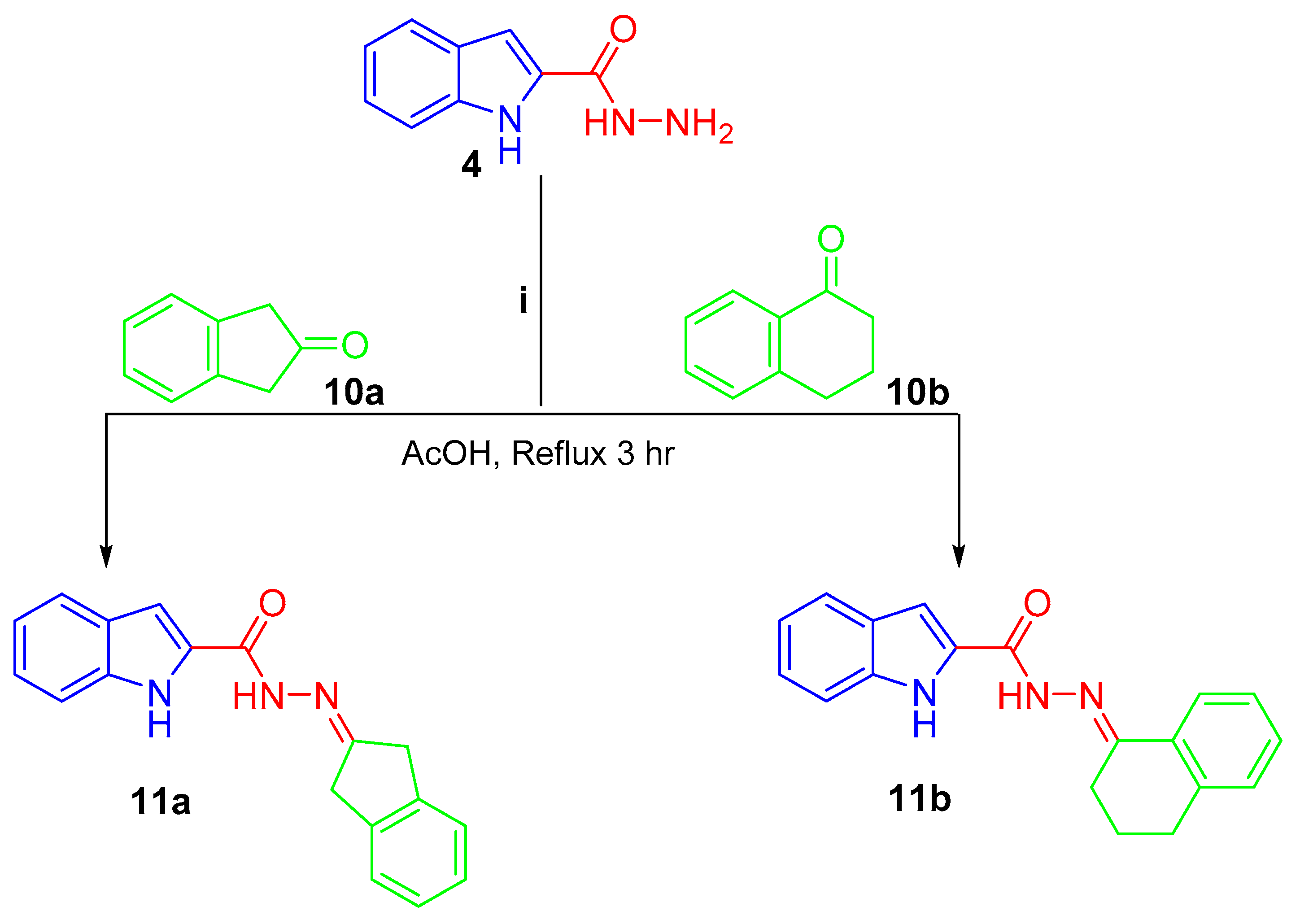
2. Results and Discussion
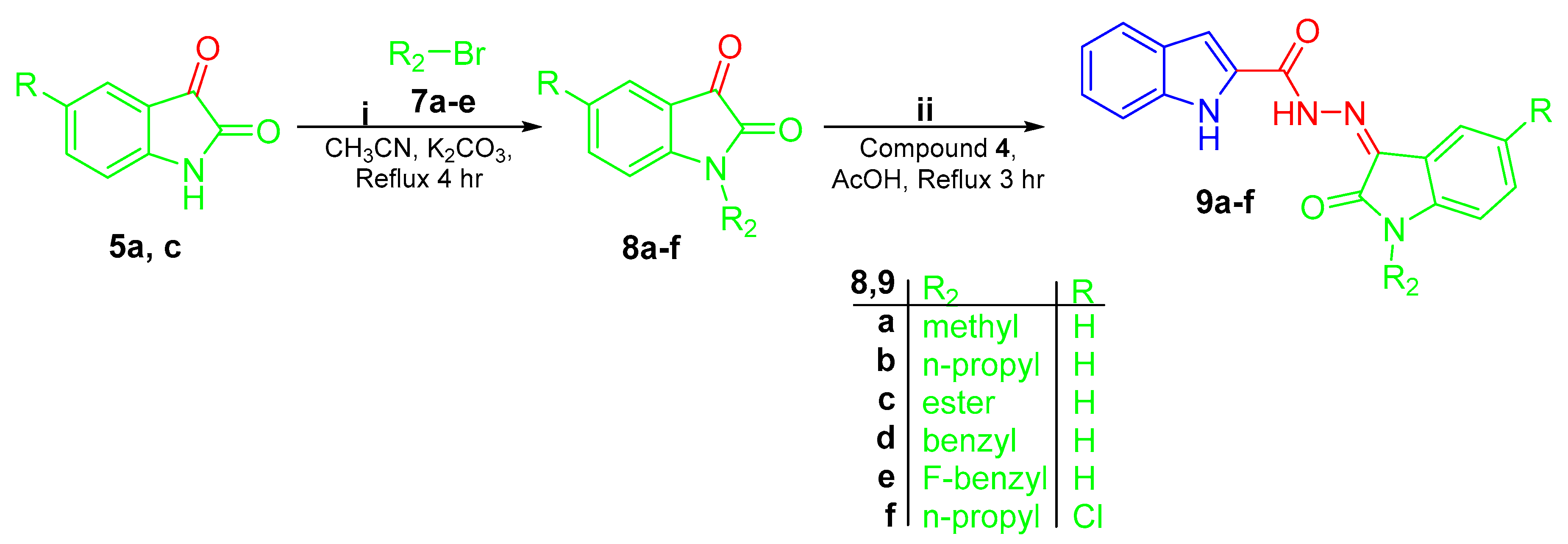
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Anti-Proliferative Activities towards Breast Cancer Cell Lines (MCF-7 and MDA-MB-231)
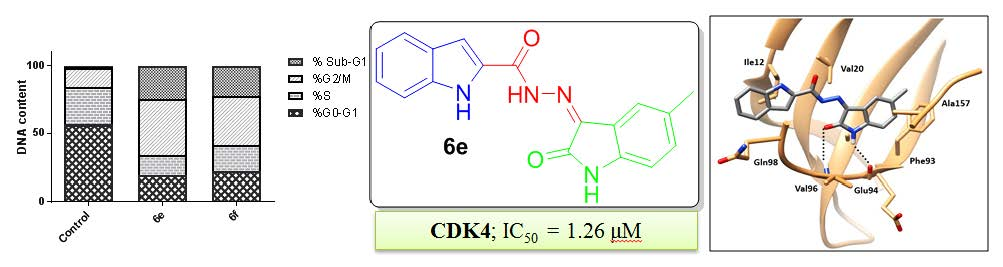
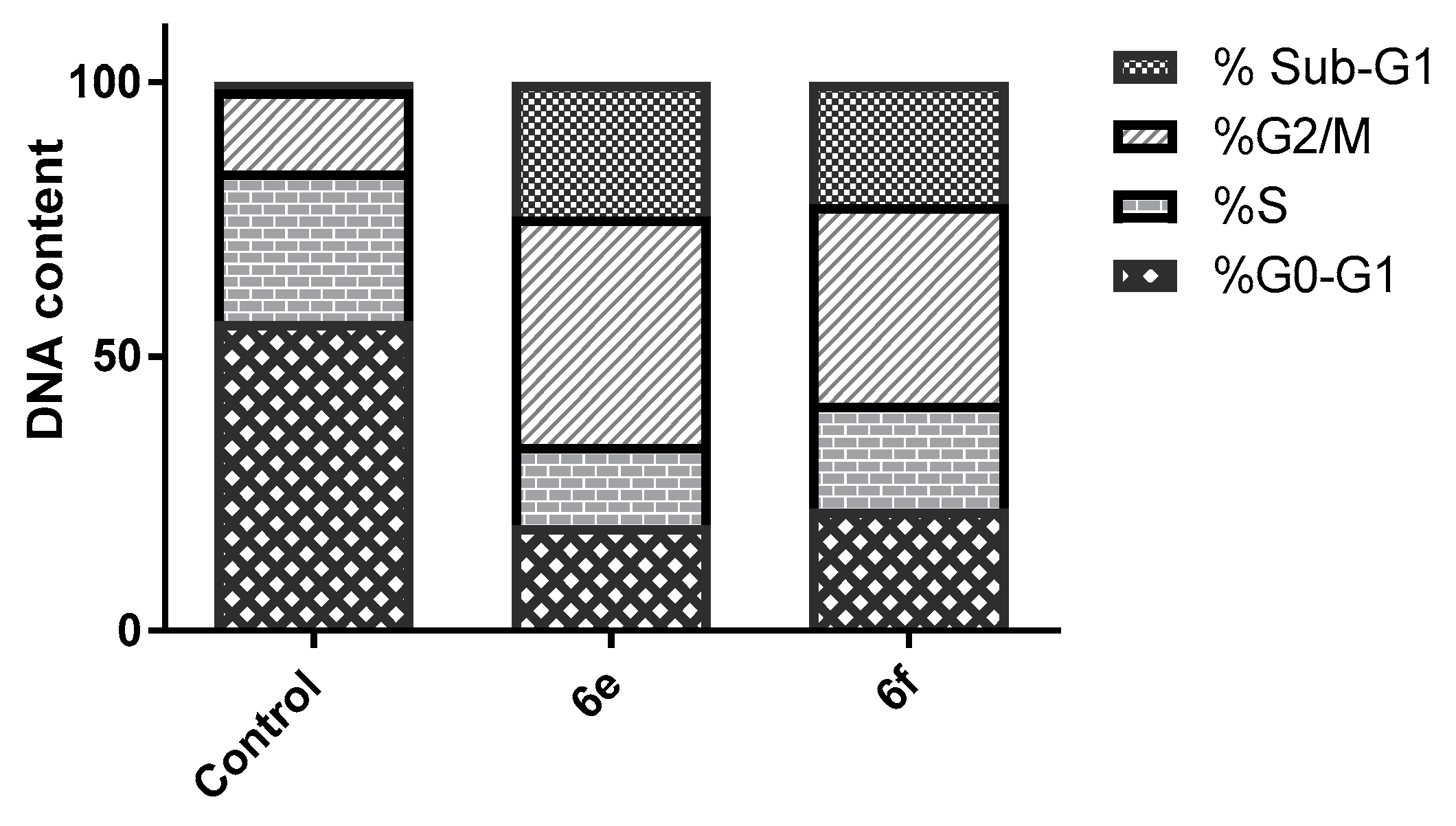
2.2.2. Cell Cycle Analysis
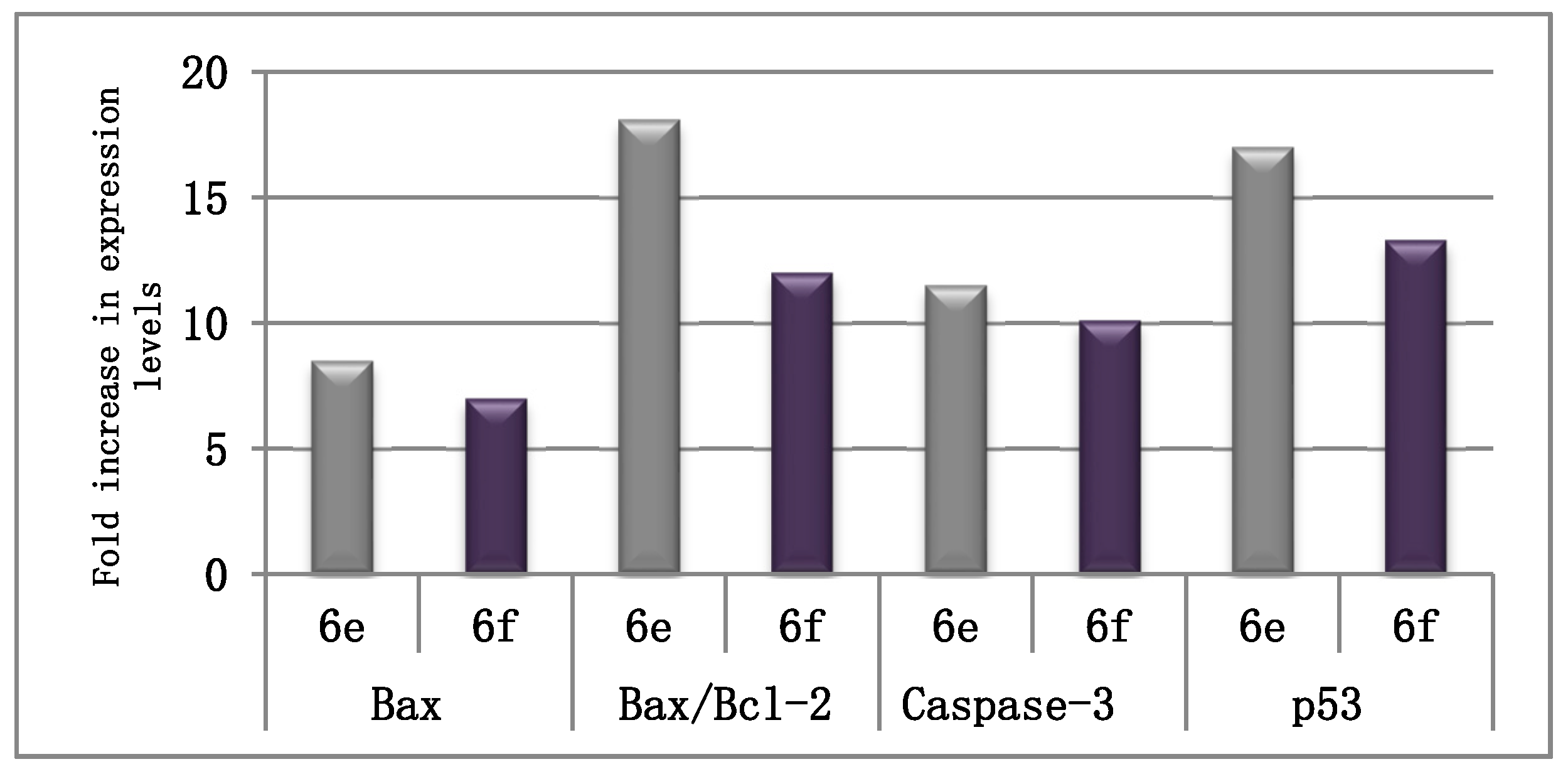
2.2.3. The Effect on the Apoptotic and Anti-Apoptotic Marker Levels
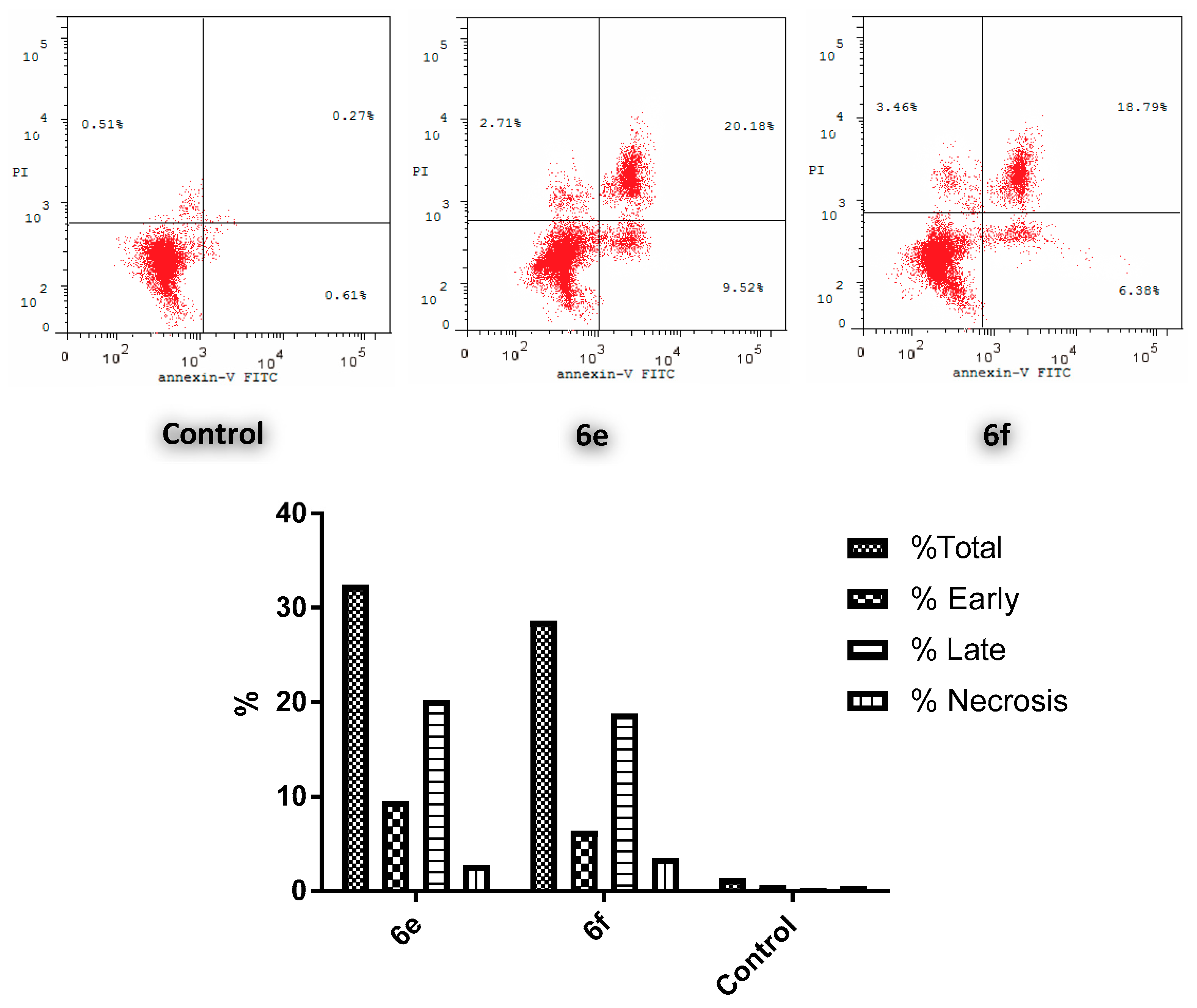
2.2.4. Annexin V-FITC (Anx V) Apoptosis Assay
2.2.5. CDK Inhibitory Activity
CDK4 Enzyme Assay
Screening of CDK2 and CDK9 Inhibitory Activities
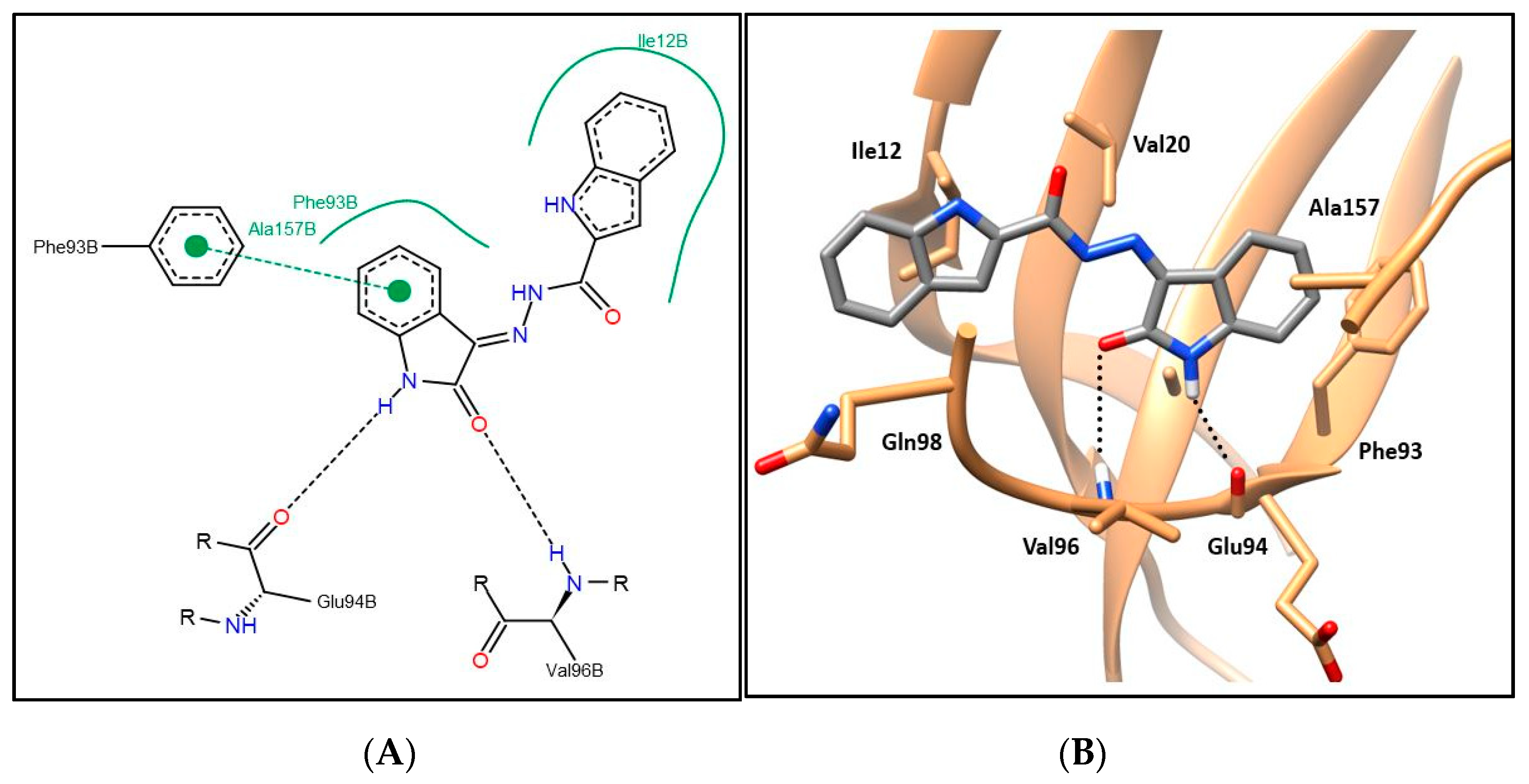
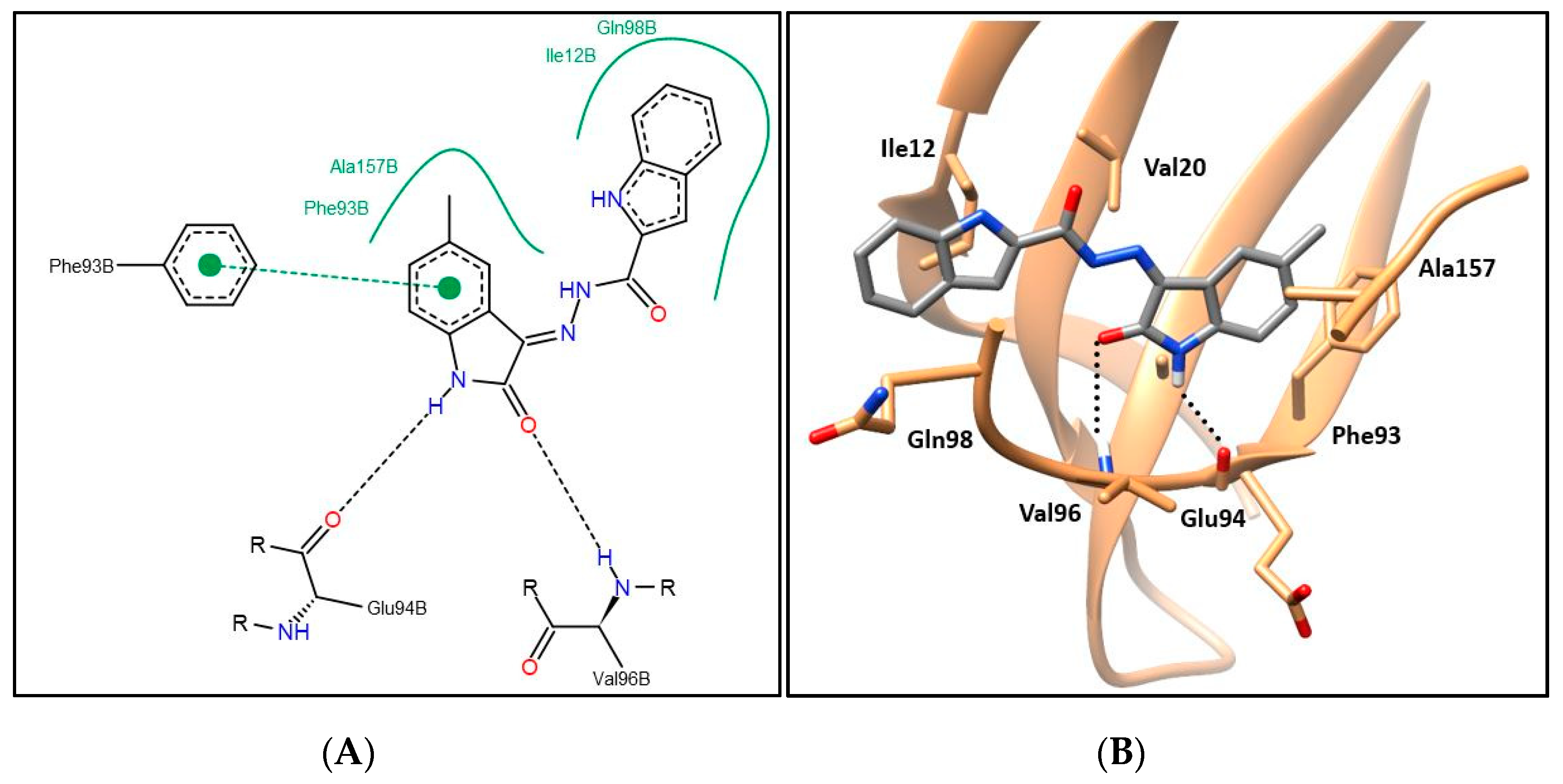
2.3. CDK4 Molecular Docking Study and Structure–Activity Relationship
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Preparation of Key Intermediate 1H-Indole-2-Carbohydrazide 4
3.1.3. Synthesis of Final Compounds 6a–i, 9a–f, and 11a,b
3.2. Biological Evaluation
3.2.1. Anti-Proliferative Activities Against Human Breast Cancer Cell Lines
3.2.2. Cell Cycle Analysis
3.2.3. Apoptosis study
3.2.4. CDK Kinase Inhibitory Activity
3.2.5. Molecular Modeling Study
Defining CDK4 Binding Site and Adjusting Its Shape and Topology
Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics 2016. CA: Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVita, V.T.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [Green Version]
- Padma, V.V. An overview of targeted cancer therapy. Biomedicine 2015, 5, 1–6. [Google Scholar] [CrossRef]
- Topcul, M.; Cetin, I. Endpoint of Cancer Treatment: Targeted Therapies. Asian Pac. J. Cancer Prev. 2014, 15, 4395–4403. [Google Scholar] [CrossRef] [Green Version]
- Figel, S.; Fenstermaker, R.A. Cell-cycle regulation. In Handbook of Brain Tumor Chemotherapy, Molecular Therapeutics, and Immunotherapy; Elsevier: London, UK, 2018; pp. 257–269. [Google Scholar]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.; Yang, Z.; Wang, S.; Li, Y.; Wei, H.; Tian, X.; Kan, Q. Recent development of CDK inhibitors: An overview of CDK/inhibitor co-crystal structures. Eur. J. Med. Chem. 2019, 164, 615–639. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Karim, S.S.; Syam, Y.M.; El Kerdawy, A.M.; Abdelghany, T.M. New thiazol-hydrazono-coumarin hybrids targeting human cervical cancer cells: Synthesis, CDK2 inhibition, QSAR and molecular docking studies. Bioorg. Chem. 2019, 86, 80–96. [Google Scholar] [CrossRef]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.; Cross, F.R. Multiple levels of cyclin specificity in cell-cycle control. Nat. Rev. Mol. Cell Boil. 2007, 8, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Ingham, M.; Schwartz, G.K. Cell-Cycle Therapeutics Come of Age. J. Clin. Oncol. 2017, 35, 2949–2959. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Beach, D.; I Shapiro, G. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2015, 6, 353–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.S.; Turner, N.C.; Yap, T.A. CDK4/6 inhibitors: promising opportunities beyond breast cancer. Cancer Discov. 2016, 6, 697–699. [Google Scholar] [CrossRef] [Green Version]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular pathways: Targeting the cyclin D–CDK4/6 axis for cancer treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parylo, S.; Vennepureddy, A.; Dhar, V.; Patibandla, P.; Sokoloff, A. Role of cyclin-dependent kinase 4/6 inhibitors in the current and future eras of cancer treatment. J. Oncol. Pharm. Pract. 2019, 25, 110–129. [Google Scholar] [CrossRef]
- Niu, Y.; Xu, J.; Sun, T. Cyclin-dependent kinases 4/6 inhibitors in breast cancer: Current status, resistance, and combination strategies. J. Cancer 2019, 10, 5504–5517. [Google Scholar] [CrossRef]
- Ribnikar, D.; Volovat, S.R.; Cardoso, F. Targeting CDK4/6 pathways and beyond in breast cancer. Breast 2019, 43, 8–17. [Google Scholar] [CrossRef]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 inhibitory strategy and combinations in breast cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, A.; Buneeva, O.; Gnedenko, O.V.; Ershov, P.V.; Ivanov, A. Isatin, an endogenous nonpeptide biofactor: A review of its molecular targets, mechanisms of actions, and their biomedical implications. BioFactors 2018, 44, 95–108. [Google Scholar] [CrossRef]
- Goodman, V.L.; Rock, E.P.; Dagher, R.; Ramchandani, R.P.; Abraham, S.; Gobburu, J.V.S.; Booth, B.P.; Verbois, S.L.; Morse, D.E.; Liang, C.Y.; et al. Approval Summary: Sunitinib for the Treatment of Imatinib Refractory or Intolerant Gastrointestinal Stromal Tumors and Advanced Renal Cell Carcinoma. Clin. Cancer Res. 2007, 13, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Dweedar, H.E.; Mahrous, H.; Ibrahim, H.; A Abdel-Aziz, H. Analogue-based design, synthesis and biological evaluation of 3-substituted-(methylenehydrazono)indolin-2-ones as anticancer agents. Eur. J. Med. Chem. 2014, 78, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, A.D.; Chang, P.-T.; Liu, J.-R.; Wang, H.-C.; Kondekar, N.B.; Shen, L.-J.; Tseng, H.-W.; Chen, G.S.; Chern, J.-W. Novel acylureidoindolin-2-one derivatives as dual Aurora B/FLT3 inhibitors for the treatment of acute myeloid leukemia. Eur. J. Med. Chem. 2014, 85, 268–288. [Google Scholar] [CrossRef]
- Laufer, R.; Forrest, B.; Li, S.-W.; Liu, Y.; Sampson, P.; Edwards, L.; Lang, Y.; Awrey, D.E.; Mao, G.; Plotnikova, O.; et al. The Discovery of PLK4 Inhibitors: (E)-3-((1H-Indazol-6-yl)methylene)indolin-2-ones as Novel Antiproliferative Agents. J. Med. Chem. 2013, 56, 6069–6087. [Google Scholar] [CrossRef]
- Lozinskaya, N.A.; Babkov, D.A.; Zaryanova, E.V.; Bezsonova, E.N.; Efremov, A.M.; Tsymlyakov, M.D.; Anikina, L.V.; Zakharyascheva, O.Y.; Borisov, A.V.; Perfilova, V.N.; et al. Synthesis and biological evaluation of 3-substituted 2-oxindole derivatives as new glycogen synthase kinase 3β inhibitors. Bioorganic Med. Chem. 2019, 27, 1804–1817. [Google Scholar] [CrossRef]
- Wang, H.-C.; Jagtap, A.D.; Chang, P.-T.; Liu, J.-R.; Liu, C.-P.; Tseng, H.-W.; Chen, G.S.; Chern, J.-W. Bioisosteric replacement of an acylureido moiety attached to an indolin-2-one scaffold with a malonamido or a 2/4-pyridinoylamido moiety produces a selectively potent Aurora-B inhibitor. Eur. J. Med. Chem. 2014, 84, 312–334. [Google Scholar] [CrossRef]
- Zhong, Y.; Xue, M.; Zhao, X.; Yuan, J.; Liu, X.; Huang, J.; Zhao, Z.; Li, H.; Xu, Y. Substituted indolin-2-ones as p90 ribosomal S6 protein kinase 2 (RSK2) inhibitors: Molecular docking simulation and structure–activity relationship analysis. Bioorganic Med. Chem. 2013, 21, 1724–1734. [Google Scholar] [CrossRef]
- Aneja, B.; Khan, N.S.; Khan, P.; Queen, A.; Hussain, A.; Rehman, T.; Alajmi, M.F.; El-Seedi, H.R.; Ali, S.; Hassan, I.; et al. Design and development of Isatin-triazole hydrazones as potential inhibitors of microtubule affinity-regulating kinase 4 for the therapeutic management of cell proliferation and metastasis. Eur. J. Med. Chem. 2019, 163, 840–852. [Google Scholar] [CrossRef]
- Hou, Y.; Shang, C.; Wang, H.; Yun, J. Isatin-azole hybrids and their anticancer activities. Arch. Pharm. 2019, 353, e1900272. [Google Scholar] [CrossRef]
- Ding, Z.; Zhou, M.; Zeng, C. Recent advances in isatin hybrids as potential anticancer agents. Arch. Pharm. 2020, 353, e1900367. [Google Scholar] [CrossRef]
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Attia, M.I.; Eldehna, W.M.; Afifi, S.A.; Keeton, A.B.; Piazza, G.; A Abdel-Aziz, H. New hydrazonoindolin-2-ones: Synthesis, exploration of the possible anti-proliferative mechanism of action and encapsulation into PLGA microspheres. PLoS ONE 2017, 12, e0181241. [Google Scholar] [CrossRef] [Green Version]
- Eldehna, W.M.; Al-Wabli, R.I.; Almutairi, M.S.; Keeton, A.B.; Piazza, G.A.; A Abdel-Aziz, H.; Attia, M.I. Synthesis and biological evaluation of certain hydrazonoindolin-2-one derivatives as new potent anti-proliferative agents. J. Enzym. Inhib. Med. Chem. 2018, 33, 867–878. [Google Scholar] [CrossRef] [PubMed]
- A Abdel-Aziz, H.; Eldehna, W.M.; Keeton, A.B.; Piazza, G.; A Kadi, A.; Attwa, M.W.; Abdelhameed, A.S. Isatin-benzoazine molecular hybrids as potential antiproliferative agents: synthesis and in vitro pharmacological profiling. Drug Des. Dev. Ther. 2017, 11, 2333–2346. [Google Scholar] [CrossRef] [Green Version]
- Eldehna, W.M.; Almahli, H.; Al-Ansary, G.H.; Ghabbour, H.A.; Aly, M.H.; Ismael, O.E.; Al-Dhfyan, A.; A Abdel-Aziz, H. Synthesis and in vitro anti-proliferative activity of some novel isatins conjugated with quinazoline/phthalazine hydrazines against triple-negative breast cancer MDA-MB-231 cells as apoptosis-inducing agents. J. Enzym. Inhib. Med. Chem. 2017, 32, 600–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Naggar, M.; Eldehna, W.M.; Almahli, H.; Elgez, A.; Fares, M.; Elaasser, M.; A Abdel-Aziz, H. Novel Thiazolidinone/Thiazolo[3,2-a]Benzimidazolone-Isatin Conjugates as Apoptotic Anti-proliferative Agents Towards Breast Cancer: One-Pot Synthesis and In Vitro Biological Evaluation. Molecules 2018, 23, 1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldehna, W.M.; Nocentini, A.; Al-Rashood, S.T.; Hassan, G.S.; Alkahtani, H.M.; Almehizia, A.A.; Reda, A.M.; Abdel-Aziz, H.A.; Supuran, C.T. Tumor-associated carbonic anhydrase isoform IX and XII inhibitory properties of certain isatin-bearing sulfonamides endowed with in vitro antitumor activity towards colon cancer. Bioorg. Chem. 2018, 81, 425–432. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Abo-Ashour, M.F.; Nocentini, A.; El-Haggar, R.S.; Bua, S.; Bonardi, A.; Al-Rashood, S.T.; Hassan, G.S.; Gratteri, P.; A Abdel-Aziz, H.; et al. Enhancement of the tail hydrophobic interactions within the carbonic anhydrase IX active site via structural extension: Design and synthesis of novel N-substituted isatins-SLC-0111 hybrids as carbonic anhydrase inhibitors and antitumor agents. Eur. J. Med. Chem. 2019, 162, 147–160. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Fares, M.; Ibrahim, H.; Aly, M.H.; Zada, S.; Ali, M.M.; Abou-Seri, S.M.; A Abdel-Aziz, H.; El Ella, D.A.A.; Ali, M.M. Indoline ureas as potential anti-hepatocellular carcinoma agents targeting VEGFR-2: Synthesis, in vitro biological evaluation and molecular docking. Eur. J. Med. Chem. 2015, 100, 89–97. [Google Scholar] [CrossRef]
- Eldehna, W.M.; El Kerdawy, A.M.; Al-Ansary, G.H.; Al-Rashood, S.T.; Ali, M.M.; Mahmoud, A.E. Type IIA-Type IIB protein tyrosine kinase inhibitors hybridization as an efficient approach for potent multikinase inhibitor development: Design, synthesis, anti-proliferative activity, multikinase inhibitory activity and molecular modeling of novel indolinone-based ureides and amides. Eur. J. Med. Chem. 2019, 163, 37–53. [Google Scholar] [CrossRef]
- Giancotti, G.; Cancellieri, M.; Balboni, A.; Giustiniano, M.; Novellino, E.; Delang, L.; Neyts, J.; Leyssen, P.; Brancale, A.; Bassetto, M. Rational modifications on a benzylidene-acrylohydrazide antiviral scaffold, synthesis and evaluation of bioactivity against Chikungunya virus. Eur. J. Med. Chem. 2018, 149, 56–68. [Google Scholar] [CrossRef]
- Skehan, P.; Scudiero, M.; Vistica, D.; Bokesch, H.; Kenney, S.; Storeng, R.; Monks, A.; McMahon, J.; Warren, J.T.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Day, P.J.; Cleasby, A.; Tickle, I.; O’Reilly, M.; Coyle, J.E.; Holding, F.P.; McMenamin, R.L.; Yon, J.; Chopra, R.; Lengauer, C.; et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc. Natl. Acad. Sci. USA 2009, 106, 4166–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, P.; Chashoo, G.; Gupta, M.; Kumar, A.; Singh, P.P.; Nargotra, A. Fusion of Structure and Ligand Based Methods for Identification of Novel CDK2 Inhibitors. J. Chem. Inf. Model. 2017, 57, 1957–1969. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, M.; Kamata, K.; Fukasawa, K.; Honma, T.; Machida, T.; Hirai, H.; Suzuki-Takahashi, I.; Hayama, T.; Nishimura, S. Crystallographic Approach to Identification of Cyclin-dependent Kinase 4 (CDK4)-specific Inhibitors by Using CDK4 Mimic CDK2 Protein. J. Boil. Chem. 2001, 276, 27548–27554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldehna, W.M.; Hassan, G.S.; Al-Rashood, S.T.; Alkahtani, H.M.; A Almehizia, A.; Al-Ansary, G.H. Marine-Inspired Bis-indoles Possessing Antiproliferative Activity against Breast Cancer; Design, Synthesis, and Biological Evaluation. Mar. Drugs 2020, 18, 190. [Google Scholar] [CrossRef] [Green Version]
- Abo-Ashour, M.F.; Eldehna, W.M.; Nocentini, A.; Bonardi, A.; Bua, S.; Ibrahim, H.; Elaasser, M.M.; Kryštof, V.; Jorda, R.; Gratteri, P.; et al. 3-Hydrazinoisatin-based benzenesulfonamides as novel carbonic anhydrase inhibitors endowed with anticancer activity: Synthesis, in vitro biological evaluation and in silico insights. Eur. J. Med. Chem. 2019, 184, 111768. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, N.M.; Noel, B.; Shah, K. A series of 2-arylamino-5-(indolyl)-1,3,4-thiadiazoles as potent cytotoxic agents. Eur. J. Med. Chem. 2012, 55, 432–438. [Google Scholar] [CrossRef]
- Al-Wabli, R.I.; AlMomen, A.A.; Almutairi, M.S.; Keeton, A.B.; Piazza, G.A.; Attia, M.I. New Isatin-Indole Conjugates: Synthesis, Characterization, and a Plausible Mechanism of Their in vitro Antiproliferative Activity. Drug Des. Dev. Ther. 2020, 14, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Sabt, A.; Abdelhafez, O.M.; El-Haggar, R.S.; Madkour, H.M.F.; Eldehna, W.M.; El-Khrisy, E.E.-D.A.M.; Abdel-Rahman, M.A.; Rashed, L.A. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: synthesis, in vitro biological evaluation, and QSAR studies. J. Enzym. Inhib. Med. Chem. 2018, 33, 1095–1107. [Google Scholar] [CrossRef] [Green Version]
- A Abdel-Aziz, H.; Ghabbour, H.A.; Eldehna, W.M.; Qabeel, M.M.; Fun, H.K. Synthesis, Crystal Structure, and Biological Activity of cis/trans Amide Rotomers of (Z)- N′-(2-Oxoindolin-3-ylidene)formohydrazide. J. Chem. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Abo-Ashour, M.F.; Ibrahim, H.; Al-Ansary, G.H.; Ghabbour, H.A.; Elaasser, M.M.; Ahmed, H.; Safwat, N.A. Novel [(3-indolylmethylene)hydrazono]indolin-2-ones as apoptotic anti-proliferative agents: design, synthesis andin vitrobiological evaluation. J. Enzym. Inhib. Med. Chem. 2018, 33, 686–700. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Discovery Studio visualized 2017R2; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2016.
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. OMEGA 3.0.0.1; OpenEye Scientific Software: Santa Fe, NM, USA, 2018. [Google Scholar]
- Hawkins, P.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- OEDOCKING 3.2.0.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2018.
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible Shape-Guided Docking For Pose Prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef]
- Stierand, K.; Rarey, M. Drawing the PDB: Protein−Ligand Complexes in Two Dimensions. ACS Med. Chem. Lett. 2010, 1, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Stierand, K.; Rarey, M. From Modeling to Medicinal Chemistry: Automatic Generation of Two-Dimensional Complex Diagrams. ChemMedChem 2007, 2, 853–860. [Google Scholar] [CrossRef]
- Stierand, K.; Rarey, M. PoseView v1.1.2; BioSolveIT GmbH: St. Augustin, Germany, 2010. [Google Scholar]
- Stierand, K.; Maaß, P.C.; Rarey, M. Molecular complexes at a glance: automated generation of two-dimensional complex diagrams. Bioinform. 2006, 22, 1710–1716. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds (6a–i, 9a–f, and 11a,b) are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Comp. | R | R1 | R2 | IC50 (µM)a | |
|---|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | ||||
| 6a | H | H | - | 3.12 ± 0.14 | 15.10 ± 0.73 |
| 6b | F | H | - | 6.0 ± 0.32 | 6.36 ± 0.29 |
| 6c | Cl | H | - | 2.72 ± 0.17 | 9.48 ± 0.44 |
| 6d | Br | H | - | 3.31 ± 0.11 | 18.33 ± 0.71 |
| 6e | CH3 | H | - | 0.39 ± 0.05 | 22.54 ± 1.67 |
| 6f | OCH3 | H | - | 1.85 ± 0.08 | 2.85 ± 0.9 |
| 6g | OCF3 | H | - | 5.07 ± 0.21 | 7.19 ± 0.38 |
| 6h | NO2 | H | - | 21.40 ± 1.58 | 1.85 ± 0.07 |
| 6i | CH3 | CH3 | - | 47.16 ± 3.02 | NAb |
| 9a | H | - | -CH3 | 5.50 ± 0.28 | 8.39 ± 0.27 |
| 9b | H | - | -CH2CH2CH3 | 40.89 ± 3.17 | 18.50 ± 0.85 |
| 9c | H | - | -CH2COOC2H5 | 67.51 ± 3.02 | NAb |
| 9d | H | - | -CH2C6H5 | 9.56 ± 0.61 | 6.50 ± 0.39 |
| 9e | H | - | -CH2C6H4-4-F | 3.70 ± 0.19 | 1.03 ± 0.04 |
| 9f | Cl | - | -CH2CH2CH3 | 19.26 ± 0.65 | 9.44 ± 0.35 |
| 11a | - | - | - | 84.70 ± 4.02 | NAb |
| 11b | - | - | - | NAb | NAb |
| Staurosporine | - | - | - | 6.81 ± 0.22 | 10.29 ± 0.72 |
| Comp. | %G0–G1 | %S | %G2/M | %Sub-G1 |
|---|---|---|---|---|
| 6e | 19.23 | 14.81 | 41.47 | 24.49 |
| 6f | 22.19 | 19.29 | 36.26 | 22.26 |
| Control | 56.45 | 27.45 | 14.73 | 1.37 |
| Cpd. | Bax (pg/mg of Total Protein) | Bcl-2 (ng/mg of Total Protein) | Bax/Bcl-2 |
|---|---|---|---|
| 6e | 325.3 ± 12.1 | 2.19 ± 0.10 | 148.5 |
| 6f | 268.7 ± 9.5 | 2.73 ± 0.13 | 98.6 |
| Control | 38.3 ± 2.2 | 4.65 ± 0.23 | 8.2 |
| Cpd. | Caspase-3 (pg/mg) | p53 (pg/mg) |
|---|---|---|
| 6e | 411.70 ± 12.3 | 704.39 ± 32.8 |
| 6f | 364.15 ± 14.8 | 547.60 ± 29.5 |
| Control | 35.92 ± 1.8 | 41.26 ± 2.7 |
| Comp. | CDK4 | |
|---|---|---|
| % Inhibition | IC50 (µM) | |
| 6a | 92 | 1.82 |
| 6b | 33 | - |
| 6c | 28 | - |
| 6d | 43 | - |
| 6e | 93 | 1.26 |
| 6f | 14 | - |
| 6g | 19 | - |
| 6h | 46 | - |
| 9a | 20 | - |
| 9e | 12 | - |
| Staurosporine | 99 | 0.017 |
| Comp. | % Enzyme Inhibitory Activity | |
|---|---|---|
| CDK2 | CDK9 | |
| 6a | 78 | 20 |
| 6b | 48 | 7 |
| 6c | 57 | 11 |
| 6d | 85 | 32 |
| 6e | 68 | 18 |
| 6f | 50 | 8 |
| 6g | 14 | 4 |
| 6h | 67 | 10 |
| 9a | 41 | 13 |
| 9e | 5 | 9 |
| Staurosporine | 99 | 98 |
| Comp. | Energy Score (S) kcal/mol |
|---|---|
| 6a | −8.60 |
| 6b | −8.43 |
| 6c | −8.27 |
| 6d | −7.83 |
| 6e | −8.36 |
| 6f | −7.62 |
| 6g | −6.61 |
| 6h | −7.65 |
| 6i | −8.04 |
| Oxindole ligand | −9.72 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Warhi, T.; El Kerdawy, A.M.; Aljaeed, N.; Ismael, O.E.; Ayyad, R.R.; Eldehna, W.M.; Abdel-Aziz, H.A.; Al-Ansary, G.H. Synthesis, Biological Evaluation and In Silico Studies of Certain Oxindole–Indole Conjugates as Anticancer CDK Inhibitors. Molecules 2020, 25, 2031. https://doi.org/10.3390/molecules25092031
Al-Warhi T, El Kerdawy AM, Aljaeed N, Ismael OE, Ayyad RR, Eldehna WM, Abdel-Aziz HA, Al-Ansary GH. Synthesis, Biological Evaluation and In Silico Studies of Certain Oxindole–Indole Conjugates as Anticancer CDK Inhibitors. Molecules. 2020; 25(9):2031. https://doi.org/10.3390/molecules25092031
Chicago/Turabian StyleAl-Warhi, Tarfah, Ahmed M. El Kerdawy, Nada Aljaeed, Omnia E. Ismael, Rezk R. Ayyad, Wagdy M. Eldehna, Hatem A. Abdel-Aziz, and Ghada H. Al-Ansary. 2020. "Synthesis, Biological Evaluation and In Silico Studies of Certain Oxindole–Indole Conjugates as Anticancer CDK Inhibitors" Molecules 25, no. 9: 2031. https://doi.org/10.3390/molecules25092031
APA StyleAl-Warhi, T., El Kerdawy, A. M., Aljaeed, N., Ismael, O. E., Ayyad, R. R., Eldehna, W. M., Abdel-Aziz, H. A., & Al-Ansary, G. H. (2020). Synthesis, Biological Evaluation and In Silico Studies of Certain Oxindole–Indole Conjugates as Anticancer CDK Inhibitors. Molecules, 25(9), 2031. https://doi.org/10.3390/molecules25092031