
Free Energies of Hydrated Halide Anions: High Through-Put Computations on Clusters to Treat Rough Energy-Landscapes



Abstract
:
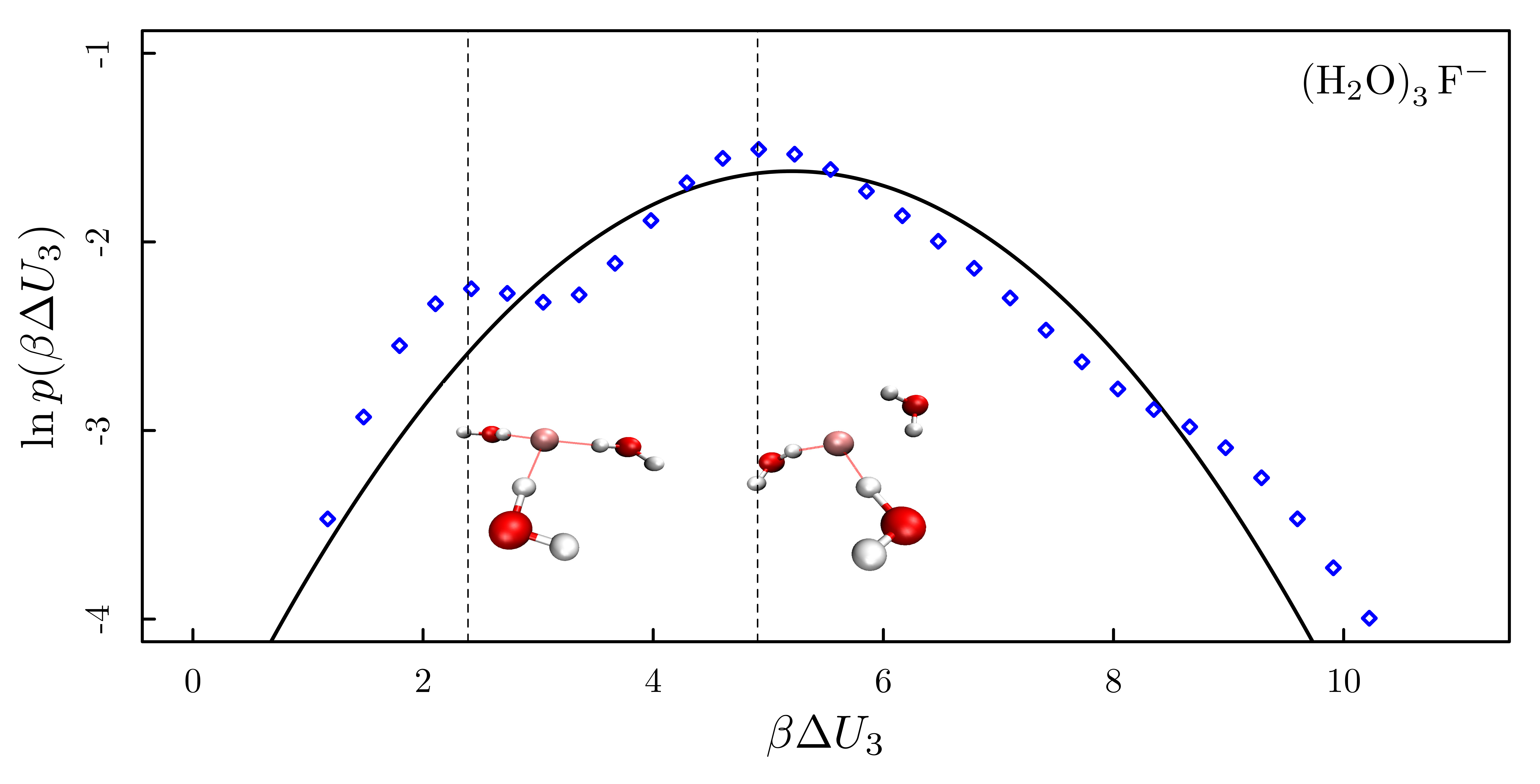{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Software and Procedures
4.2. Theory
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| QCT | Quasi-chemical theory |
| AIMD | ab initio molecular dynamics |
References
- Hofmeister, F. Zur Lehre von der Wirkung der Salze. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1888, 24, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Kunz, W.; Henle, J.; Ninham, B.W. ‘Zur Lehre von der Wirkung der Salze’ (about the science of the effect of salts): Franz Hofmeister’s historical papers. Curr. Opin. Colloid Interface Sci. 2004, 9, 19–37. [Google Scholar] [CrossRef]
- Zhang, Y.; Cremer, P. Chemistry of Hofmeister anions and osmolytes. Annu. Rev. Phys. Chem. 2010, 61, 63–83. [Google Scholar] [CrossRef] [Green Version]
- Kunz, W. Specific ion effects, evidences. In Encyclopedia of Applied Electrochemistry; Kreysa, G., Ota, K.I., Savinell, R.F., Eds.; Springer: New York, NY, USA, 2014; pp. 2050–2053. [Google Scholar] [CrossRef]
- Okur, H.I.; Hladílková, J.; Rembert, K.B.; Cho, Y.; Heyda, J.; Dzubiella, J.; Cremer, P.S.; Jungwirth, P. Beyond the Hofmeister series: Ion-specific effects on proteins and their biological functions. J. Phys. Chem. B 2017, 121, 1997–2014. [Google Scholar] [CrossRef]
- Mazzini, V.; Liu, G.; Craig, V.S.J. Probing the Hofmeister series beyond water: Specific-ion effects in non-aqueous solvents. J. Chem. Phys. 2018, 148, 222805. [Google Scholar] [CrossRef] [PubMed]
- Hribar-Lee, B.; Vlachy, V.; Dill, K. Modeling Hofmeister effects. Acta Chim Slov. 2009, 56, 196–202. [Google Scholar]
- Horinek, D. Specific ion effects, theory. In Encyclopedia of Applied Electrochemistry; Kreysa, G., Ota, K.I., Savinell, R.F., Eds.; Springer: New York, NY, USA, 2014; pp. 2050–2053. [Google Scholar] [CrossRef]
- Pollard, T.P.; Beck, T.L. Toward a quantitative theory of Hofmeister phenomena: From quantum effects to thermodynamics. Curr. Opin. Colloid Interface Sci. 2016, 23, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Varma, S.; Rogers, D.M.; Pratt, L.R.; Rempe, S.B. Design principles for K+ selectivity in membrane transport. J. Gen. Physiol. 2011, 137, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.J.; Rempe, S.L.B. Ion-specific effects in carboxylate binding sites. J. Phys. Chem. B 2016, 120, 12519–12530. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, M.I.; Vanegas, J.M.; Pratt, L.R.; Muralidharan, A.; Rempe, S.B. Hydration Mimicry by Membrane Ion Channels. Ann. Rev. Phys. Chem. 2019, 71, 461–484. [Google Scholar] [CrossRef] [Green Version]
- Pratt, L.R.; Rempe, S.B. Quasi-chemical theory and implicit solvent models for simulations. In AIP Conference Proceedings; American Institute of Physics: College Park, MD, USA, 1999; Volume 492, pp. 172–201. [Google Scholar]
- Asthagiri, D.; Dixit, P.; Merchant, S.; Paulaitis, M.; Pratt, L.; Rempe, S.; Varma, S. Ion selectivity from local configurations of ligands in solutions and ion channels. Chem. Phys. Letts. 2010, 485, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Muralidharan, A.; Pratt, L.R.; Chaudhari, M.I.; Rempe, S.B. Quasi-chemical theory for anion hydration and specific ion effects: Cl−(aq) vs. F−(aq). Chem. Phys. Letts. X 2019, 4, 100037. [Google Scholar] [CrossRef]
- Frenkel, J. Statistical Theory of Condensation Phenomena. J. Chem. Phys. 1939, 7, 200–201. [Google Scholar] [CrossRef]
- Band, W. Dissociation Treatment of Condensing Systems. J. Chem. Phys. 1939, 7, 324–326. [Google Scholar] [CrossRef]
- Band, W. Dissociation Treatment of Condensing Systems. II. J. Chem. Phys. 1939, 7, 927–931. [Google Scholar] [CrossRef]
- Stillinger, F.H. Rigorous Basis of the Frenkel-Band Theory of Association Equilibrium. J. Chem. Phys. 1963, 38, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Stillinger, F.H. Physical Clusters, Surface Tension, and Critical Phenomena. J. Chem. Phys. 1967, 47, 2513–2533. [Google Scholar] [CrossRef]
- Chandler, D.; Pratt, L.R. Statistical mechanics of chemical equilibria and intramolecular structures of nonrigid molecules in condensed phases. J. Chem. Phys. 1976, 65, 2925–2940. [Google Scholar] [CrossRef]
- Lockett, A.M. A theory of homogeneous condensation from small nuclei. I. Modified Mayer theory of physical clusters. J. Chem. Phys. 1980, 72, 4822–4831. [Google Scholar] [CrossRef]
- Pratt, L.R.; LaViolette, R.A. Quasi-chemical theories of associated liquids. Mol. Phys. 1998, 94, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.E. The Theory of Condensation and the Critical Point. Physics 1967, 3, 255–283. [Google Scholar] [CrossRef] [Green Version]
- Pratt, L.R.; Asthagiri, D. Potential distribution methods and free energy models of molecular solutions. In Free Energy Calculations; Springer: Berlin/Heidelberg, Germany, 2007; pp. 323–351. [Google Scholar]
- Gadre, S.R.; Yeole, S.D.; Sahu, N. Quantum Chemical Investigations on Molecular Clusters. Chem. Rev. 2014, 114, 12132–12173. [Google Scholar] [CrossRef] [PubMed]
- Rempe, S.; Jonsson, H. A computational exercise illustrating molecular vibrations and normal modes. Chem. Educat. 1998, 3, 1–17. [Google Scholar] [CrossRef]
- Rogers, D.M.; Rempe, S.B. Probing the thermodynamics of competitive ion binding using minimum energy structures. J. Phys. Chem. B 2011, 115, 9116–9129. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Glezakou, V.A. Global optimization of chemical cluster structures: Methods, applications, and challenges. Int. J. Quant. Chem. 2021, 121, e26553. [Google Scholar] [CrossRef]
- Jesus, W.; Prudente, F.; Marques, J.; Pereira, F. Modeling microsolvation clusters with electronic-structure calculations guided by analytical potentials and predictive machine learning techniques. Phys. Chem. Chem. Phys. 2021, 23, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.M.; Jiao, D.; Pratt, L.R.; Rempe, S.B. Chapter Four—Structural Models and Molecular Thermodynamics of Hydration of Ions and Small Molecules. In Annual Reports in Computational Chemistry; Wheeler, R.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 8, pp. 71–127. [Google Scholar] [CrossRef]
- Sabo, D.; Varma, S.; Martin, M.G.; Rempe, S.B. Studies of the thermodynamic properties of hydrogen gas in bulk water. J. Phys. Chem. B 2008, 112, 867–876. [Google Scholar] [CrossRef]
- Perera, L.; Berkowitz, M.L. Structure and dynamics of Cl−(H2O)n clusters: The effect of the polarizability and the charge of the ion. J. Chem. Phys. 1992, 96, 8288–8294. [Google Scholar] [CrossRef] [Green Version]
- Perera, L.; Berkowitz, M.L. Stabilization energies of Cl−, Br−, and I− ions in water clusters. J. Chem. Phys. 1993, 99, 4222–4224. [Google Scholar] [CrossRef] [Green Version]
- Perera, L.; Berkowitz, M.L. Structures of Cl−(H2O)n and F−(H2O)n (n = 2, 3, ..., 15) clusters. Molecular dynamics computer simulations. J. Chem. Phys. 1994, 100, 3085–3093. [Google Scholar] [CrossRef]
- Sremaniak, L.S.; Perera, L.; Berkowitz, M.L. Enthalpies of formation and stabilization energies of Br−(H2O)n. Chem. Phys. Letts. 1994, 218, 377–382. [Google Scholar] [CrossRef]
- Combariza, J.E.; Kestner, N.R.; Jortner, J. Microscopic solvation of anions in water clusters. Chem. Phys. Lett. 1993, 203, 423–428. [Google Scholar] [CrossRef]
- Combariza, J.E.; Kestner, N.R. Microscopic Study of Fluoride-Water Clusters. J. Phys. Chem. 1994, 98, 3513–3517. [Google Scholar] [CrossRef]
- Combariza, J.E.; Kestner, N.R.; Jortner, J. Energy-structure relationships for microscopic solvation of anions in water clusters. J. Chem. Phys. 1994, 100, 2851–2864. [Google Scholar] [CrossRef]
- Combariza, J.E.; Kestner, N.R.; Jortner, J. Surface and interior states of iodide—water clusters. Chem. Phys. Letts. 1994, 221, 156–160. [Google Scholar] [CrossRef]
- Combariza, J.E.; Kestner, N.R. Density Functional Study of Short-Range Interaction Forces between Ions and Water Molecules. J. Phys. Chem. 1995, 99, 2717–2723. [Google Scholar] [CrossRef]
- Cheshnovsky, O.; Giniger, R.; Markovich, G.; Makov, G.; Nitzan, A.; Jortner, J. Surface and interior anion solvation in water clusters. J. Chim. Phys. 1995, 92, 397–408. [Google Scholar] [CrossRef]
- Choi, J.H.; Kuwata, K.T.; Cao, Y.B.; Okumura, M. Vibrational Spectroscopy of the Cl−(H2O)n Anionic Clusters, n = 1–5. J. Phys. Chem. A 1998, 102, 503–507. [Google Scholar] [CrossRef]
- Basdogan, Y.; Groenenboom, M.C.; Henderson, E.; De, S.; Rempe, S.B.; Keith, J.A. Machine learning-guided approach for studying solvation environments. J. Chem. Theory Comput. 2020, 16, 633–642. [Google Scholar] [CrossRef]
- Tissandier, M.D.; Cowen, K.A.; Feng, W.Y.; Gundlach, E.; Cohen, M.H.; Earhart, A.D.; Coe, J.V.; Tuttle, T.R. The proton’s absolute aqueous enthalpy and Gibbs free energy of solvation from cluster-ion solvation data. J. Phys. Chem. A 1998, 102, 7787–7794. [Google Scholar] [CrossRef]
- Rogers, D.M.; Beck, T.L. Quasichemical and structural analysis of polarizable anion hydration. J. Chem. Phys. 2010, 132, 014505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, M.I.; Rempe, S.B.; Pratt, L.R. Quasi-chemical theory of F − (aq): The “no split occupancies rule” revisited. J. Chem. Phys. 2017, 147, 161728. [Google Scholar] [CrossRef]
- Muralidharan, A.; Pratt, L.R.; Chaudhari, M.I.; Rempe, S.B. Quasi-Chemical Theory with Cluster Sampling from Ab Initio Molecular Dynamics: Fluoride (F−) Anion Hydration. J. Phys. Chem. A 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rempe, S.B.; Pratt, L.R.; Hummer, G.; Kress, J.D.; Martin, R.L.; Redondo, A. The hydration number of Li+ in liquid water. J. Am. Chem. Soc. 2000, 122, 966–967. [Google Scholar] [CrossRef] [Green Version]
- Rempe, S.B.; Pratt, L.R. The hydration number of Na+ in liquid water. Fluid Phase Equilibria 2001, 183–184, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Rempe, S.B.; Asthagiri, D.; Pratt, L.R. Inner shell definition and absolute hydration free energy of K+(aq) on the basis of quasi-chemical theory and ab initio molecular dynamics. Phys. Chem. Chem. Phys. 2004, 6, 1966–1969. [Google Scholar] [CrossRef] [Green Version]
- Sabo, D.; Jiao, D.; Varma, S.; Pratt, L.R.; Rempe, S.B. Case study of Rb+(aq), quasi-chemical theory of ion hydration, and the no split occupancies rule. Annu. Rep. Prog. Chem. Sect. C Phys. Chem. 2013, 109, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, M.I.; Soniat, M.; Rempe, S.B. Octa-Coordination and the aqueous Ba2+ Ion. J. Phys. Chem. B 2015, 119, 8746–8753. [Google Scholar] [CrossRef]
- Chaudhari, M.I.; Rempe, S.B. Strontium and barium in aqueous solution and a potassium channel binding site. J. Chem. Phys. 2018, 148, 222831. [Google Scholar] [CrossRef] [Green Version]
- Soniat, M.; Rogers, D.M.; Rempe, S.B. Dispersion- and exchange-corrected density functional theory for sodium ion hydration. J. Chem. Theory Comput. 2015, 11, 2958–2967. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- Kühne, T.D.; Iannuzzi, M.; Ben, M.D.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703. [Google Scholar] [CrossRef] [Green Version]
- Lippert, G.; Hutter, J.; Parrinello, M. The Gaussian and augmented-plane-wave density functional method for ab initio molecular dynamics simulations. Theor. Chem. Acc. 1999, 103, 124–140. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.; Peralta, J.; Ogliaro, F.; Bearpark, M.; Heyd, J.; Brothers, E.; Kudin, K.; et al. Gaussian 09, Revision A. 1; Gaussian Inc.: Wallingford, UK, 2009. [Google Scholar]
- Jain, R.; Ahuja, B.; Sharma, B. Density-Functional Thermochemistry. III. The Role of Exact Exchange. Indian J. Pure Appl. Phys. 2004, 42, 43–48. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; et al. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.E.; Paulaitis, M.E.; Pratt, L.R. Potential Distribution Theorem and Statistical Thermodynamics of Molecular Solutions; Cambridge University Press: New York, NY, USA, 2006. [Google Scholar]
Short Biography of Authors
 | Diego Gomez is a graduate student at Tulane University in the Department of Chemical Engineering. He graduated from New Mexico State University in 2017 with a B.S. in Chemical Engineering. After graduation, he interned at Sandia National Labs as a researcher for High-Performance Computing in Livermore, CA. He is a member of the American Institute of Chemical Engineers. His current research interests are statistical mechanics, large-scale computing and stochastic processes. |
 | Lawrence Pratt is Professor Emeritus at Tulane University, where he was the Herman and George R. Brown Chair in Chemical Engineering. He spent his earlier career as a Staff Scientist at Los Alamos National Lab. In 2018, he won the American Chemical Society’s Joel Henry Hildebrand Award in Theoretical and Experimental Chemistry of Liquids. |
 | David Rogers is a Staff Scientist specializing in high-performance computing for chemistry and materials at the ORNL National Center for Computational Sciences. He obtained his Ph.D. in physical chemistry from the University of Cincinnati in 2009 for work on applying Bayes’ theorem to the free energy problem with applications to multiscale modeling of fluids and interface chemistry. After a post-doc at Sandia National Labs, he joined the USF Department of Chemistry as an Assistant Professor in 2013, and moved to ORNL in 2020. His current research interests include mathematical and computational theory and methods for multiscale modeling, with an emphasis on developing more powerful and general libraries and interfaces for modeling. |
 | Susan Rempe is a Staff Scientist at Sandia National Labs and a Research Professor in Chemical & Biological Engineering at the University of New Mexico. She earned her PhD at the University of Washington, and engaged in postdoc studies at Los Alamos National Lab. Her work focuses on theoretical molecular studies of solutions, materials, and biomolecules. Insights from those studies have informed the synthesis of new materials for water purification, and CO2 separation and capture. |





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez, D.T.; Pratt, L.R.; Rogers, D.M.; Rempe, S.B. Free Energies of Hydrated Halide Anions: High Through-Put Computations on Clusters to Treat Rough Energy-Landscapes. Molecules 2021, 26, 3087. https://doi.org/10.3390/molecules26113087
Gomez DT, Pratt LR, Rogers DM, Rempe SB. Free Energies of Hydrated Halide Anions: High Through-Put Computations on Clusters to Treat Rough Energy-Landscapes. Molecules. 2021; 26(11):3087. https://doi.org/10.3390/molecules26113087
Chicago/Turabian StyleGomez, Diego T., Lawrence R. Pratt, David M. Rogers, and Susan B. Rempe. 2021. "Free Energies of Hydrated Halide Anions: High Through-Put Computations on Clusters to Treat Rough Energy-Landscapes" Molecules 26, no. 11: 3087. https://doi.org/10.3390/molecules26113087
APA StyleGomez, D. T., Pratt, L. R., Rogers, D. M., & Rempe, S. B. (2021). Free Energies of Hydrated Halide Anions: High Through-Put Computations on Clusters to Treat Rough Energy-Landscapes. Molecules, 26(11), 3087. https://doi.org/10.3390/molecules26113087