
Composition Profiling and Authenticity Assessment of Camellia Oil Using High Field and Low Field 1H NMR

 and
and 
Abstract
:1. Introduction
2. Results and Discussion
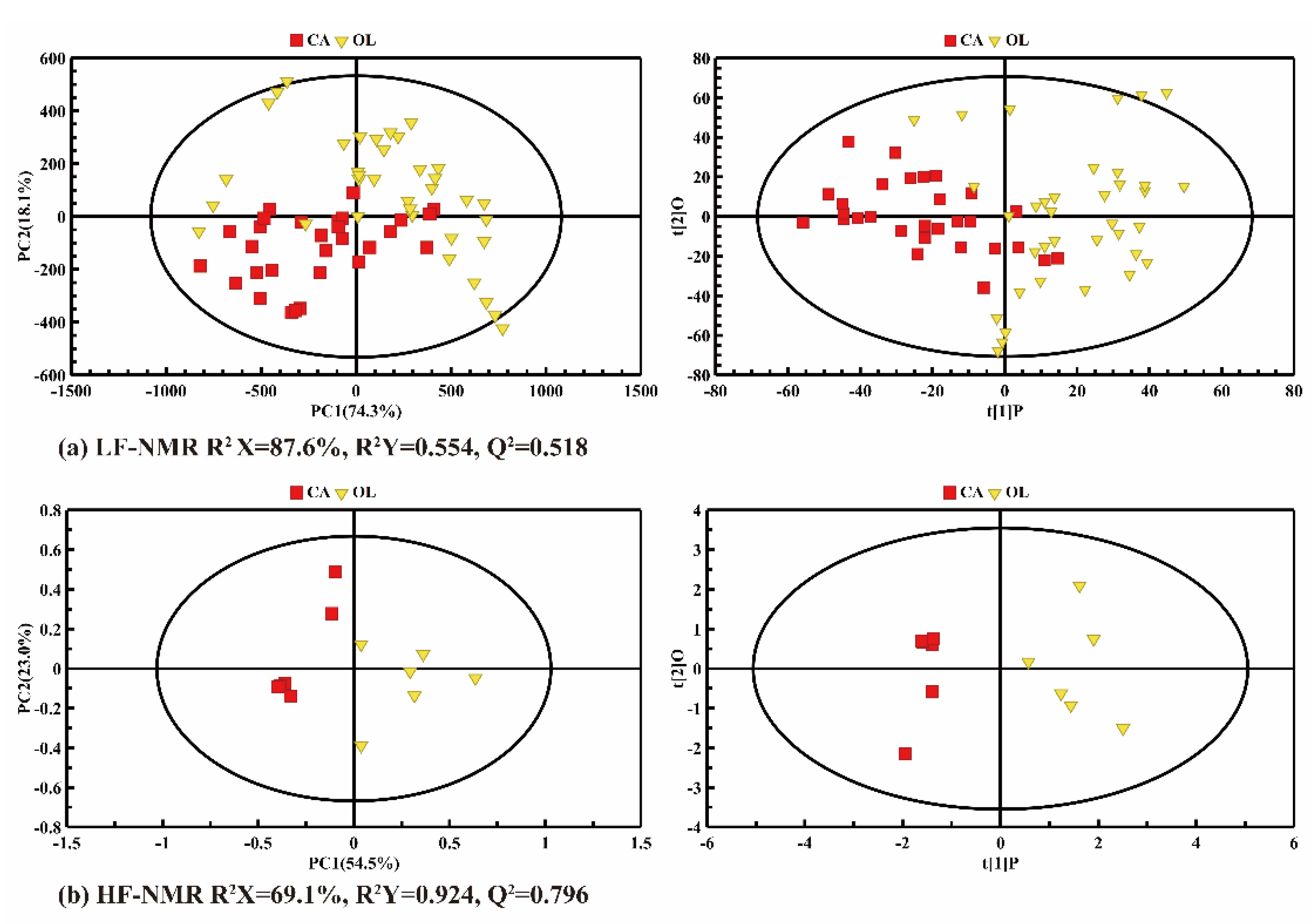
2.1. Nutritional Compositional Difference between CA and OL
2.2. T2 Distribution of CA and OL
2.3. Identification of CA Adulteration by LF-NMR
2.3.1. T2 Distribution of the Adulterated CA
2.3.2. Identification of Adulterated CA with Pattern Recognition Analysis
2.3.3. Prediction of Adulterated CA by Discriminant Analysis
3. Materials and Methods
3.1. Experimental Oil Sample
3.1.1. Sample Preparation for HF-NMR
3.1.2. Sample Preparation for LF-NMR
3.2. NMR Detection
3.2.1. HF-NMR Detection
3.2.2. LF-NMR Detection
3.3. Preprocessing of NMR Spectra
3.3.1. HF-NMR Spectral Preprocessing
3.3.2. LF-NMR Spectral Preprocessing
3.4. Quantitative Calculation of Nutritional Components of Oil Samples
3.5. Multivariate Statistical Analysis
3.6. Discriminant Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cheng, X.X.; Yang, T.; Wang, Y.H.; Zhou, B.Q.; Yan, L.; Teng, L.Z.; Wang, F.B.; Chen, L.L.; He, Y.; Guo, K.P.; et al. New method for effective identification of adulterated Camellia oil basing on Camellia oleifera-specific DNA. Arab. J. Chem. 2018, 11, 815–826. [Google Scholar] [CrossRef]
- Su, M.H.; Shih, M.C.; Lin, K.H. Chemical composition of seed oils in native Taiwanese Camellia species. Food Chem. 2014, 156, 369–373. [Google Scholar] [CrossRef]
- Wang, X.Q.; Zeng, Q.M.; Contreras, M.D.; Wang, L.J. Profiling and quantification of phenolic compounds in Camellia seed oils: Natural tea polyphenols in vegetable oil. Food Res. Int. 2017, 102, 184–194. [Google Scholar] [CrossRef]
- Zhou, D.; Shi, Q.L.; Pan, J.B.; Liu, M.; Long, Y.; Ge, F.H. Effectively improve the quality of camellia oil by the combination of supercritical fluid extraction and molecular distillation (SFE-MD). LWT Food Sci. Technol. 2019, 110, 175–181. [Google Scholar] [CrossRef]
- Zeb, A. Triacylglycerols composition, oxidation and oxidation compounds in camellia oil using liquid chromatography-mass spectrometry. Chem. Phys. Lipids 2012, 165, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Rahiman, S.; El-Metwally, T.H.; Shrivastava, D.; Tantry, M.N.; Tantry, B.A. Oleuropein and oleic acid: A novel emerging dietary target for human chronic diseases. Indian J. Biochem. Biophys. 2019, 56, 263–268. [Google Scholar]
- Zhu, M.T.; Shi, T.; Chen, Y.; Luo, S.H.; Leng, T.; Wang, Y.L.; Guo, C.; Xie, M.Y. Prediction of fatty acid composition in camellia oil by 1H NMR combined with PLS regression. Food Chem. 2019, 279, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lee, F.S.C.; Wang, X.R.; He, Y. Feasibility study of quantifying and discriminating soybean oil adulteration in camellia oils by attenuated total reflectance MIR and fiber optic diffuse reflectance NIR. Food Chem. 2006, 95, 529–536. [Google Scholar] [CrossRef]
- Li, R.F.; Huang, J.L.; Huang, L.; Teng, J.W.; Xia, N.; Wei, B.Y.; Zhao, M.M. Comparison of GC and DSC monitoring the adulteration of camellia oil with selected vegetable oils. J. Therm. Anal. Calorim. 2016, 126, 1735–1746. [Google Scholar] [CrossRef]
- Dais, P.; Hatzakis, E. Quality assessment and authentication of virgin olive oil by NMR spectroscopy: A critical review. Anal. Chim. Acta 2013, 765, 1–27. [Google Scholar] [CrossRef]
- Cataldo, A.; Piuzzi, E.; Cannazza, G.; De Benedetto, E.; Tarricone, L. Quality and anti-adulteration control of vegetable oils through microwave dielectric spectroscopy. Measurement 2010, 43, 1031–1039. [Google Scholar] [CrossRef]
- Aparicio, R.; Morales, M.T.; Alonso, V. Authentication of European virgin olive oils by their chemical compounds, sensory attributes, and consumers’ attitudes. J. Agric. Food Chem. 1997, 45, 1076–1083. [Google Scholar] [CrossRef]
- Cao, G.D.; Ruan, D.L.; Chen, Z.B.; Hong, Y.J.; Cai, Z.W. Recent developments and applications of mass spectrometry for the quality and safety assessment of cooking oil. TrAC Trends Anal. Chem. 2017, 96, 201–211. [Google Scholar] [CrossRef]
- Ellis, D.I.; Brewster, V.L.; Dunn, W.B.; Allwood, J.W.; Golovanov, A.P.; Goodacre, R. Fingerprinting food: Current technologies for the detection of food adulteration and contamination. Chem. Soc. Rev. 2012, 41, 5706–5727. [Google Scholar] [CrossRef]
- Aparicio, R.; Aparicio-Ruiz, R. Authentication of vegetable oils by chromatographic techniques. J. Chromatogr. A 2000, 881, 93–104. [Google Scholar] [CrossRef]
- Gomez-Caravaca, A.M.; Maggio, R.M.; Cerretani, L. Chemometric applications to assess quality and critical parameters of virgin and extra-virgin olive oil. A review. Anal. Chim. Acta 2016, 913, 1–21. [Google Scholar] [CrossRef]
- Nunes, C.A. Vibrational spectroscopy and chemometrics to assess authenticity, adulteration and intrinsic quality parameters of edible oils and fats. Food Res. Int. 2014, 60, 255–261. [Google Scholar] [CrossRef]
- Shi, T.; Wu, G.C.; Jin, Q.Z.; Wang, X.G. Camellia oil authentication: A comparative analysis and recent analytical techniques developed for its assessment. A review. Trends Food Sci. Technol. 2020, 97, 88–99. [Google Scholar] [CrossRef]
- Jiang, X.Y.; Li, C.; Chen, Q.Q.; Weng, X.C. Comparison of 19F and 1H NMR spectroscopy with conventional methods for the detection of extra virgin olive oil adulteration. Grasas Y Aceites 2018, 69, 249. [Google Scholar] [CrossRef] [Green Version]
- Rotondo, A.; Mannina, L.; Salvo, A. Multiple Assignment Recovered Analysis (MARA) NMR for a Direct Food Labeling: The Case Study of Olive Oils. Food Anal. Methods 2019, 12, 1238–1245. [Google Scholar] [CrossRef]
- Rotondo, A.; Torre, G.L.L.; Dugo, G.; Cicero, N.; Santini, A.; Salvo, A. Oleic Acid Is not the Only Relevant Mono-Unsaturated Fatty Ester in Olive Oil. Foods 2020, 9, 384. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.T.; Shi, T.; Luo, X.; Tang, L.J.; Liao, H.X.; Chen, Y. Determination of the oxidative stability of camellia oils using a chemometrics tool based on 1H NMR spectra and alpha-tocopherol content. Anal. Chem. 2020, 92, 932–939. [Google Scholar] [CrossRef]
- Zhu, M.T.; Shi, T.; Guo, Z.Y.; Liao, H.X.; Chen, Y. Comparative study of the oxidation of cold-pressed and commercial refined camellia oil during storage with 1H and 31P NMR spectroscopy. Food Chem. 2020, 321, 126640. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.R.; Wang, X.; Chen, L.H. Rapid detection of peanut oil adulteration using low-field nuclear magnetic resonance and chemometrics. Food Chem. 2017, 216, 268–274. [Google Scholar] [CrossRef] [PubMed]
- He, C.X.; Liu, Y.; Liu, H.L.; Zheng, X.; Shen, G.P.; Feng, J.H. Compositional identification and authentication of Chinese honeys by 1H NMR combined with multivariate analysis. Food Res. Int. 2020, 130, 108936. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tu, C.H.; Rui, X.; Gao, Y.W.; Li, W.; Wang, K.; Xiao, Y.; Dong, M.S. Study of Water Dynamics in the Soaking, Steaming, and Solid-State Fermentation of Glutinous Rice by LF-NMR: A Novel Monitoring Approach. J. Agric. Food Chem. 2015, 63, 3261–3270. [Google Scholar] [CrossRef]
- Pitombo, R.N.M.; Lima, G. Nuclear magnetic resonance and water activity in measuring the water mobility in Pintado (Pseudoplatystoma corruscans) fish. J. Food Eng. 2003, 58, 59–66. [Google Scholar] [CrossRef]
- Shao, J.H.; Deng, Y.M.; Song, L.; Batur, A.; Jia, N.; Liu, D.-Y. Investigation the effects of protein hydration states on the mobility water and fat in meat batters by LF-NMR technique. LWT Food Sci. Technol. 2016, 66, 1–6. [Google Scholar] [CrossRef]
- Hou, X.W.; Wang, G.L.; Su, G.Q.; Wang, X.; Nie, S.D. Rapid identification of edible oil species using supervised support vector machine based on low-field nuclear magnetic resonance relaxation features. Food Chem. 2019, 280, 139–145. [Google Scholar] [CrossRef]
- Ingallina, C.; Cerreto, A.; Mannina, L.; Circi, S.; Vista, S.; Capitani, D.; Spano, M.; Sobolev, A.P.; Marini, F. Extra-virgin olive oils from nine Italian regions: An 1H NMR-chemometric characterization. Metabolites 2019, 9, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ok, S. Detection of olive oil adulteration by low-field NMR relaxometry and UV-Vis spectroscopy upon mixing olive oil with various edible oils. Grasas Y Aceites 2017, 68, e173. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, Y.R.; Shen, G.P.; Zhong, S.; Feng, J.H. NMR spectroscopy in conjugation with multivariate statistical analysis for distinguishing plant origin of edible oils. J. Food Compos. Anal. 2018, 69, 140–148. [Google Scholar] [CrossRef]
- He, W.X.; Lei, T.X. Identification of camellia oil using FT-IR spectroscopy and chemometrics based on both isolated unsaponifiables and vegetable oils. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 228, 117839. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Lacatusu, I.; Badea, G.; Grafu, I.A.; Istrati, D.; Babeanu, N.; Stan, R.; Badea, N.; Meghea, A. Exploitation of amaranth oil fractions enriched in squalene for dual delivery of hydrophilic and lipophilic actives. Ind. Crop. Prod. 2015, 77, 342–352. [Google Scholar] [CrossRef]
- Nam, A.-M.; Bighelli, A.; Tomi, F.; Casanova, J.; Paoli, M. Quantification of Squalene in Olive Oil Using 13C Nuclear Magnetic Resonance Spectroscopy. Magnetochemistry 2017, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, N.; Ito, J.; Kato, S.; Eitsuka, T.; Miyazawa, T.; Nakagawa, K. Significance of Squalene in Rice Bran Oil and Perspectives on Squalene Oxidation. J. Nutr. Sci. Vitaminol. 2019, 65, S62–S66. [Google Scholar] [CrossRef] [Green Version]
- Pan, A.; Chen, M.; Chowdhury, R.; Wu, J.H.Y.; Sun, Q.; Campos, H.; Mozaffarian, D.; Hu, F.B. alpha-Linolenic acid and risk of cardiovascular disease: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2012, 96, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wang, X.; Liu, B.L. Low-field Nuclear Magnetic Resonance Relaxation Characteristics of Fatty Acids with Different Chain Lengths and Degree of Unsaturation. Chin. J. Anal. Chem. 2018, 46, 281–287. [Google Scholar]
- Lu, Y.S.; Larock, R.C. Novel Polymeric Materials from Vegetable Oils and Vinyl Monomers: Preparation, Properties, and Applications. ChemSusChem 2009, 2, 136–147. [Google Scholar] [CrossRef]
- Cao, X.H.; Zhang, M.; Mujumdar, A.S.; Zhong, Q.F.; Wang, Z.S. Measurement of water mobility and distribution in vacuum microwave-dried barley grass using Low-Field-NMR. Dry. Technol. 2018, 36, 1892–1899. [Google Scholar] [CrossRef]
- Sun, Y.N.; Zhang, M.; Fan, D.C. Effect of ultrasonic on deterioration of oil in microwave vacuum frying and prediction of frying oil quality based on low field nuclear magnetic resonance (LF-NMR). Ultrason. Sonochem. 2019, 51, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Meiri, N.; Berman, P.; Colnago, L.A.; Moraes, T.B.; Linder, C.; Wiesman, Z. Liquid-phase characterization of molecular interactions in polyunsaturated and n-fatty acid methyl esters by H-1 low-field nuclear magnetic resonance. Biotechnol. Biofuels 2015, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.S.; Zhou, X.L.; Wu, W.P.; Zhang, Y.Y.; Ni, Z.H. Development of the Miniature NMR Apparatus for Edible Oil Quality Control. Appl. Magn. Reson. 2014, 45, 461–469. [Google Scholar] [CrossRef]
- Wang, S.H.; Lai, G.Y.; Lin, J.Z.; Xia, F.; Ding, Z.N.; Feng, J.H.; Xu, J.J.; Shen, G.P. Rapid Detection of Adulteration in Extra Virgin Olive Oil by Low-Field Nuclear Magnetic Resonance Combined with Pattern Recognition. Food Anal. Methods 2021, 14, 1322–1335. [Google Scholar] [CrossRef]
- Shi, T.; Wu, G.C.; Jin, Q.Z.; Wang, X.G. Detection of camellia oil adulteration using chemometrics based on fatty acids GC fingerprints and phytosterols GC-MS fingerprints. Food Chem. 2021, 352, 129422. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Zhu, M.T.; Chen, Y.; Yan, X.L.; Chen, Q.; Wu, X.L.; Lin, J.N.; Xie, M.Y. 1H NMR combined with chemometrics for the rapid detection of adulteration in camellia oils. Food Chem. 2018, 242, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Zailer, E.; Holzgrabe, U.; Diehl, B.W.K. Interlaboratory Comparison Test as an Evaluation of Applicability of an Alternative Edible Oil Analysis by 1H NMR Spectroscopy. J. AOAC Int. 2017, 100, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types of Oil Sample | Linolenic Acid (%) | Linoleic Acid (%) | Oleic Acid (%) | Saturated Fatty Acid (%) | Acid Value (%) | Squalene (%) | |
|---|---|---|---|---|---|---|---|
| Camellia oil | DY | 2.54 | 6.51 | 75.31 | 15.64 | 0.36 | 3.45 |
| JH | 1.03 | 5.08 | 81.42 | 12.47 | 0.45 | 2.00 | |
| QY | 2.37 | 5.45 | 76.69 | 15.50 | 0.19 | 3.96 | |
| RX | 2.60 | 6.31 | 75.27 | 15.82 | 0.55 | 1.88 | |
| YL | 2.34 | 4.31 | 77.15 | 16.21 | 0.76 | 1.69 | |
| DL | 2.09 | 5.08 | 77.02 | 15.81 | 1.98 | 1.23 | |
| Mean ± SD a | 2.16 ± 0.53 | 5.46 ± 0.76 | 77.14 ± 2.06 | 15.24 ± 1.26 | 0.72 ± 0.59 | 2.36 ± 0.98 | |
| Olive oil | AN | 2.03 | 5.53 | 77.17 | 15.26 | 1.75 | 0.78 |
| AG | 1.56 | 8.12 | 72.99 | 17.33 | 0.85 | 0.27 | |
| OG | 1.07 | 8.17 | 71.12 | 19.65 | 0.98 | 0.53 | |
| OV | 2.17 | 2.63 | 77.38 | 17.81 | 1.50 | 0.51 | |
| DE | 2.10 | 5.01 | 74.18 | 18.71 | 1.21 | 0.49 | |
| QI | 1.42 | 5.12 | 77.55 | 15.91 | 0.77 | 0.36 | |
| Mean ± SD | 1.73 ± 0.41 | 5.76 ± 1.92 | 75.07± 2.47 | 17.44 ± 1.51 | 1.18 ± 0.35 | 0.49 ± 0.16 | |
| p values | 0.014 | 0.827 | 0.364 | 0.057 | 0.005 | 0.002 | |
| Types of Oil Sample | T21 (ms) | T22 (ms) | (ms) | S21 (%) | S22 (%) | |
|---|---|---|---|---|---|---|
| Camellia oil | DY | 69.38 ± 2.35 | 241.63 ± 4.27 | 119.88 ± 3.25 | 70.68 ± 0.46 | 29.32 ± 0.46 |
| DL | 87.35 ± 2.96 | 304.21 ± 11.67 | 137.00 ± 7.37 | 77.17 ± 1.43 | 22.83 ± 1.46 | |
| JH | 68.02 ± 2.65 | 241.63 ± 3.27 | 121.18 ± 2.43 | 69.43 ± 0.92 | 30.57 ± 0.92 | |
| QY | 73.45 ± 2.53 | 250.90 ± 4.22 | 124.75 ± 0.73 | 70.98 ± 0.41 | 29.02 ± 0.41 | |
| RX | 76.38 ± 2.93 | 250.91 ± 3.54 | 129.54 ± 3.01 | 71.19 ± 0.12 | 28.81 ± 0.12 | |
| YL | 69.99 ± 4.47 | 243.05 ± 5.87 | 118.51 ± 5.54 | 71.69 ± 2.60 | 28.31 ± 2.60 | |
| Mean ± SD a | 70.94 ± 5.98 | 246.12 ± 7.86 | 122.81 ± 4.91 | 70.90 ± 1.63 | 29.10 ± 1.63 | |
| Olive oil | AN | 66.35 ± 1.37 | 238.54 ± 3.74 | 117.48 ± 2.35 | 70.32 ± 1.04 | 29.68 ± 1.04 |
| AG | 70.74 ± 2.71 | 234.07 ± 2.32 | 114.53 ± 9.40 | 73.18 ± 0.23 | 26.82 ± 0.23 | |
| OG | 68.02 ± 2.33 | 232.36 ± 3.46 | 114.80 ± 2.83 | 71.53 ± 1.72 | 28.47 ± 1.72 | |
| OV | 68.02 ± 1.25 | 250.90 ± 2.54 | 122.59 ± 1.26 | 70.16 ± 0.69 | 29.84 ± 0.69 | |
| DE | 66.35 ± 2.37 | 244.72 ± 3.74 | 118.26 ± 3.02 | 70.90 ± 0.25 | 29.10 ± 0.25 | |
| QI | 68.02 ± 1.28 | 241.63 ± 4.27 | 118.01 ± 1.47 | 71.17 ± 0.75 | 28.83 ± 0.75 | |
| Mean ± SD | 67.67 ± 2.46 | 241.18 ± 4.31 | 117.61 ± 3.95 | 71.09 ± 1.28 | 28.91 ± 1.28 | |
| p values | 0.023 | 0.359 | 0.049 | 0.694 | 0.694 | |
| Adulteration Ratio | OPLS-DA Model | True Camellia Oil | Adulterated Camellia Oil | Total Accuracy |
|---|---|---|---|---|
| 20–100% | Training set (n = 158) | 84.1% (37/44) | 90.3% (103/114) | 88.6% (140/158) |
| Prediction set (n = 44) | 68.8% (11/16) | 85.7% (24/28) | 79.5% (35/44) | |
| 30–100% | Training set (n = 136) | 100% (44/44) | 100% (92/92) | 100% (136/136) |
| Prediction set (n = 36) | 100% (16/16) | 100% (20/20) | 100% (36/36) | |
| 40–100% | Training set (n = 113) | 100% (44/44) | 100% (69/69) | 100% (113/113) |
| Prediction set (n = 29) | 100% (16/16) | 100% (13/13) | 100% (29/29) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, M.; Wang, S.; Lin, J.; Xia, F.; Feng, J.; Shen, G. Composition Profiling and Authenticity Assessment of Camellia Oil Using High Field and Low Field 1H NMR. Molecules 2021, 26, 4738. https://doi.org/10.3390/molecules26164738
Xing M, Wang S, Lin J, Xia F, Feng J, Shen G. Composition Profiling and Authenticity Assessment of Camellia Oil Using High Field and Low Field 1H NMR. Molecules. 2021; 26(16):4738. https://doi.org/10.3390/molecules26164738
Chicago/Turabian StyleXing, Meijun, Shenghao Wang, Jianzhong Lin, Feng Xia, Jianghua Feng, and Guiping Shen. 2021. "Composition Profiling and Authenticity Assessment of Camellia Oil Using High Field and Low Field 1H NMR" Molecules 26, no. 16: 4738. https://doi.org/10.3390/molecules26164738
APA StyleXing, M., Wang, S., Lin, J., Xia, F., Feng, J., & Shen, G. (2021). Composition Profiling and Authenticity Assessment of Camellia Oil Using High Field and Low Field 1H NMR. Molecules, 26(16), 4738. https://doi.org/10.3390/molecules26164738