
Potential Anti-Alzheimer Agents from Guanidinyl Tryptophan Derivatives with Activities of Membrane Adhesion and Conformational Transition Inhibitions

 ,
,  , ,
, , 

Abstract
:1. Introduction
2. Results and Discussion
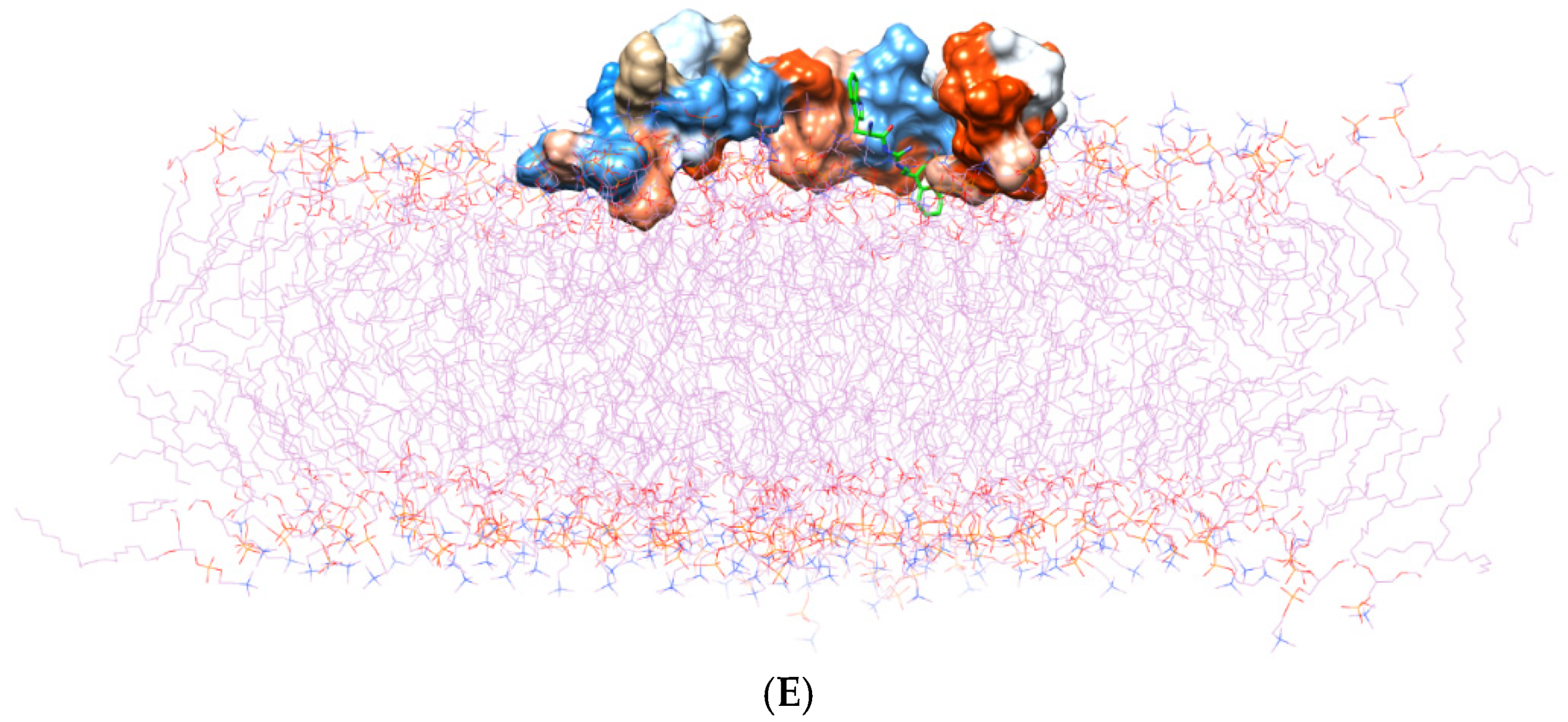
2.1. Adsorption of the Aβ1–42 Monomers on the Lipid Bilayer
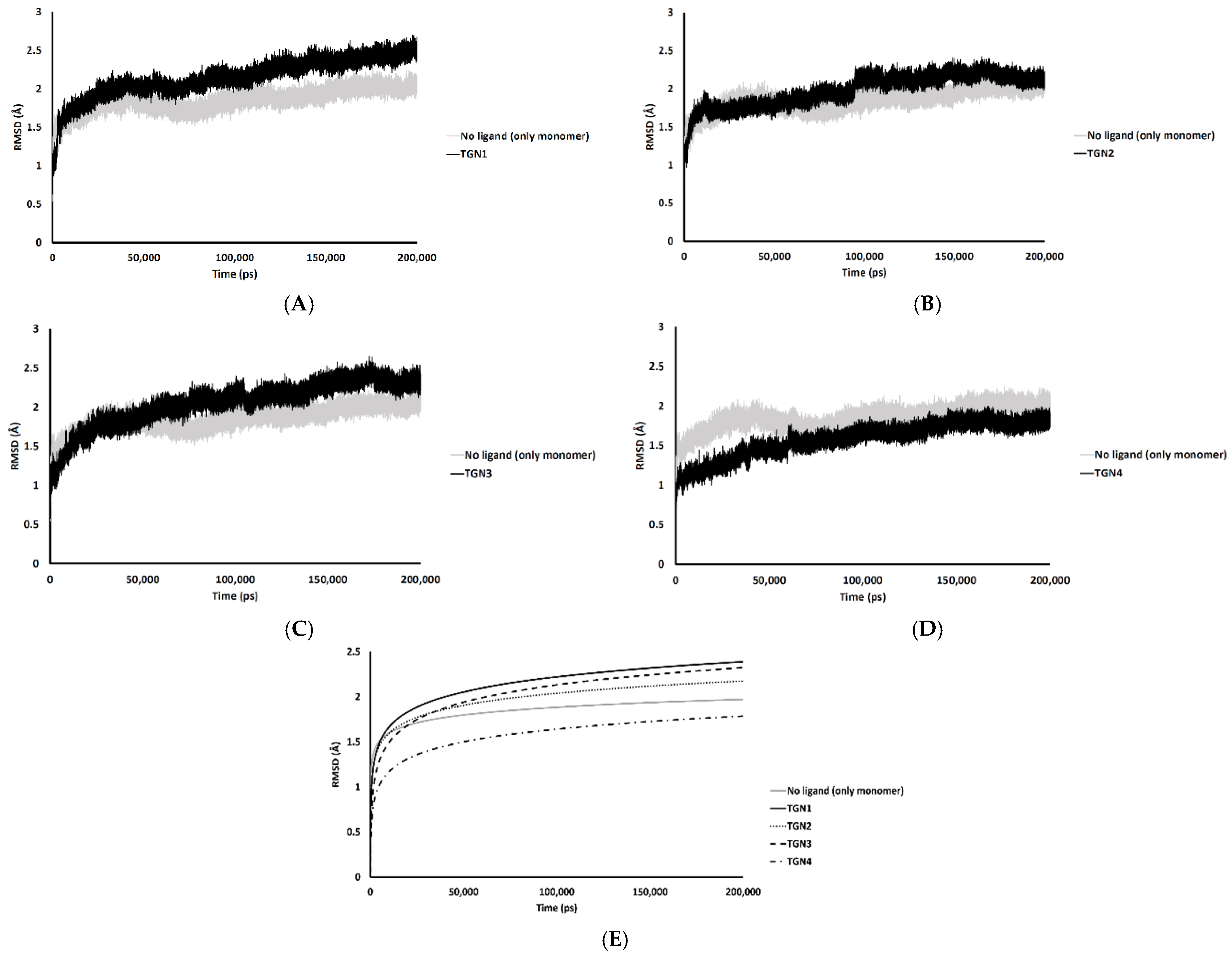
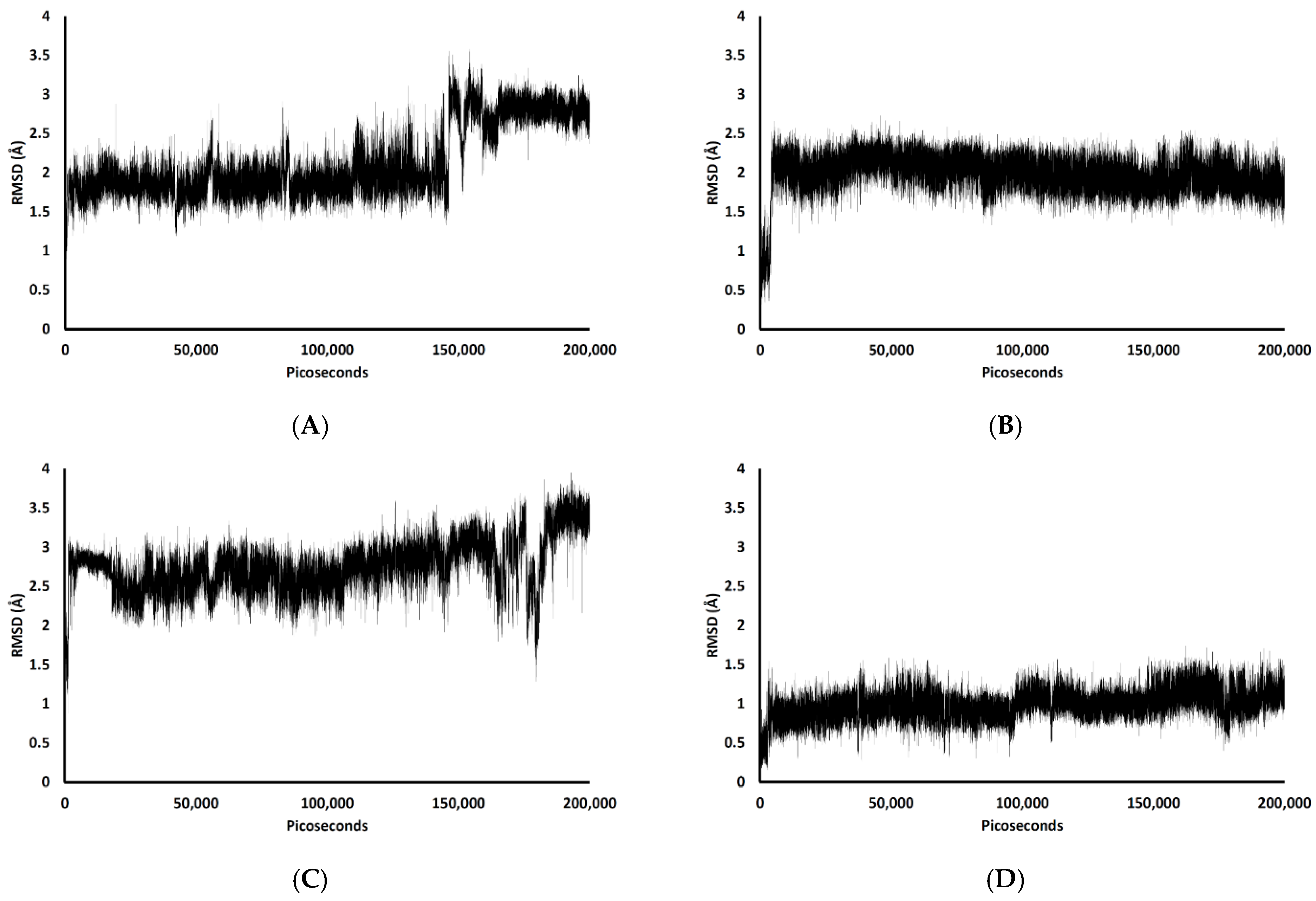
2.2. Conformational Dynamics of the Aβ1–42 Monomer at the Water-Lipid Interface
2.3. Binding Affinity Analysis
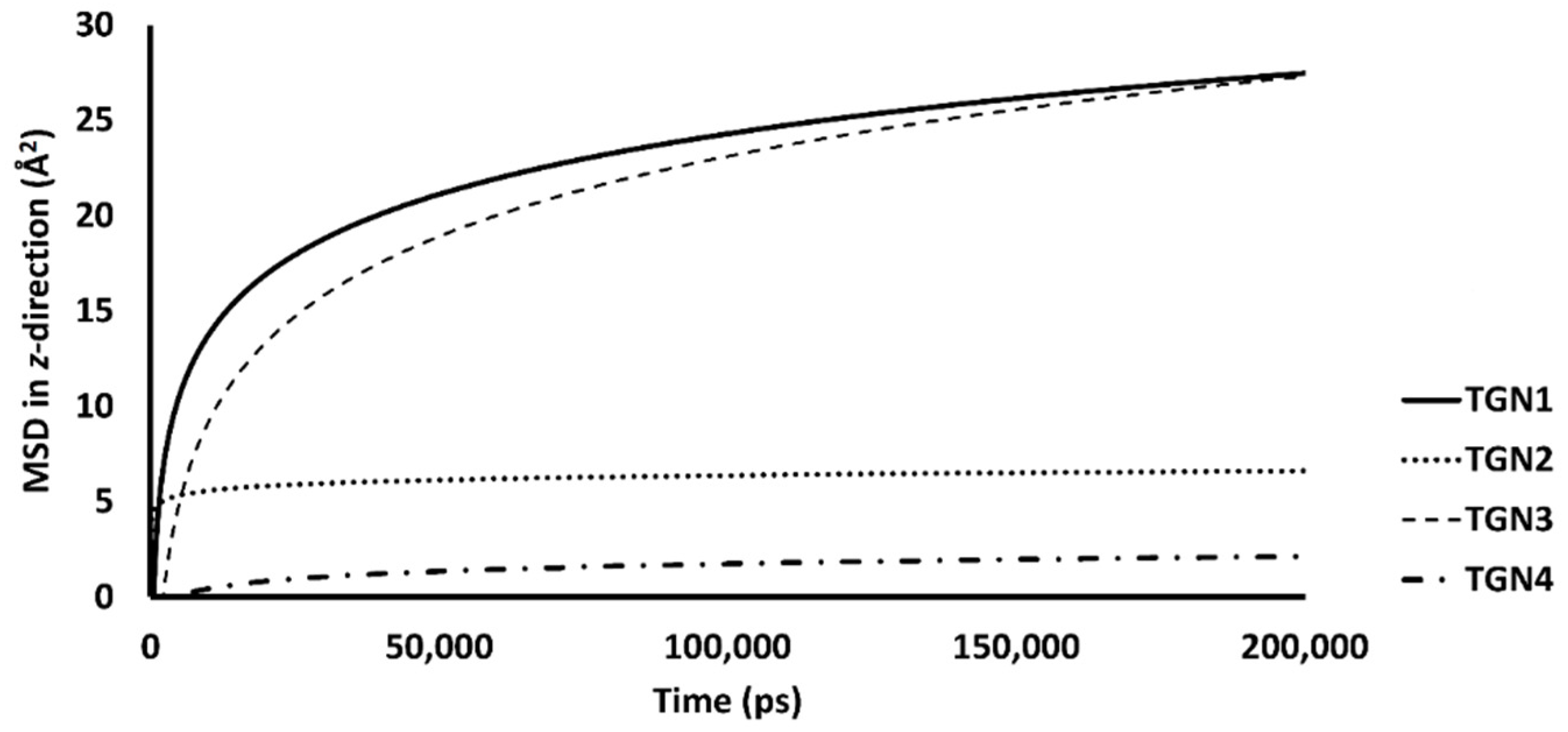
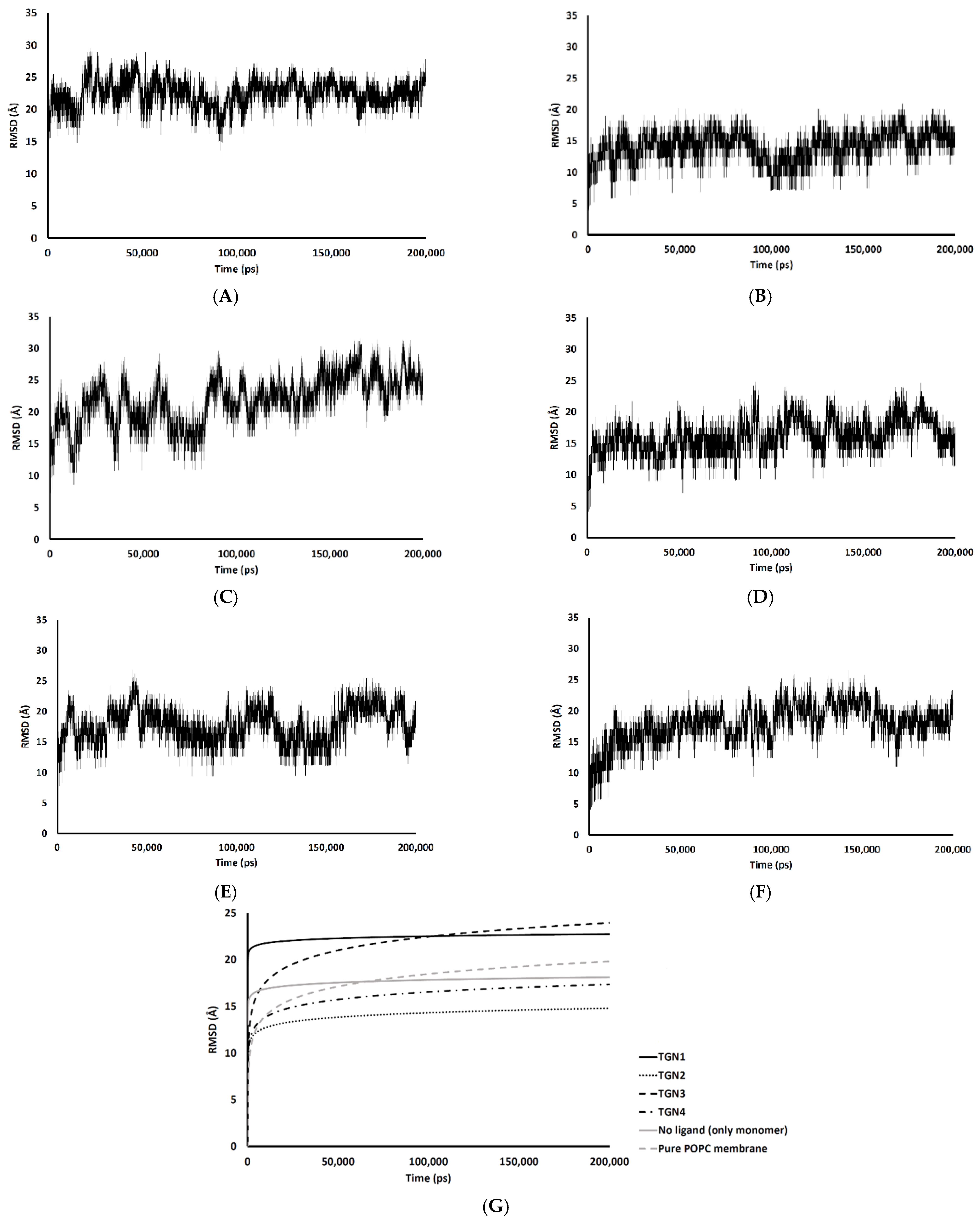
2.4. Conformational Dynamics and Movement of the Ligands into the POPC Bilayer
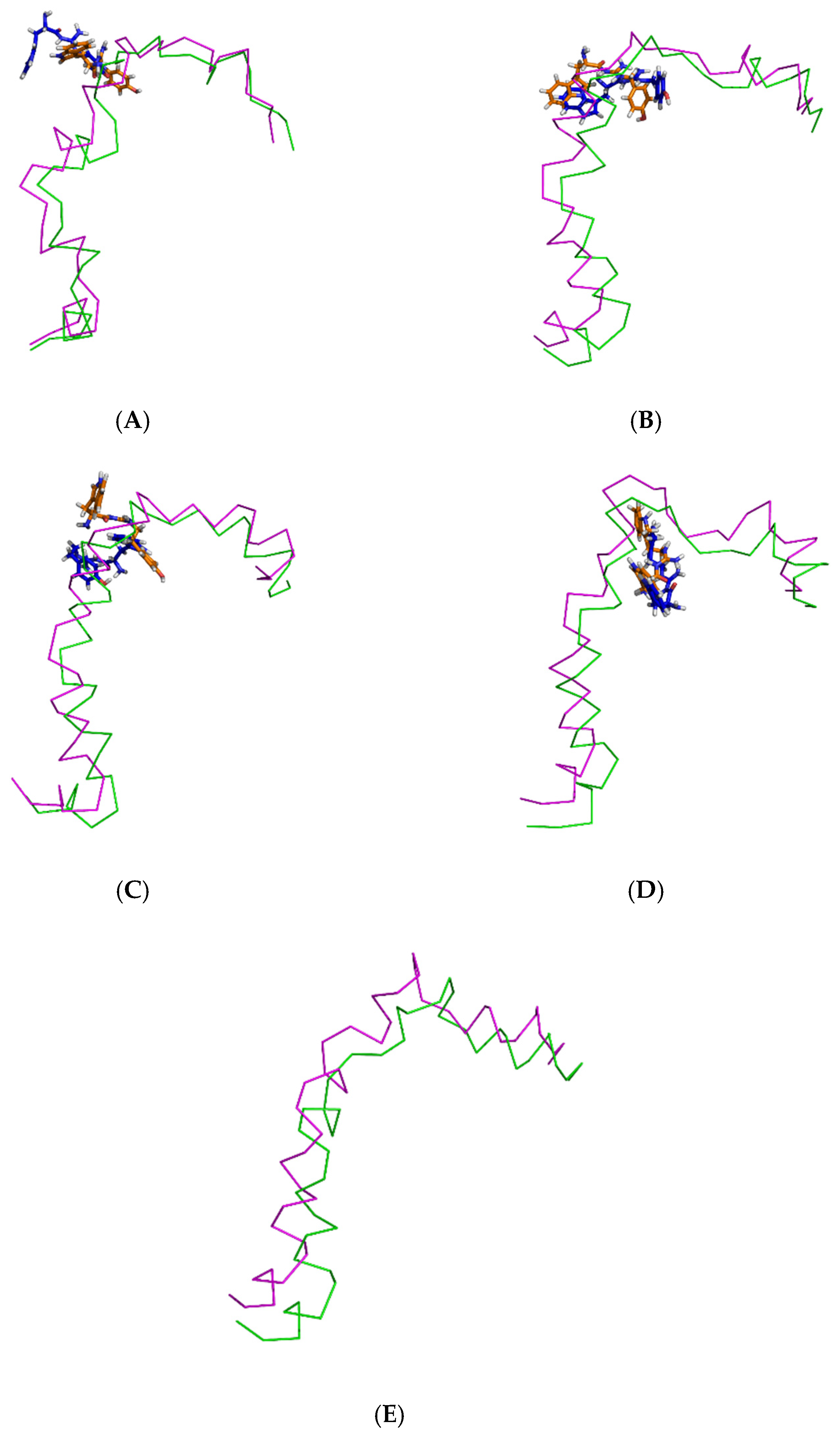
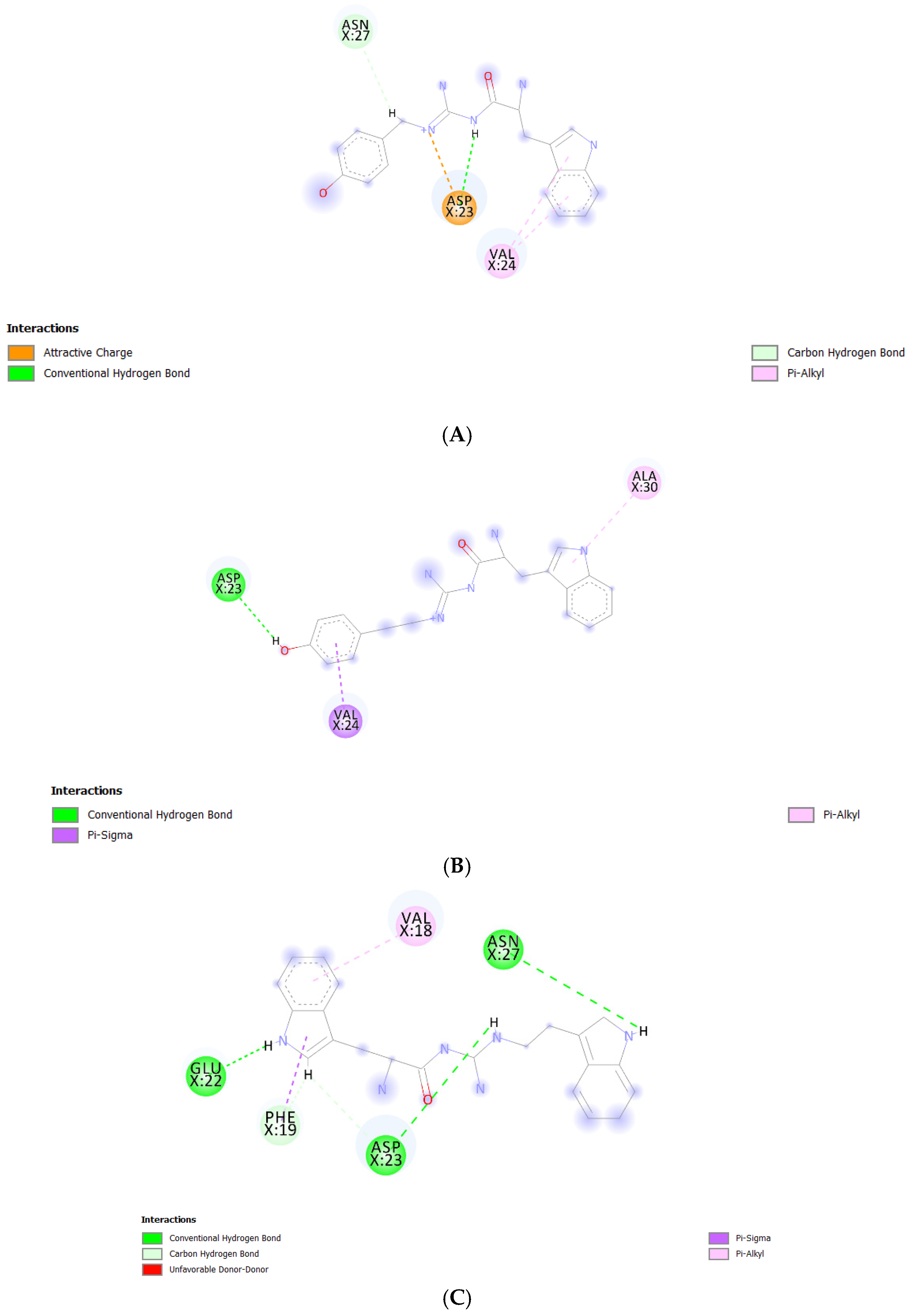
2.5. Binding Modes of the Post-MD Simulation Structures of the Ligand-Aβ1–42 Monomer Complexes
3. Conclusions
4. Materials and Methods
4.1. Lipid Bilayer Construction
4.2. Ligand and Protein Preparation
4.3. Molecular Docking
4.4. MD Simulation Protocol
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Semba, R.D. Perspective: The Potential Role of Circulating Lysophosphatidylcholine in Neuroprotection against Alzheimer Disease. Adv. Nutr. 2020, 11, 760–772. [Google Scholar] [CrossRef]
- Wongta, A.; Hongsibsong, S.; Chantara, S.; Pattarawarapan, M.; Sapbamrer, R.; Sringarm, K.; Xu, Z.-L.; Wang, H. Development of an Immunoassay for the Detection of Amyloid Beta 1–42 and Its Application in Urine Samples. J. Immunol. Res. 2020, 2020, 8821181. [Google Scholar] [CrossRef]
- Liu, F.; Ma, Z.; Sang, J.; Lu, F. Edaravone inhibits the conformational transition of amyloid-β42: Insights from molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 38, 2377–2388. [Google Scholar] [CrossRef]
- Mrdenovic, D.; Zarzycki, P.; Majewska, M.; Pieta, I.S.; Nowakowski, R.; Kutner, W.; Lipkowski, J.; Pieta, P. Inhibition of Amyloid β-Induced Lipid Membrane Permeation and Amyloid β Aggregation by K162. ACS Chem. Neurosci. 2021, 12, 531–541. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Solomon, T.; Malajczuk, C.J.; Mancera, R.L.; Howard, M.; Arrigan, D.W.M.; Newsholme, P.; Martins, R.N. Role of the cell membrane interface in modulating production and uptake of Alzheimer’s beta amyloid protein. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1639–1651. [Google Scholar] [CrossRef]
- Suwanttananuruk, P.; Jiaranaikulwanitch, J.; Waiwut, P.; Vajragupta, O. Lead discovery of a guanidinyl tryptophan derivative on amyloid cascade inhibition. Open Chem. 2020, 18, 546–558. [Google Scholar] [CrossRef]
- Dong, X.; Tang, Y.; Zhan, C.; Wei, G. Green tea extract EGCG plays a dual role in Aβ(42) protofibril disruption and membrane protection: A molecular dynamic study. Chem. Phys. Lipids 2021, 234, 105024. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Tempra, C.; Scollo, F.; Milardi, D.; La Rosa, C. Amyloid growth and membrane damage: Current themes and emerging perspectives from theory and experiments on Aβ and hIAPP. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1625–1638. [Google Scholar] [CrossRef]
- Brown, A.M.; Bevan, D.R. Molecular Dynamics Simulations of Amyloid β-Peptide (1-42): Tetramer Formation and Membrane Interactions. Biophys. J. 2016, 111, 937–949. [Google Scholar] [CrossRef] [Green Version]
- De Groot, N.S.; Aviles, F.X.; Vendrell, J.; Ventura, S. Mutagenesis of the central hydrophobic cluster in Abeta42 Alzheimer’s peptide. Side-chain properties correlate with aggregation propensities. FEBS J. 2006, 273, 658–668. [Google Scholar] [CrossRef]
- Ambadi Thody, S.; Mathew, M.K.; Udgaonkar, J.B. Mechanism of aggregation and membrane interactions of mammalian prion protein. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Q.; Pan, Q.; Zhou, F.; Zheng, J. Molecular interactions of Alzheimer amyloid-β oligomers with neutral and negatively charged lipid bilayers. Phys. Chem. Chem. Phys. PCCP 2013, 15, 8878–8889. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.; Wereszczynski, J. Probing the disparate effects of arginine and lysine residues on antimicrobial peptide/bilayer association. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. PCCP 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Pestana-Nobles, R.; Leyva-Rojas, J.A.; Yosa, J. Searching Hit Potential Antimicrobials in Natural Compounds Space against Biofilm Formation. Molecules 2020, 25, 5334. [Google Scholar] [CrossRef] [PubMed]
- Wongrattanakamon, P.; Yooin, W.; Sirithunyalug, B.; Nimmanpipug, P.; Jiranusornkul, S. Tentative Peptide‒Lipid Bilayer Models Elucidating Molecular Behaviors and Interactions Driving Passive Cellular Uptake of Collagen-Derived Small Peptides. Molecules 2021, 26, 710. [Google Scholar] [CrossRef] [PubMed]
- Tofoleanu, F.; Buchete, N.V. Molecular interactions of Alzheimer’s Aβ protofilaments with lipid membranes. J. Mol. Biol. 2012, 421, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Narang, S.S.; Goyal, D.; Goyal, B. Inhibition of Alzheimer’s amyloid-β(42) peptide aggregation by a bi-functional bis-tryptoline triazole: Key insights from molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 38, 1598–1611. [Google Scholar] [CrossRef]
- Jiaranaikulwanitch, J.; Tadtong, S.; Govitrapong, P.; Fokin, V.V.; Vajragupta, O. Neuritogenic activity of bi-functional bis-tryptoline triazole. Bioorg. Med. Chem. 2017, 25, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.F.; Liu, Z.; Bai, S.; Dong, X.Y.; Sun, Y. Exploring the inter-molecular interactions in amyloid-β protofibril with molecular dynamics simulations and molecular mechanics Poisson-Boltzmann surface area free energy calculations. J. Chem. Phys. 2012, 136, 145101. [Google Scholar] [CrossRef]
- Niu, Z.; Zhang, Z.; Zhao, W.; Yang, J. Interactions between amyloid β peptide and lipid membranes. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1663–1669. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Bueno Franco Salla, G.; Bracht, L.; Valderrama Parizotto, A.; Comar, J.F.; Peralta, R.M.; Bracht, F.; Bracht, A. Kinetics of the metabolic effects, distribution spaces and lipid-bilayer affinities of the organo-chlorinated herbicides 2,4-D and picloram in the liver. Toxicol. Lett. 2019, 313, 137–149. [Google Scholar] [CrossRef]
- Xiang, N.; Lyu, Y.; Zhu, X.; Narsimhan, G. Investigation of the interaction of amyloid β peptide (11–42) oligomers with a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane using molecular dynamics simulation. Phys. Chem. Chem. Phys. PCCP 2018, 20, 6817–6829. [Google Scholar] [CrossRef]
- Ntarakas, N.; Ermilova, I.; Lyubartsev, A.P. Effect of lipid saturation on amyloid-beta peptide partitioning and aggregation in neuronal membranes: Molecular dynamics simulations. Eur. Biophys. J. EBJ 2019, 48, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision D. 01. 2009; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tomaselli, S.; Esposito, V.; Vangone, P.; van Nuland, N.A.; Bonvin, A.M.; Guerrini, R.; Tancredi, T.; Temussi, P.A.; Picone, D. The alpha-to-beta conformational transition of Alzheimer’s Abeta-(1-42) peptide in aqueous media is reversible: A step by step conformational analysis suggests the location of beta conformation seeding. Chembiochem 2006, 7, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhou, X.; Wang, D.; Yin, D.; Lin, Z. Using molecular docking between organic chemicals and lipid membrane to revise the well known octanol-water partition coefficient of the mixture. Environ. Toxicol. Pharmacol. 2012, 34, 59–66. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 1–5. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IUPAC Name | 2D Structure | Anti-Amyloid Aggregation % Inhibition at 100 μM (±SD) |
|---|---|---|---|
| TGN1 | (S)-2-Amino-N-(N-(4-hydroxyphenyl)carbamimidoyl)-3-(1H-indol-3-yl)propanamide |  | 36.1 (±0.5) * |
| TGN2 | (S)-2-Amino-N-(N-(4-hydroxybenzyl)carbamimidoyl)-3-(1H-indol-3-yl)propanamide |  | 87.0 (±0.8) |
| TGN3 | (S)-2-Amino-N-(N-(4-hydroxyphenethyl)carbamimidoyl)-3-(1H-indol-3-yl)propanamide |  | 54.3 (±2.4) * |
| TGN4 | (S)-N-(N-(2-(1H-Indol-2-yl)ethyl)carbamimidoyl)-2-amino-3-(1H-indol-3-yl))propanamide |  | 49. 8 (±1.5) * |
| Model | MM-GBSA Binding Energy in kcal/mol (Standard Deviation) | ||
|---|---|---|---|
| Monomer-Membrane | Ligand-Monomer | Ligand-Membrane | |
| TGN1 | −85.9 (12.2) | −0.7 (3.3382) | −28.5 (4.3) |
| TGN2 | −129.8 (17.3) | −14.7 (2.4) | −11.3 (4.0) |
| TGN3 | −89.0 (12.0) | −9.2 (4.2) | −12.7 (8.2) |
| TGN4 | −34.6 (8.6) | −17.8 (2.6) | −2.9 (6.2) |
| No ligand (only monomer) | −109.3 (10.3) | - | - |
| Compound | Interaction and Occupied Residue | Residue of the Central Hydrophobic Cluster (CHC) |
|---|---|---|
| TGN1 | Undetectable | - |
| TGN2 |
| - |
| TGN3 |
| - |
| TGN4 |
| Val18 and Phe19 |
| Compound | Visualized Helical Cluster | Conserved Helical Cluster | New Helical Cluster |
|---|---|---|---|
| as Compared to the Native NMR Structure | |||
| TGN1 |
| 3 residues of the matched helical cluster:
| - |
| TGN2 |
| 14 residues of the matched helical cluster:
| 3 residues of the new helical cluster:
|
| TGN3 |
| 8 residues of the matched helical cluster:
| - |
| TGN4 |
| 11 residues of the matched helical cluster:
| 3 residues of the new helical cluster:
|
| No ligand (only monomer) |
| 7 residues of the matched helical cluster:
| 2 residues of the new helical cluster:
|
| NMR structure | Native helical clusters:
| - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wongrattanakamon, P.; Jiaranaikulwanitch, J.; Vajragupta, O.; Jiranusornkul, S.; Saenjum, C.; Yooin, W. Potential Anti-Alzheimer Agents from Guanidinyl Tryptophan Derivatives with Activities of Membrane Adhesion and Conformational Transition Inhibitions. Molecules 2021, 26, 4863. https://doi.org/10.3390/molecules26164863
Wongrattanakamon P, Jiaranaikulwanitch J, Vajragupta O, Jiranusornkul S, Saenjum C, Yooin W. Potential Anti-Alzheimer Agents from Guanidinyl Tryptophan Derivatives with Activities of Membrane Adhesion and Conformational Transition Inhibitions. Molecules. 2021; 26(16):4863. https://doi.org/10.3390/molecules26164863
Chicago/Turabian StyleWongrattanakamon, Pathomwat, Jutamas Jiaranaikulwanitch, Opa Vajragupta, Supat Jiranusornkul, Chalermpong Saenjum, and Wipawadee Yooin. 2021. "Potential Anti-Alzheimer Agents from Guanidinyl Tryptophan Derivatives with Activities of Membrane Adhesion and Conformational Transition Inhibitions" Molecules 26, no. 16: 4863. https://doi.org/10.3390/molecules26164863
APA StyleWongrattanakamon, P., Jiaranaikulwanitch, J., Vajragupta, O., Jiranusornkul, S., Saenjum, C., & Yooin, W. (2021). Potential Anti-Alzheimer Agents from Guanidinyl Tryptophan Derivatives with Activities of Membrane Adhesion and Conformational Transition Inhibitions. Molecules, 26(16), 4863. https://doi.org/10.3390/molecules26164863