
A “Pretender” Croconate-Bridged Macrocyclic Tetraruthenium Complex: Sizable Redox Potential Splittings despite Electronically Insulated Divinylphenylene Diruthenium Entities

 ,
, 
Abstract
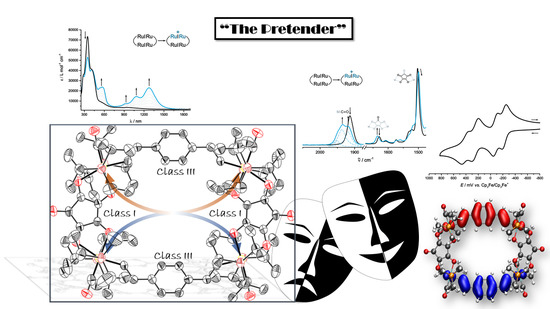
:
1. Introduction
2. Results and Discussion
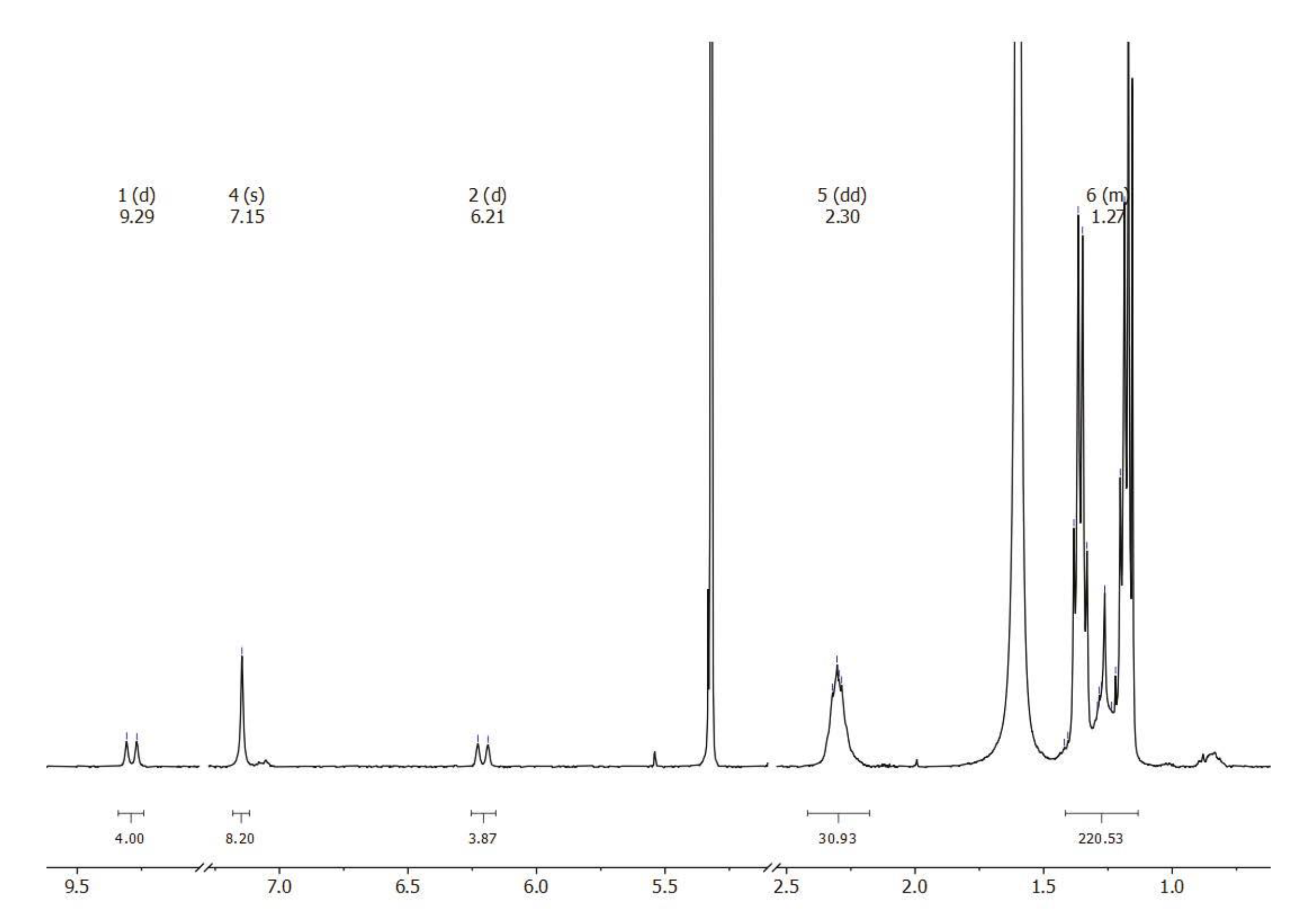
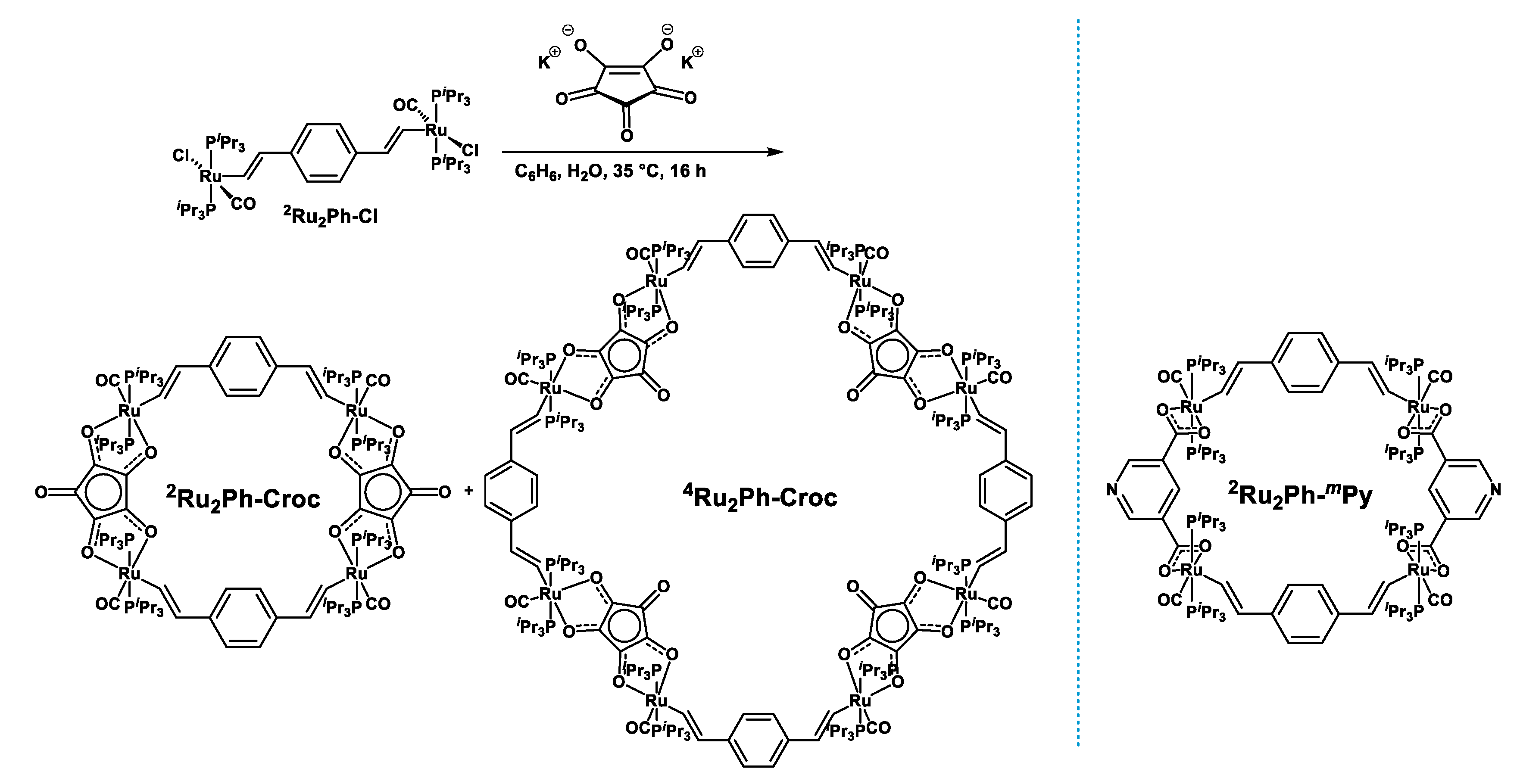
2.1. Synthesis, Spectroscopic Identification, and X-ray Crystallography
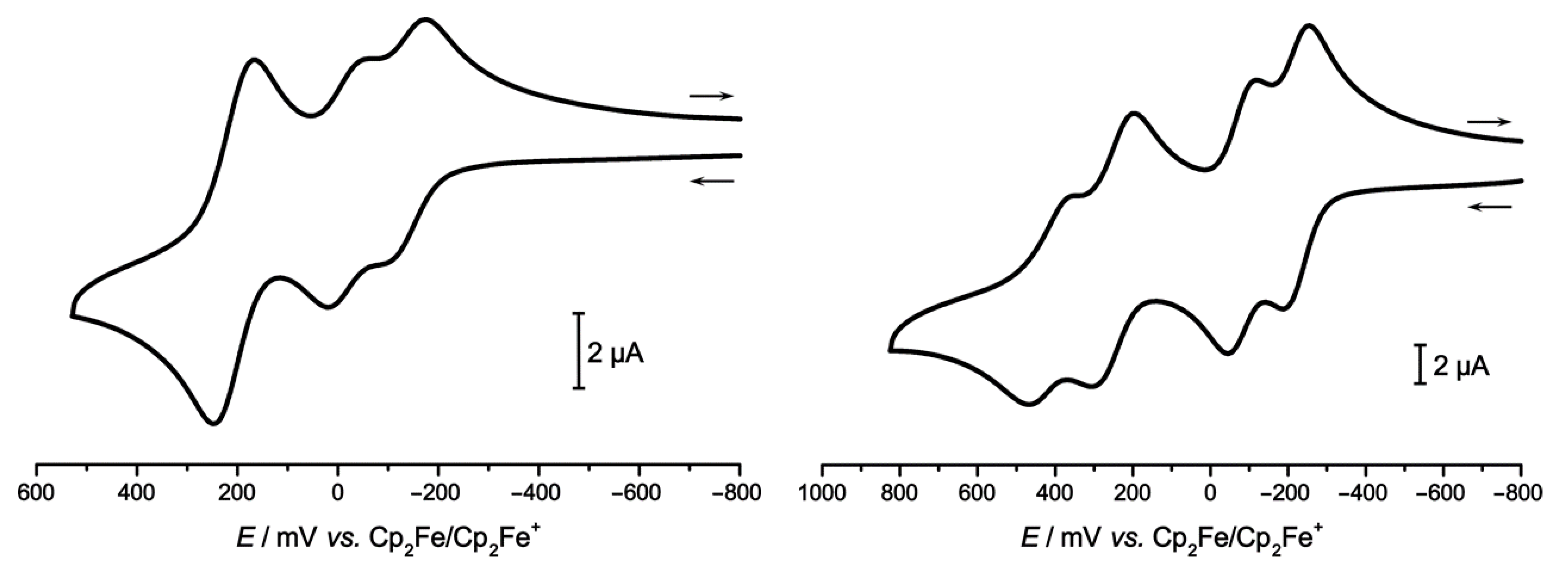
2.2. Electrochemistry
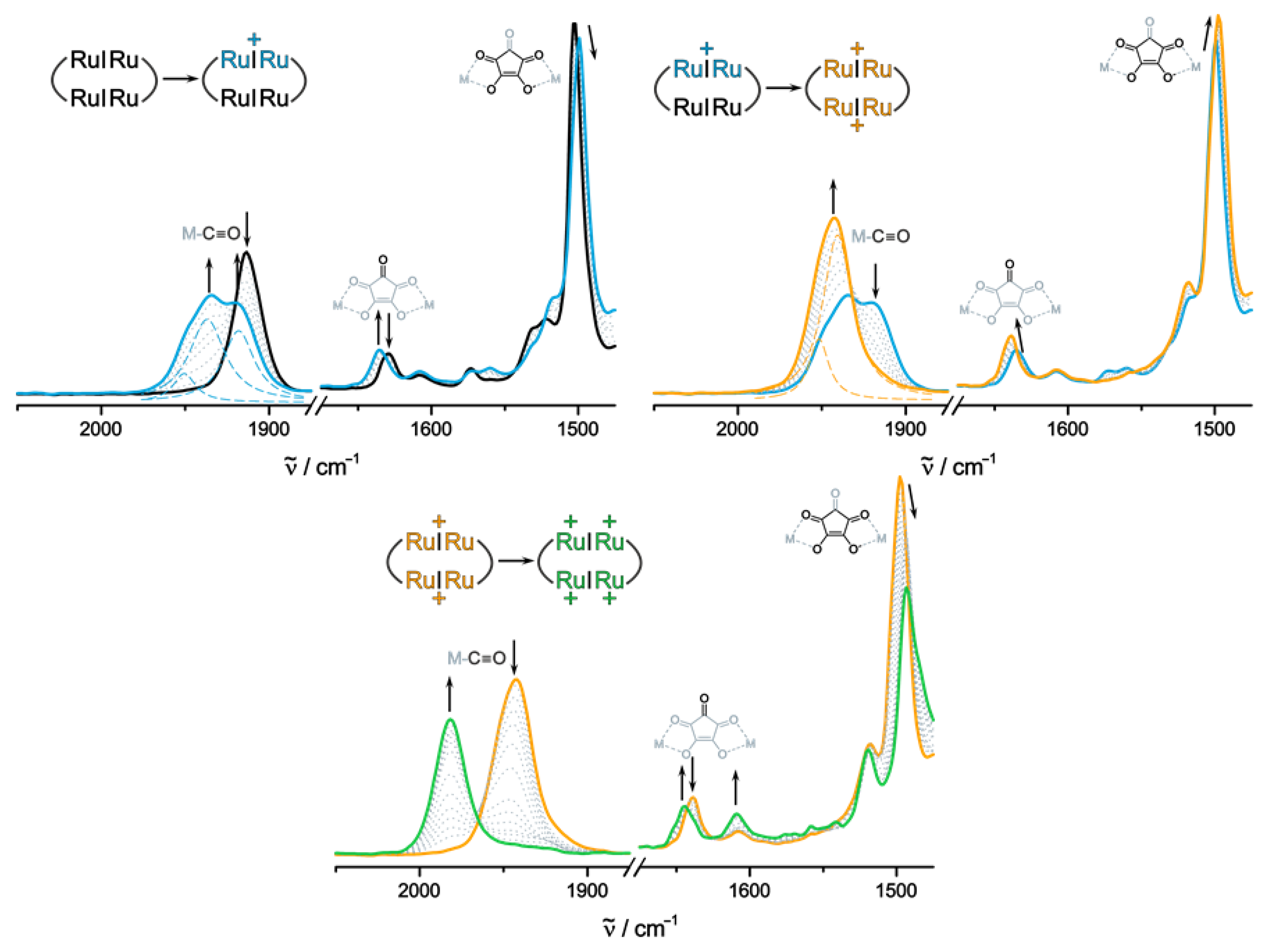
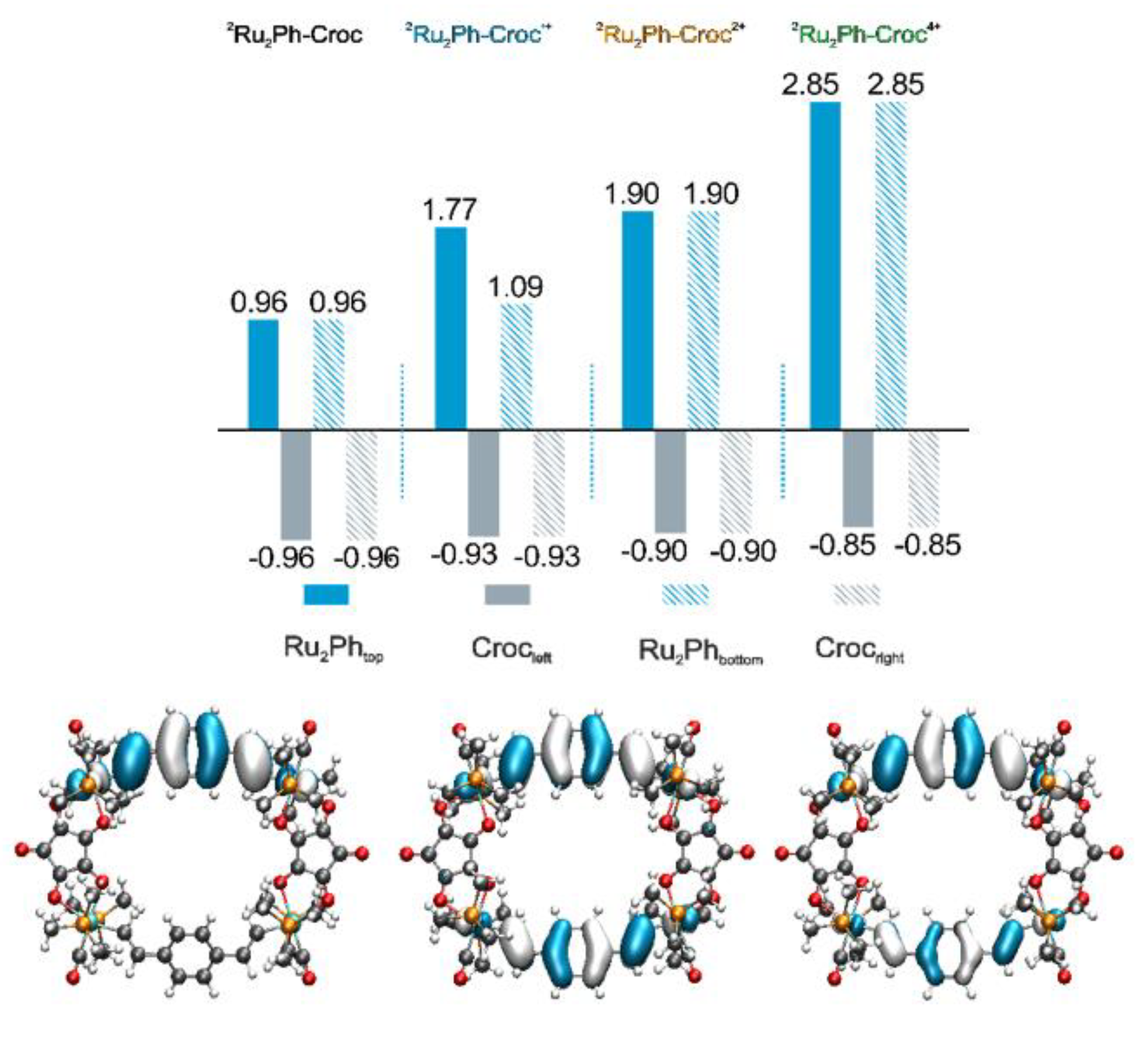
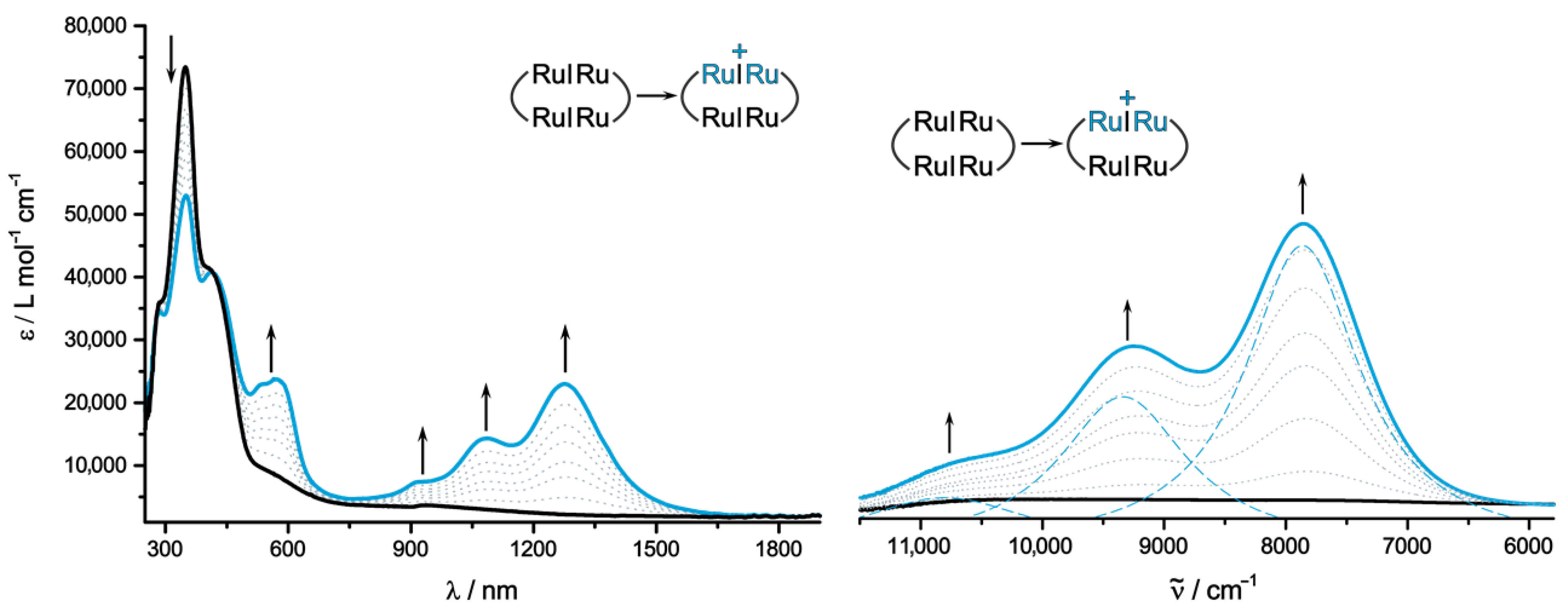
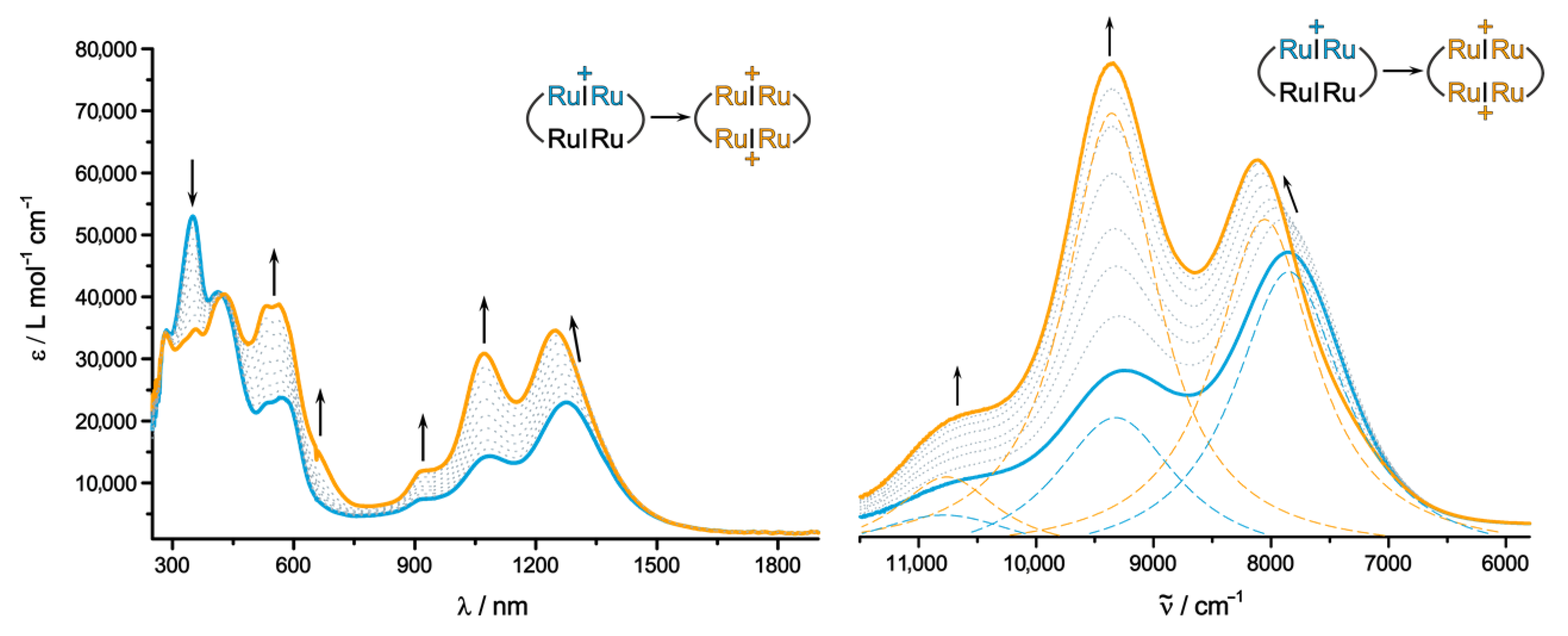
2.3. IR and UV/Vis/NIR Spectra of the Oxidized Forms and Insights from Quantum Chemistry
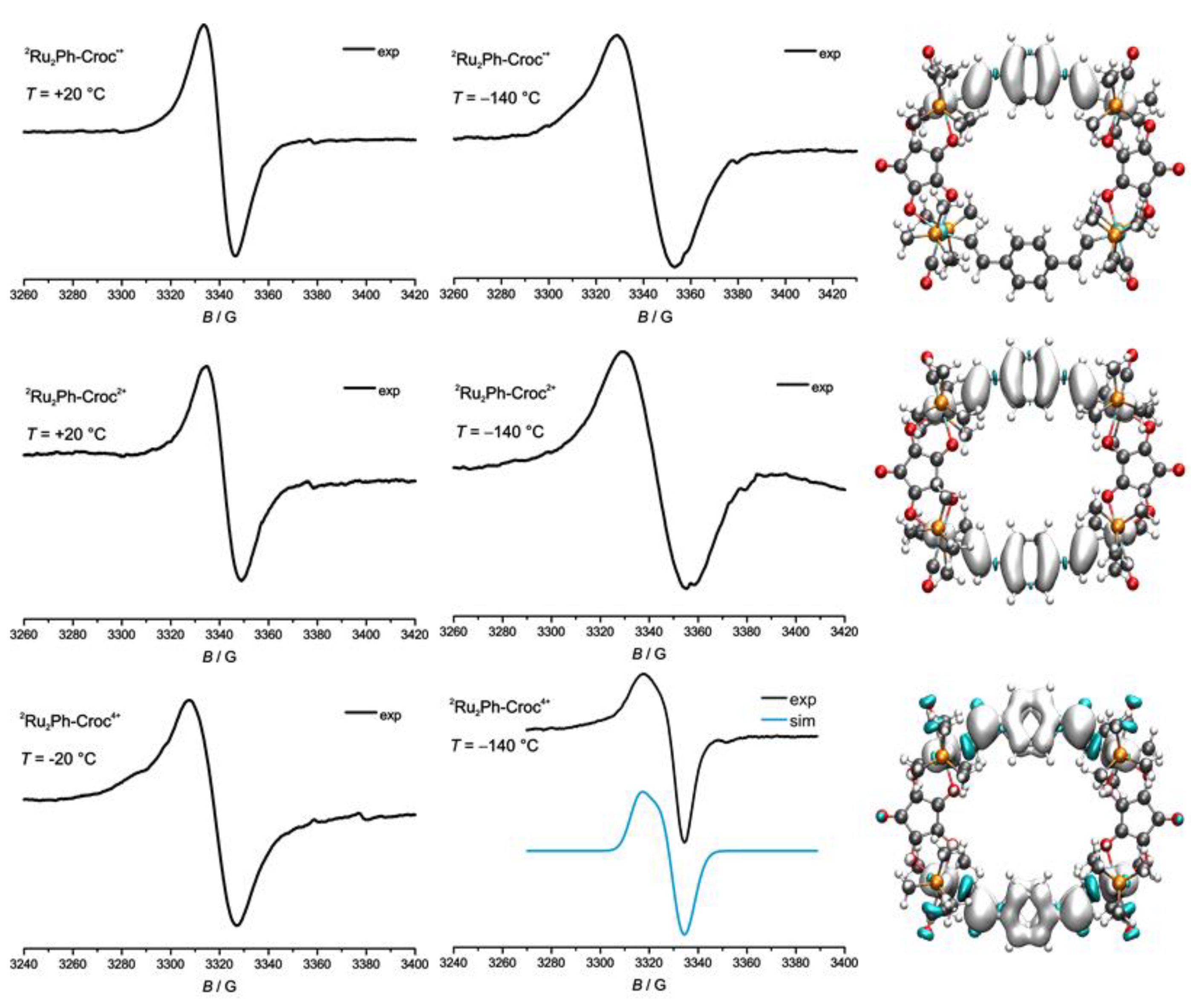
2.4. EPR Spectroscopy
3. Conclusions
4. Experimental Section
4.1. Computational Details
4.2. Materials and Methods
4.3. Synthesis
4.3.1. Synthesis and Isolation of Pure 2Ru2Ph-Croc
4.3.2. Isolation of Mixtures of 2Ru2Ph-Croc and 4Ru2Ph-Croc
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| [9]aneS3 | 1,4,7-trithiacyclononane |
| BArF24 | [B{C6H3(2,5-CF3)}]− |
| CT | charge transfer |
| EPR | electron paramagnetic resonance |
| FcH/FcH+ | ferrocene/ferrocenium |
| FMO | frontier molecular orbital |
| HOMO | energetically highest occupied molecular orbital |
| IR | infrared |
| IVCT | intervalence charge transfer |
| LUMO | energetically lowest unoccupied molecular orbital |
| LMCT | ligand-to-metal charge transfer |
| MLCT | metal-to-ligand charge transfer |
| NBO | natural bond order |
| NBu4 | tetranbutylammonium |
| NIR | near infrared |
| NMR | nuclear magnetic resonance |
| Ru2Ph | {Ru(CO)(PiPr3)2}(1,4-CH=CH-C6H4-CH=CH) |
| UV/Vis/NIR | electron spectroscopy in the ultraviolet, visible and near infrared |
| TD-DFT | time-dependent density functional theory |
References
- Khoshbin, M.S.; Ovchinnikov, M.V.; Mirkin, C.A.; Zakharov, L.N.; Rheingold, A.L. Binuclear Ruthenium Macrocycles Formed via the Weak-Link Approach. Inorg. Chem. 2005, 44, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.; Mukherjee, P.S.; Stang, P.J. Supramolecular Coordination: Self-Assembly of Finite Two- and Three-Dimensional Ensembles. Chem. Rev. 2011, 111, 6810–6918. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Saha, M.L.; Stang, P.J. Hierarchical Assemblies of Supramolecular Coordination Complexes. Acc. Chem. Res. 2018, 51, 2047–2063. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-F.; Jin, G.-X. Half-Sandwich Iridium- and Rhodium-based Organometallic Architectures: Rational Design, Synthesis, Characterization, and Applications. Acc. Chem. Res. 2014, 47, 3571–3579. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.R.; Stang, P.J. Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Gao, W.-X.; Lin, L.; Jin, G.-X. Recent advances in the construction and applications of heterometallic macrocycles and cages. Coord. Chem. Rev. 2017, 344, 323–344. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, H.-N.; Jin, G.-X. Molecular Borromean Rings Based on Half-Sandwich Organometallic Rectangles. Acc. Chem. Res. 2018, 51, 2148–2158. [Google Scholar] [CrossRef] [PubMed]
- Croue, V.; Goeb, S.; Szaloki, G.; Allain, M.; Salle, M. Reversible Guest Uptake/Release by Redox-Controlled Assembly/Disassembly of a Coordination Cage. Angew. Chem. Int. Ed. 2016, 55, 1746–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalóki, G.; Croué, V.; Carré, V.; Aubriet, F.; Alévêque, O.; Levillain, E.; Allain, M.; Aragó, J.; Ortí, E.; Goeb, S.; et al. Controlling the Host–Guest Interaction Mode through a Redox Stimulus. Angew. Chem. Int. Ed. 2017, 56, 16272–16276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goeb, S.; Sallé, M. Electron-rich Coordination Receptors Based on Tetrathiafulvalene Derivatives: Controlling the Host–Guest Binding. Acc. Chem. Res. 2021, 54, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.; Kunz, V.; Frischmann, P.D.; Würthner, F. A supramolecular ruthenium macrocycle with high catalytic activity for water oxidation that mechanistically mimics photosystem II. Nat. Chem. 2016, 8, 576. [Google Scholar] [CrossRef] [PubMed]
- Kunz, V.; Schulze, M.; Schmidt, D.; Würthner, F. Trinuclear Ruthenium Macrocycles: Toward Supramolecular Water Oxidation Catalysis in Pure Water. ACS Energy Lett. 2017, 2, 288–293. [Google Scholar] [CrossRef]
- Kunz, V.; Lindner, J.O.; Schulze, M.; Roehr, M.I.S.; Schmidt, D.; Mitric, R.; Würthner, F. Cooperative water oxidation catalysis in a series of trinuclear metallosupramolecular ruthenium macrocycles. Energy Environ. Sci. 2017, 10, 2137–2153. [Google Scholar] [CrossRef]
- Schindler, D.; Meza-Chincha, A.-L.; Roth, M.; Würthner, F. Structure-Activity Relationship for Di- up to Tetranuclear Macrocyclic Ruthenium Catalysts in Homogeneous Water Oxidation. Chem.-Eur. J. 2021. [Google Scholar] [CrossRef]
- Dinolfo, P.H.; Hupp, J.T. Tetra-Rhenium Molecular Rectangles as Organizational Motifs for the Investigation of Ligand-Centered Mixed-Valency: Three Examples of Full Delocalization. J. Am. Chem. Soc. 2004, 126, 16814–16819. [Google Scholar] [CrossRef] [PubMed]
- Dinolfo, P.H.; Williams, M.E.; Stern, C.L.; Hupp, J.T. Rhenium-Based Molecular Rectangles as Frameworks for Ligand-Centered Mixed-Valency and Optical Electron Transfer. J. Am. Chem. Soc. 2004, 126, 12989–13001. [Google Scholar] [CrossRef]
- Dinolfo, P.H.; Lee, S.J.; Coropceanu, V.; Brédas, J.-L.; Hupp, J.T. Borderline Class II/III Ligand-Centered Mixed Valency in a Porphyrinic Molecular Rectangle. Inorg. Chem. 2005, 44, 5789–5797. [Google Scholar] [CrossRef] [PubMed]
- Dinolfo, P.H.; Coropceanu, V.; Brédas, J.-L.; Hupp, J.T. A New Class of Mixed-Valence Systems with Orbitally Degenerate Organic Redox Centers. Examples Based on Hexa-Rhenium Molecular Prisms. J. Am. Chem. Soc. 2006, 128, 12592–12593. [Google Scholar] [CrossRef]
- Piotrowski, H.; Polborn, K.; Hilt, G.; Severin, K. A self-Assembled Metallamacrocyclic Ionophore with High Affinity and Selectivity for Li+ and Na+. J. Am. Chem. Soc. 2001, 123, 2699–2700. [Google Scholar] [CrossRef] [PubMed]
- Shan, N.; Vickers, S.J.; Adams, H.; Ward, M.D.; Thomas, J.A. Switchable Electron-Transfer Processes in a Mixed-Valence, Kinetically Locked, Trinuclear RuII Metallamacrocycle. Angew. Chem. Int. Ed. Eng. 2004, 43, 3938–3941. [Google Scholar] [CrossRef]
- Shan, N.; Ingram, J.D.; Easun, T.L.; Vickers, S.J.; Adams, H.; Ward, M.D.; Thomas, J.A. Kinetically locked, trinuclear RuII metallo-macrocycles—synthesis, electrochemical, and optical properties. Dalton Trans. 2006, 23, 2900–2906. [Google Scholar] [CrossRef]
- Maurer, J.; Linseis, M.; Sarkar, B.; Schwederski, B.; Niemeyer, M.; Kaim, W.; Záliš, S.; Anson, C.; Zabel, M.; Winter, R.F. Ruthenium Complexes with Vinyl, Styryl, and Vinylpyrenyl Ligands: A Case of Non-Innocence in Organometallic Chemistry. J. Am. Chem. Soc. 2008, 130, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuttke, E.; Hervault, Y.-M.; Polit, W.; Linseis, M.; Erler, P.; Rigaut, S.; Winter, R.F. Divinylphenylene- and Ethynylvinylphenylene-Bridged Mono-, Di-, and Triruthenium Complexes for Covalent Binding to Gold Electrodes. Organometallics 2014, 33, 4672–4686. [Google Scholar] [CrossRef]
- Abdel-Rahman, O.S.; Maurer, J.; Záliš, S.; Winter, R.F. Ruthenium Styryl Complexes with Ligands Derived from 2-Hydroxy- and 2-Mercaptopyridine and 2-Hydroxy- and 2-Mercaptoquinoline. Organometallics 2015, 34, 3611–3628. [Google Scholar] [CrossRef]
- Fink, D.; Linseis, M.; Winter, R.F. Constitutional Isomers of Macrocyclic Tetraruthenium Complexes with Vastly Different Spectroscopic and Electrochemical Properties. Organometallics 2018, 37, 1817–1820. [Google Scholar] [CrossRef]
- Fink, D.; Bodensteiner, M.; Linseis, M.; Winter, R.F. Macrocyclic Triruthenium Complexes Having Electronically Coupled Mixed-Valent States. Chem. Eur. J. 2018, 24, 992–996. [Google Scholar] [CrossRef]
- Scheerer, S.; Linseis, M.; Wuttke, E.; Weickert, S.; Drescher, M.; Tröppner, O.; Ivanović-Burmazović, I.; Irmler, A.; Pauly, F.; Winter, R.F. Redox-Active Tetraruthenium Macrocycles Built from 1,4-Divinylphenylene-Bridged Diruthenium Complexes. Chem. Eur. J. 2016, 22, 9574–9590. [Google Scholar] [CrossRef]
- Fink, D.; Weibert, B.; Winter, R.F. Redox-active tetraruthenium metallacycles: Reversible release of up to eight electrons resulting in strong electrochromism. Chem. Commun. 2016, 52, 6103–6106. [Google Scholar] [CrossRef] [Green Version]
- Anders, P.; Rapp, M.; Linseis, M.; Winter, R. Tetraruthenium Metallamacrocycles with Potentially Coordinating Appended Functionalities. Inorganics 2018, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Fink, D.; Orth, N.; Linseis, M.; Ivanović-Burmazović, I.; Winter, R.F. Ring size matters: Supramolecular isomerism in self-assembled redox-active tetra- and hexaruthenium macrocycles. Chem. Commun. 2020, 56, 1062–1065. [Google Scholar] [CrossRef]
- Fink, D.; Orth, N.; Linseis, M.; Ivanović-Burmazović, I.; Winter, R.F. Structural Versatility and Supramolecular Isomerism in Redox-Active Tetra- and Hexaruthenium Macrocycles. Eur. J. Inorg. Chem. 2020, 2020, 2816–2829. [Google Scholar] [CrossRef]
- Fink, D.; Staiger, A.; Orth, N.; Linseis, M.; Ivanović-Burmazović, I.; Winter, R.F. Redox-Induced Hydrogen Bond Reorientation Mimicking Electronic Coupling in Mixed-Valent Diruthenium and Macrocyclic Tetraruthenium Complexes. Inorg. Chem. 2020, 59, 16703–16715. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W. Manifestations of Noninnocent Ligand Behavior. Inorg. Chem. 2011, 50, 9752–9765. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W. The Shrinking World of Innocent Ligands: Conventional and Non-Conventional Redox-Active Ligands. Eur. J. Inorg. Chem. 2012, 2012, 343–348. [Google Scholar] [CrossRef]
- Kaim, W. Electron Transfer Reactivity of Organometallic Compounds Involving Radical-Forming Noninnocent Ligands. Proc. Nat. Acad. Sci. Ind. A 2016, 86, 445–457. [Google Scholar] [CrossRef]
- Ghumaan, S.; Mukherjee, S.; Kar, S.; Roy, D.; Mobin, S.M.; Sunoj, R.B.; Lahiri, G.K. An Experimental and Density Functional Theory Approach Towards the Establishment of Preferential Metal- or Ligand-Based Electron-Transfer Processes in Large Quinonoid-Bridged Diruthenium Complexes [{(aap)2Ru}2(μ-BL2–)]n+ (aap = 2-Arylazopyridine). Eur. J. Inorg. Chem. 2006, 2006, 4426–4441. [Google Scholar] [CrossRef]
- Ghumaan, S.; Sarkar, B.; Maji, S.; Puranik, V.G.; Fiedler, J.; Urbanos, F.A.; Jimenez-Aparicio, R.; Kaim, W.; Lahiri, G.K. Valence-State Analysis through Spectroelectrochemistry in a Series of Quinonoid-Bridged Diruthenium Complexes [(acac)2Ru(μ-L)Ru(acac)2]n (n=+2, +1, 0, −1, −2). Chem. Eur. J. 2008, 14, 10816–10828. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Sarkar, B.; Mobin, S.M.; Fiedler, J.; Urbanos, F.A.; Jimenez-Aparicio, R.; Kaim, W.; Lahiri, G.K. Valence-State Alternatives in Diastereoisomeric Complexes [(acac)2Ru(μ-QL)Ru(acac)2]n (QL2− = 1,4-Dioxido-9,10-anthraquinone,n = +2, +1, 0, −1, −2). Inorganic Chemistry 2008, 47, 5204–5211. [Google Scholar] [CrossRef]
- Mandal, A.; Agarwala, H.; Ray, R.; Plebst, S.; Mobin, S.M.; Priego, J.L.; Jiménez-Aparicio, R.; Kaim, W.; Lahiri, G.K. Sensitivity of the Valence Structure in Diruthenium Complexes as a Function of Terminal and Bridging Ligands. Inorg. Chem. 2014, 53, 6082–6093. [Google Scholar] [CrossRef]
- Ansari, M.A.; Mandal, A.; Paretzki, A.; Beyer, K.; Kaim, W.; Lahiri, G.K. Isomeric Diruthenium Complexes of a Heterocyclic and Quinonoid Bridging Ligand: Valence and Spin Alternatives for the Metal/Ligand/Metal Arrangement. Inorg. Chem. 2016, 55, 12357–12365. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W.; Lahiri, G.K. The coordination potential of indigo, anthraquinone and related redox-active dyes. Coord. Chem. Rev. 2019, 393, 1–8. [Google Scholar] [CrossRef]
- Gmelin, L. Über einige merkwürdige, bei der Darstellung des Kaliums nach der Brunner’schen Methode erhaltene Substanzen. Poggendorfs Ann. Phys. 1825, 4, 31–62. [Google Scholar] [CrossRef] [Green Version]
- Braga, D.; Maini, L.; Grepioni, F. Croconic Acid and Alkali Metal Croconate Salts: Some New Insights into an Old Story. Chem. Eur. J. 2002, 8, 1804–1812. [Google Scholar] [CrossRef]
- Ranganathan, A.; Kulkarni, G.U. An Experimental Electron Density Investigation of Squarate and Croconate Dianions. J. Phys. Chem. A 2002, 106, 7813–7819. [Google Scholar] [CrossRef]
- Georgopoulos, S.L.; Garcia, H.C.; Edwards, H.G.M.; Cappa de Oliveira, L.F. Spectroscopic and structural investigation of oxocarbon salts with tetraalkylammonium ions. J. Mol. Struct. 2016, 1108, 542–551. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Najafian, K.; Kiran, B.; Jiao, H. Are Oxocarbon Dianions Aromatic? J. Org. Chem. 2000, 65, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Maurer, J.; Sarkar, B.; Schwederski, B.; Kaim, W.; Winter, R.F.; Záliš, S. Divinylphenylene bridged diruthenium complexes bearing Ru(CO)Cl(PiPr3)2 entities. Organometallics 2006, 25, 3701–3712. [Google Scholar] [CrossRef] [Green Version]
- Linseis, M.; Winter, R.F.; Sarkar, B.; Kaim, W.; Záliš, S. Multistep Electrochromic Behavior from an Organometallic Tetranuclear Complex of a Tetradonor-Substituted Olefin. Organometallics 2008, 27, 3321–3324. [Google Scholar] [CrossRef] [Green Version]
- Linseis, M.; Záliš, S.; Zabel, M.; Winter, R.F. Ruthenium Stilbenyl and Diruthenium Distyrylethene Complexes: Aspects of Electron Delocalization and Electrocatalyzed Isomerization of the Z-Isomer. J. Am. Chem. Soc. 2012, 134, 16671–16692. [Google Scholar] [CrossRef]
- Pfaff, U.; Hildebrandt, A.; Korb, M.; Oßwald, S.; Linseis, M.; Schreiter, K.; Spange, S.; Winter, R.F.; Lang, H. Electronically Strongly Coupled Divinylheterocyclic-Bridged Diruthenium Complexes. Chem. Eur. J. 2016, 22, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, E.; Fink, D.; Anders, P.; Maria Hoyt, A.-L.; Polit, W.; Linseis, M.; Winter, R.F. Homo- and heterobimetallic 1,4-divinylphenylene- and naphthalene-1,8-divinyl-bridged diruthenium, diosmium and ruthenium osmium complexes. J. Organomet. Chem. 2016, 821, 4–18. [Google Scholar] [CrossRef]
- Rotthowe, N.; Zwicker, J.; Winter, R.F. Influence of Quinoidal Distortion on the Electronic Properties of Oxidized Divinylarylene-Bridged Diruthenium Complexes. Organometallics 2019, 38, 2782–2799. [Google Scholar] [CrossRef]
- Doane, L.M.; Fatiadi, A.J. Electrochemical oxidation of several oxocarbon salts in N,N-dimethylformamide. J. Electroanal. Chem. Interfacial Electrochem. 1982, 135, 193–209. [Google Scholar] [CrossRef]
- Fabre, P.-L.; Dumestre, F.; Soula, B.; Galibert, A.-M. Spectroelectrochemical behavior in dimethylformamide of pseudo-oxocarbons dianions derived from the croconate dianion. Electrochim. Acta 2000, 45, 2697–2705. [Google Scholar] [CrossRef]
- Dumestre, F.; Soula, B.; Galibert, A.-M.; Fabre, P.-L.; Bernardinelli, G.; Donnadieu, B.; Castan, P. Synthesis and characterization of cobalt(II) complexes of croconate and dicyanomethylene-substituted derivatives. J. Chem. Soc. Dalton Trans. 1998, 24, 4131–4138. [Google Scholar] [CrossRef]
- Massoud, S.S.; Vicente, R.; Fontenot, P.R.; Gallo, A.A.; Mikuriya, M.; Albering, J.H.; Mautner, F.A. Polynuclear croconato-bridged-copper(II) complexes derived from tri- and tetra-dentate amines. Polyhedron 2012, 46, 66–73. [Google Scholar] [CrossRef]
- Carranza, J.; Sletten, J.; Lloret, F.; Julve, M. Manganese(II) complexes with croconate and 2-(2-pyridyl)imidazole ligands: Syntheses, X-ray structures and magnetic properties. Inorg. Chim. Acta 2009, 362, 2636–2642. [Google Scholar] [CrossRef]
- Brouca-Cabarrecq, C.; Trombe, J.-C. f Element croconates 1. Lanthanide croconates—Synthesis, crystal structure and thermal behavior. Inorg. Chim. Acta 1992, 191, 227–240. [Google Scholar] [CrossRef]
- Castro, I.; Calatayud, M.L.; Lloret, F.; Sletten, J.; Julve, M. Syntheses, crystal structures and magnetic properties of di- and trinuclear croconato-bridged copper(ii) complexes. J. Chem. Soc. Dalton Trans. 2002, 11, 2397–2403. [Google Scholar] [CrossRef]
- Carranza, J.; Brennan, C.; Sletten, J.; Vangdal, B.; Rillema, P.; Lloret, F.; Julve, M. Syntheses, crystal structures and magnetic properties of new oxalato-, croconato- and squarato-containing copper(ii) complexes. New J. Chem. 2003, 27, 1775–1783. [Google Scholar] [CrossRef]
- Wang, C.-C.; Ke, M.-J.; Tsai, C.-H.; Chen, I.H.; Lin, S.-I.; Lin, T.-Y.; Wu, L.-M.; Lee, G.-H.; Sheu, H.-S.; Fedorov, V.E. [M(C5O5)2(H2O)n]2− as a Building Block for Hetero- and Homo-bimetallic Coordination Polymers: From 1D Chains to 3D Supramolecular Architectures. Cryst. Growth Des. 2009, 9, 1013–1019. [Google Scholar] [CrossRef]
- Mautner, F.A.; Albering, J.H.; Vicente, R.; Louka, F.R.; Gallo, A.A.; Massoud, S.S. Copper(II) complexes derived from tripodal tris[(2-ethyl-(1-pyrazolyl)]amine. Inorg. Chim. Acta 2011, 365, 290–296. [Google Scholar] [CrossRef]
- Zheng, Y.-R.; Stang, P.J. Direct and Quantitative Characterization of Dynamic Ligand Exchange between Coordination-Driven Self-Assembled Supramolecular Polygons. J. Am. Chem. Soc. 2009, 131, 3487–3489. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-R.; Zhou, H.-C. Bridging-ligand-substitution strategy for the preparation of metal–organic polyhedra. Nat. Chem. 2010, 2, 893–898. [Google Scholar] [CrossRef]
- Neogi, S.; Lorenz, Y.; Engeser, M.; Samanta, D.; Schmittel, M. Heteroleptic Metallosupramolecular Racks, Rectangles, and Trigonal Prisms: Stoichiometry-Controlled Reversible Interconversion. Inorg. Chem. 2013, 52, 6975–6984. [Google Scholar] [CrossRef] [PubMed]
- Garci, A.; Gupta, G.; Dalvit, C.; Therrien, B. Investigating the Formation Mechanism of Arene Ruthenium Metallacycles by NMR Spectroscopy. Eur. J. Inorg. Chem. 2014, 2014, 5651–5661. [Google Scholar] [CrossRef]
- Garci, A.; Marti, S.; Schurch, S.; Therrien, B. Insight into the dynamic ligand exchange process involved in bipyridyl linked arene ruthenium metalla-rectangles. RSC Adv. 2014, 4, 8597–8604. [Google Scholar] [CrossRef]
- Burgstahler, A.W.; Barkhurst, R.C. Preparation of Leuconic Acid from Inositol. Trans. Kansas Acad. Sci. 1968, 71, 150–153. [Google Scholar] [CrossRef]
- Williams, R.F.X. Transition Metal Complexes with Organo-Chalcogen Ligands. I. Synthesis of the Dithoiocroconate Dianion. Phosphorus Sulf. Rel. Elem. 1976, 2, 141–146. [Google Scholar] [CrossRef]
- Spek, A. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. C Cryst. Struct. Commun. 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Seiler, P.; Czechtizky, W. Crystal Structure of Potassium Croconate Dihydrate, after 175 Years. Angew. Chem. Int. Ed. 2001, 40, 1779–1780. [Google Scholar] [CrossRef]
- Richardson, D.E.; Taube, H. Mixed-valence molecules: Electronic delocalization and stabilization. Coord. Chem. Rev. 1984, 60, 107–129. [Google Scholar] [CrossRef]
- Crutchley, R.J. Intervalence Charge Transfer and Electron Exchange Studies of Dinuclear Ruthenium Complexes. In Advances in Inorganic Chemistry; Sykes, A.G., Ed.; Academic Press: Cambridge, MA, USA, 1994; Volume 41, pp. 273–325. [Google Scholar]
- Evans, C.E.B.; Naklicki, M.L.; Rezvani, A.R.; White, C.A.; Kondratiev, V.V.; Crutchley, R.J. An Investigation of Superexchange in Dinuclear Mixed-Valence Ruthenium Complexes. J. Am. Chem. Soc. 1998, 120, 13096–13103. [Google Scholar] [CrossRef]
- D’Alessandro, D.M.; Keene, F.R. A cautionary warning on the use of electrochemical measurements to calculate comproportionation constants for-mixed-valence compounds. Dalton Trans. 2004, 3950–3954. [Google Scholar] [CrossRef] [PubMed]
- Fellows, E.A.; Keene, F.R. Influence of Anions on Intervalence Charge Transfer (IVCT) in Mixed-Valence Dinuclear Complexes. J. Phys. Chem. B 2007, 111, 6667–6675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, R.F. Half-Wave Potential Splittings ΔE1/2 as a Measure of Electronic Coupling in Mixed-Valent Systems: Triumphs and Defeats. Organometallics 2014, 33, 4517–4536. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Miesel, D.; Lang, H. Electrostatic interactions within mixed-valent compounds. Coord. Chem. Rev. 2018, 371, 56–66. [Google Scholar] [CrossRef]
- Ribeiro, M.C.; de Oliveira, L.F.; Gonçalves, N.S. Boson peak in the room-temperature molten salt tetra(n-butyl)ammonium croconate. Phys. Rev. B 2001, 63, 104303. [Google Scholar] [CrossRef]
- Krejcik, M.; Danek, M.; Hartl, F. Simple construction of an infrared optically transparent thin-layer cell: Applications to the redox reactions of ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-di-t-butyl-catecholate)−. J. Electroanal. Chem. 1991, 317, 179–187. [Google Scholar] [CrossRef]
- Hassenrück, C.; Mücke, P.; Scheck, J.; Demeshko, S.; Winter, R.F. Oxidized Styrylruthenium–Ferrocene Conjugates: From Valence Localization to Valence Tautomerism. Eur. J. Inorg. Chem. 2017, 2017, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Záliš, S.; Winter, R.F.; Kaim, W. Quantum chemical interpretation of redox properties of ruthenium complexes with vinyl and TCNX type non-innocent ligands. Coord. Chem. Rev. 2010, 254, 1383–1396. [Google Scholar]
- Scheerer, S.; Rotthowe, N.; Abdel-Rahman, O.S.; He, X.; Rigaut, S.; Kvapilová, H.; Záliš, S.; Winter, R.F. Vinyl Ruthenium-Modified Biphenyl and 2,2′-Bipyridines. Inorg. Chem. 2015, 54, 3387–3402. [Google Scholar] [CrossRef]
- Chen, J.; Winter, R.F. Studies on a Vinyl Ruthenium-Modified Squaraine Dye: Multiple Vis/NIR Absorbance Switching through Dye and Substituent-Based Redox Processes. Chem. Eur. J. 2012, 18, 10733–10741. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Orth, N.; Ebel, V.; Gogesch, F.S.; Staiger, A.; Linseis, M.; Ivanović-Burmazović, I.; Winter, R.F. Self-Assembled Redox-Active Tetraruthenium Macrocycles with Large Intracyclic Cavities. Organometallics 2020, 39, 1861–1880. [Google Scholar] [CrossRef]
- Petrov, A.I.; Lutoshkin, M.A. TD-DFT assessment of UV-vis spectra palladium and platinum complexes with thiols and disulfides. J. Mol. Model. 2021, 27, 152. [Google Scholar] [CrossRef] [PubMed]
- Gąsiorski, P.; Matusiewicz, M.; Gondek, E.; Uchacz, T.; Danel, A.; Wojtasik, K.; Vlokh, R.; Kityk, A.V. Synthesis, UV–Vis spectroscopy and DFT/TDDFT calculations on 6-substituted halogen derivatives of 1,3-diphenyl-1H-pyrazolo [3,4−b] quinoxalines dyes. J. Lumin. 2017, 192, 288–296. [Google Scholar] [CrossRef]
- Amat, A.; Miliani, C.; Romani, A.; Fantacci, S. DFT/TDDFT investigation on the UV-vis absorption and fluorescence properties of alizarin dye. Phys. Chem. Chem. Phys. 2015, 17, 6374–6382. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, M.; Gückel, S.; Renz, M.; Klawohn, S.; Theilacker, K.; Parthey, M.; Lambert, C. Electron transfer pathways in mixed-valence paracyclophane-bridged bis-triarylamine radical cations. J. Comput. Chem. 2016, 37, 93–102. [Google Scholar] [CrossRef]
- Sakamaki, D.; Ito, A.; Tsutsui, Y.; Seki, S. Tetraaza[14]- and Octaaza[18]paracyclophane: Synthesis and Characterization of Their Neutral and Cationic States. J. Org. Chem. 2017, 82, 13348–13358. [Google Scholar] [CrossRef]
- Kaupp, M.; Renz, M.; Parthey, M.; Stolte, M.; Würthner, F.; Lambert, C. Computational and spectroscopic studies of organic mixed-valence compounds: Where is the charge? Phys. Chem. Chem. Phys. 2011, 13, 16973–16986. [Google Scholar] [CrossRef] [Green Version]
- Parthey, M.; Kaupp, M. Quantum-chemical insights into mixed-valence systems: Within and beyond the Robin-Day scheme. Chem. Soc. Rev. 2014, 43, 5067–5088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian, Inc., Wallingford CT, Gaussian 09, Revision D.01. 2016. Available online: https://gaussian.com/ (accessed on 28 August 2021).
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. J. Chem. Phys. 1994, 100, 7532–7535. [Google Scholar] [CrossRef]
- Andrae, D.; Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Hariharan, P.H.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Enzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tange, O. GNU Parallel 2018. 2018. Available online: https://www.gnu.org/software/parallel/ (accessed on 28 August 2021). [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Persistence of Vision Pty. Ltd. Persistence of Vision Raytracer (Version 3.7). 2004. Available online: https://www.povray.org/ (accessed on 28 August 2021).
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Res. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrolyte | E1/20/+ [mV] | E1/2+/2+ [mV] | ΔE1/20/+//+/2+ [mV] | E1/22+/3+ [mV] | E1/23+/4+ [mV] | ΔE1/22+/3+//3+/4+ [mV] | |
|---|---|---|---|---|---|---|---|
| 2Ru2Ph-Croc | NBu4PF6 | −134 | −17 | 117 | 207 | 0 | |
| NBu4BArF24 | −226 | −85 | 141 | 245 | 410 | 165 | |
| Ru2Ph-Cl a | NBu4PF6 | −75 | 175 | 250 | - | - | - |
| NBu4BArF24 | −180 | 130 | 310 | - | - | - | |
| 2Ru2Ph-mPy a | NBu4BArF24 | −431 | −334 | 97 | −39 | 42 | 81 |
| [cm−1] | |||||||
|---|---|---|---|---|---|---|---|
| 2Ru2Ph-Croc | IR Label | Neutral | Monocation | Dication (Singlet) | Dication (Triplet) | Tetracation (Singlet) | Tetracation (Quintet) |
| Calculated a,b | Ru(CO) | 1913, 1914 | 1918, 1920, 1934, 1942 | 1939, 1942, 1942, 1949 | 1939, 1939, 1945, 1948 | 1977, 1978, 1981, 1983 | 1983, 1984, 1988, 1989 |
| C=OCroc | 1652 | 1656 | 1655 | 1659 | 1664 | 1661 | |
| C-Crocking | 1485 | 1482 | 1486 | 1479 | 1477 | 1473 | |
| Experimental | Ru(CO) | 1913 | 1917, 1935, 1949 | 1941, 1952 | 1982 | ||
| C=OCroc | 1630 | 1636 | 1639 | 1644 | |||
| C-Crocking | 1503 | 1499 | 1498 | 1493 | |||
| λmax [nm] (ε [M−1 cm−1]) | TD-DFT calc. Transitions b | |||
|---|---|---|---|---|
| λcalc [nm] | Contribution (Minor) | Character | ||
| 0 | 550 (8900), 410 (41,100), 350 (73,400) | 626–608 431 329 | H → L, (H-1 → L+1) H-2 → L H → L+4 | ML-L′CT π-π*Croc π-π*Ru2Ph |
| 1+ | 1275 (23,000), 1085 (14,300), 930 (7300), 570 (23,700), 535 (22,900), 410 (41,100), 350 (53,000) | 2171 1011 663 505 | βH → βL βH-1 → βL βH → βL+2 αH-1 → αL | IVCT (n.o.) ML-L′CT ML-L′CT ML-L′CT |
| 2+ b | 1240 (34,600), 1067 (30,800), 930 (11,900), 570 (38,800), 535 (38,800), 410 (41,100), 350 (34,600) | 928, 923 529 | βH → βL, (βH-1 → βL+1) αH-1 → αL | CR ML-L′CT |
| 4+ c | 650 (sh), 605 (7,6500), 410 (48,400) | 940–923 574 400 | H → L, (H-1 → L+1) H-2 → L, (H-3 → L+1) H → L+2 | L′-MLCT (n.o.) π-π*Ru2Ph π-π*Croc |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotthowe, N.; Linseis, M.; Vogelsang, L.; Orth, N.; Ivanović-Burmazović, I.; Winter, R.F. A “Pretender” Croconate-Bridged Macrocyclic Tetraruthenium Complex: Sizable Redox Potential Splittings despite Electronically Insulated Divinylphenylene Diruthenium Entities. Molecules 2021, 26, 5232. https://doi.org/10.3390/molecules26175232
Rotthowe N, Linseis M, Vogelsang L, Orth N, Ivanović-Burmazović I, Winter RF. A “Pretender” Croconate-Bridged Macrocyclic Tetraruthenium Complex: Sizable Redox Potential Splittings despite Electronically Insulated Divinylphenylene Diruthenium Entities. Molecules. 2021; 26(17):5232. https://doi.org/10.3390/molecules26175232
Chicago/Turabian StyleRotthowe, Nils, Michael Linseis, Lars Vogelsang, Nicole Orth, Ivana Ivanović-Burmazović, and Rainer F. Winter. 2021. "A “Pretender” Croconate-Bridged Macrocyclic Tetraruthenium Complex: Sizable Redox Potential Splittings despite Electronically Insulated Divinylphenylene Diruthenium Entities" Molecules 26, no. 17: 5232. https://doi.org/10.3390/molecules26175232
APA StyleRotthowe, N., Linseis, M., Vogelsang, L., Orth, N., Ivanović-Burmazović, I., & Winter, R. F. (2021). A “Pretender” Croconate-Bridged Macrocyclic Tetraruthenium Complex: Sizable Redox Potential Splittings despite Electronically Insulated Divinylphenylene Diruthenium Entities. Molecules, 26(17), 5232. https://doi.org/10.3390/molecules26175232