
Adulteration of Essential Oils: A Multitask Issue for Quality Control. Three Case Studies: Lavandula angustifolia Mill., Citrus limon (L.) Osbeck and Melaleuca alternifolia (Maiden & Betche) Cheel

 ,
,  ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Reference Chemical Profiles of Genuine Essential Oils
2.2. Adulteration with Cheaper Essential Oils or Synthetic Compounds
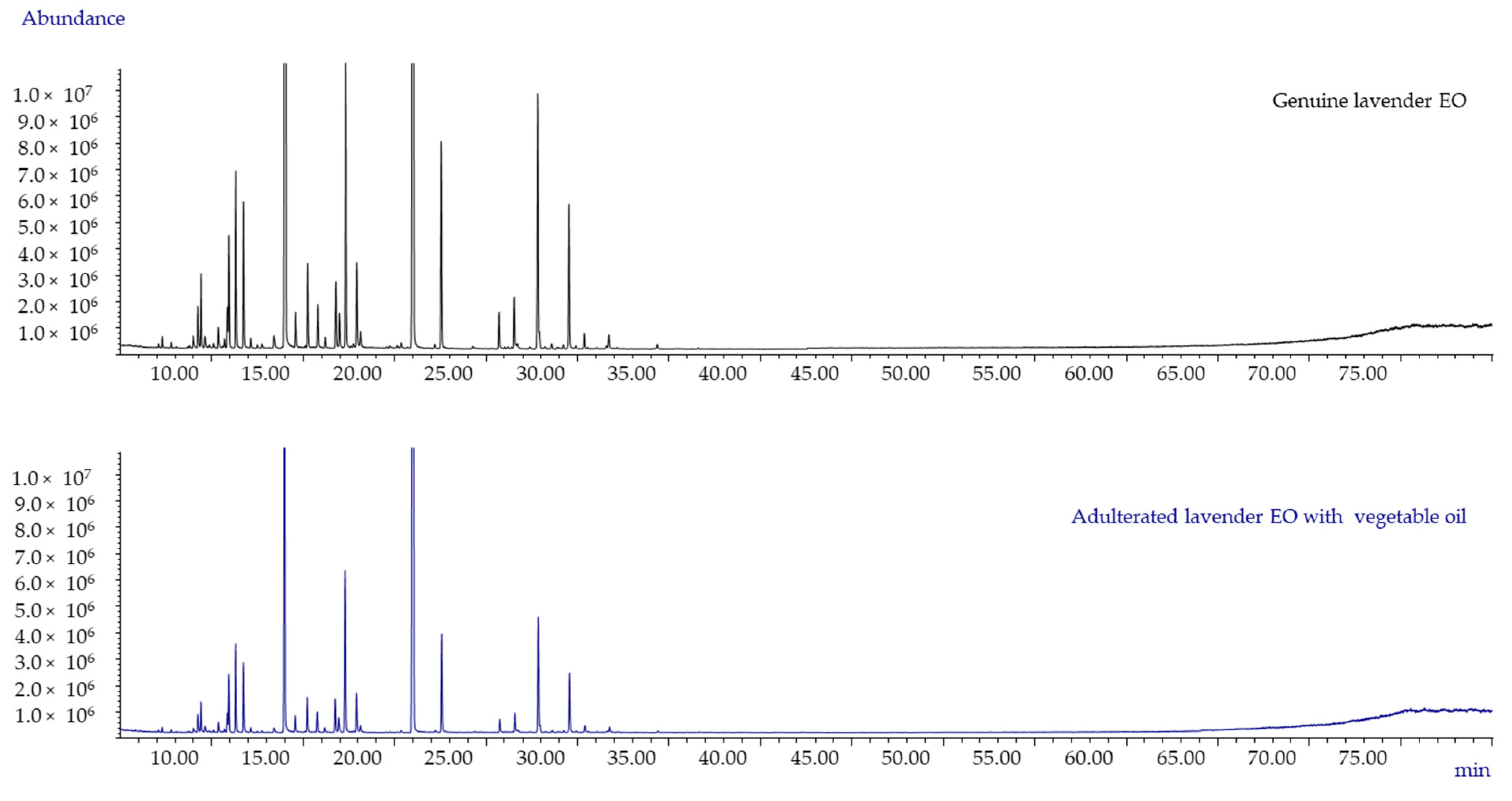
2.3. Commercial EO Adulterated with Vegetable Oils
3. Materials and Methods
3.1. Essential Oils, Standards and Materials
3.2. GC Analysis Conditions
3.3. HS-SPME Sampling Conditions
3.4. Quantitative Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- European Directorate for the Quality of Medicines & HealthCare (EDQM), European Pharmacopoeia, 10th ed.; EDQM Council of Europe: Strasbourg, France, 2021.
- Aromatic Natural Raw Materials—Vocabulary; ISO NORM 9235:2013; International Organization for Standardization: Geneva, Switzerland, 2013.
- Bicchi, C.; Liberto, E.; Matteodo, M.; Sgorbini, B.; Mondello, L.; d’Acampora Zellner, B.; Costa, R.; Rubiolo, P. Quantitative analysis of essential oils: A complex task. Flavour Fragr. J. 2008, 23, 382–391. [Google Scholar] [CrossRef]
- Can Baser, K.H.; Buchbauer, G. (Eds.) Handbook of Essential Oilss—Science, Technology and Applications; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Cagliero, C.; Sgorbini, B.; Cordero, C.; Liberto, E.; Rubiolo, P.; Bicchi, C. Enantioselective Gas Chromatography with Derivatized Cyclodextrins in the Flavour and Fragrance Field. Isr. J. Chem. 2016, 56, 925–939. [Google Scholar] [CrossRef]
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach; Wiley: Chirchester, UK, 2009. [Google Scholar]
- Do, T.K.T.; Hadji-Minaglou, F.; Antoniotti, S.; Fernandez, X. Authenticity of essential oils. Trends Anal. Chem. 2015, 66, 146–157. [Google Scholar] [CrossRef]
- Boren, K.E.; Young, D.G.; Woolley, C.L.; Smith, B.L.; Carlson, R.E. Detecting Essential Oil Adulteration. J. Environ. Anal. Chem. 2015, 2, 1000132. [Google Scholar]
- Feudjio, W.M.; Ghalila, H.; Nsangou, M.; Majdi, Y.; Kongbonga, Y.M.; Jaïdane, N. Fluorescence Spectroscopy Combined with Chemometrics for the Investigation of the Adulteration of Essential Oils. Food Anal. Methods 2017, 10, 2539–2548. [Google Scholar] [CrossRef]
- Vargas Jentzsch, P.; Gualpa, F.; Ramos, L.A.; Ciobotă, V. Adulteration of clove essential oil: Detection using a handheld Raman spectrometer. Flavour Fragr. J. 2017, 33, 184–190. [Google Scholar] [CrossRef]
- Truzzi, E.; Marchetti, L.; Bertelli, D.; Benvenuti, S. Attenuated Total Reflectance−Fourier Transform Infrared (ATR−FTIR) Spectroscopy Coupled with Chemometric Analysis for Detection and Quantification of Adulteration in Lavender and Citronella Essential Oils. Phytochem. Anal. 2021, No. pca.3034. [Google Scholar] [CrossRef]
- Truzzi, E.; Marchetti, L.; Benvenuti, S.; Ferroni, A.; Rossi, M.C.; Bertelli, D. Novel Strategy for the Recognition of Adulterant Vegetable Oils in Essential Oils Commonly Used in Food Industries by Applying (13)C NMR Spectroscopy. J. Agric. Food Chem. 2021, 69, 8276–8286. [Google Scholar] [CrossRef]
- Essential Oils Market. Available online: https://www.statista.com/study/60656/essential-oils-market/ (accessed on 1 July 2021).
- Verzera, A.; Lamonica, G.; Mondello, L.; Trozzi, A.; Dugo, G. The composition of bergamot oil. Perf. Flavor. 1996, 21, 19–35. [Google Scholar]
- Mondello, L.; Verzera, A.; Previti, P.; Crispo, F.; Dugo, G. Multidimensional capillary GC-GC for the analysis of complex samples. Enantiomeric distribution of monoterpene hydrocarbons, monoterpene alcohols and linalyl acetate of bergamot (Citrus bergamia Risso et Poiteau) oils. J. Agric. Food Chem. 1998, 46, 4275–4282. [Google Scholar] [CrossRef]
- ISO 4730:2017. Essential Oil of Melaleuca, Terpinen-4-ol Type (Tea Tree Oil). Available online: www.iso.org (accessed on 1 July 2021).
- Wong, Y.F.; Davies, N.W.; Chin, S.T.; Larkman, T.; Marriott, P.J. Enantiomeric distribution of selected terpenes for authenticity assessment of Australian Melaleuca alternifolia oil. Ind. Crops Prod. 2015, 67, 475–483. [Google Scholar] [CrossRef]
- Sgorbini, B.; Cagliero, C.; Boggia, L.; Liberto, E.; Reichenbach, S.E.; Rubiolo, P.; Cordero, C.; Bicchi, C. Parallel dual secondary-column-dual detection comprehensive two-dimensional gas chromatography: A flexible and reliable analytical tool for essential oils quantitative profiling. Flavour Fragr. J. 2015, 30, 366–380. [Google Scholar] [CrossRef]
- Agnel, R.; Teisseire, P. Essential Oil of French Lavender—Its Composition and Its Adulteration. Perf. Flavor. 1984, 9, 53–56. [Google Scholar]
- Bicchi, C.; D’Amato, A.; Manzin, V.; Galli, A.; Galli, M. Cyclodextrin derivatives in the gas chromatographic separation of racemic mixtures of volatile compounds X. 2,3-Di-O-ethyl-6-O-tert-butyldimethylsilyl-β- and -γ-cyclodextrins. J. Chromatogr. A 1996, 742, 161–173. [Google Scholar] [CrossRef]
- Adams, R.P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry, 4th ed.; Allured Publ.: Carol Stream, IL, USA, 2007. [Google Scholar]
- Liberto, E.; Cagliero, C.; Sgorbini, B.; Bicchi, C.; Sciarrone, D.; D’Acampora Zellner, B.; Mondello, L.; Rubiolo, P. Enantiomer identification in the flavour and fragrance fields by “interactive” combination of linear retention indices from enantioselective gas chromatography and mass spectrometry. J. Chromatogr. A 2008, 1195, 117–126. [Google Scholar] [CrossRef]
- Costa, R.; d’Acampora Zellner, B.; Crupi, M.L.; De Fina, M.R.; Valentino, M.R.; Dugo, P.; Dugo, G.; Mondello, L. GC-MS, GC-O and enantio-GC investigation of the essential oil of Tarchonanthus camphoratus L. Flavour Fragr. J. 2008, 23, 40–48. [Google Scholar] [CrossRef]
- Wang, Y.; O’Reilly, J.; Chen, Y.; Pawliszyn, J. Equilibrium in-fibre standardisation technique for solid-phase microextraction. J. Chromatogr. A 2005, 1072, 13–17. [Google Scholar] [CrossRef]
- Bicchi, C.; Cordero, C.; Liberto, E.; Sgorbini, B.; Rubiolo, P. Reliability of fibres insolid phase microextraction for routine analysis of the headspace of aromaticand medicinal plants. J. Chromatogr. A 2007, 1152, 138–149. [Google Scholar] [CrossRef]
- Markelov, M.; Guzowski, J.P. Matrix independent headspace gas chromatographic analysis. This full evaporation technique. Anal. Chim. Acta 1993, 276, 235–245. [Google Scholar] [CrossRef]
- Kolb, B.; Ettre, L.S. Static Headspace-Gas Chromatography, Theory and Practice; Wiley–VCH: New York, NY, USA, 1997. [Google Scholar]
- Ezquerro, O.; Ortiz, G.; Pons, B.; Tena, M.T. Determination of benzene, toluene, ethylbenzene and xylenes in soils by multiple headspace solid-phase microextraction. J. Chromatogr. A 2004, 1035, 17–22. [Google Scholar] [CrossRef]
- Bicchi, C.; Ruosi, M.R.; Cagliero, C.; Cordero, C.; Liberto, E.; Rubiolo, P.; Sgorbini, B. Quantitative analysis of volatiles from solid matrices of vegetable origin by high concentration capacity headspace techniques: Determination of furan in roasted coffee. J. Chromatogr. A 2011, 1218, 753–762. [Google Scholar] [CrossRef]
- Sgorbini, B.; Bicchi, C.; Cagliero, C.; Cordero, C.; Liberto, E.; Rubiolo, P. Herbs and spices: Characterization and quantitation of biologically-active markers for routine quality control by multiple headspace solid-phase microextraction combined with separative or non-separative analysis. J. Chromatogr. A 2015, 1376, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Klee, M.S.; Blumberg, L.M. Theoretical and practical aspects of fast gas chromatography and method translation. J. Chromatogr. Sci. 2002, 40, 234–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GC Method Translation Software. Available online: https://www.agilent.com/en-us/support/gas-chromatography/gcmethodtranslation?searchTermRedirect=gc%20method%20translation (accessed on 1 July 2021).




{kind=link}
{kind=link}
{kind=link}
| Citrus limon(L.) Osbeck | ||||||||
| Compounds | Itsexp | Itslit | Min | Max | Average | σ | Ital. Ph. | |
| 2 | sabinene | 976 | 976 | 0.8 | 1.0 | 0.9 | 0.1 | 0.5–2.0 |
| 3 | β-pinene | 978 | 980 | 5.6 | 6.9 | 6.5 | 0.4 | 5.0–10.0 |
| 5 | β-myrcene | 992 | 991 | 0.7 | 1.5 | 1.0 | 0.2 | |
| 7 | α-terpinene | 1018 | 1018 | 0.1 | 0.2 | 0.1 | 0.1 | |
| 8 | p-cymene | 1028 | 1026 | 0.2 | 0.7 | 0.4 | 0.2 | |
| 9 | limonene | 1031 | 1031 | 34.3 | 40.9 | 37.8 | 2.3 | 30.0–50.0 |
| 12 | trans-β-ocimene | 1051 | 1050 | 0.1 | 0.2 | 0.1 | 0.1 | |
| 13 | γ-terpinene | 1061 | 1062 | 5.3 | 8.0 | 6.7 | 0.9 | 6.0–18.5 |
| 14 | α-terpinolene | 1089 | 1088 | 0.3 | 0.6 | 0.3 | 0.1 | |
| 15 | linalool | 1100 | 1098 | 9.6 | 12.3 | 10.7 | 0.8 | 6.0–15.0 |
| 21 | neral | 1243 | 1240 | 0.1 | 0.3 | 0.2 | 0.1 | |
| 22 | linalyl acetate | 1264 | 1257 | 30.0 | 35.8 | 31.5 | 1.6 | 23.0–35.0 |
| 23 | geranial | 1273 | 1270 | 0.1 | 0.5 | 0.3 | 0.1 | max 0.5 |
| 25 | neryl acetate | 1369 | 1365 | 0.3 | 0.8 | 0.5 | 0.1 | |
| 26 | geranyl acetate | 1386 | 1383 | 0.3 | 0.7 | 0.4 | 0.1 | 0.1–0.7 |
| 27 | trans-β-caryophyllene | 1419 | 1418 | 0.1 | 0.6 | 0.3 | 0.1 | 0.2–0.5 |
| 30 | β-bisabolene | 1510 | 1509 | 0.1 | 0.4 | 0.2 | 0.1 | |
| Lavandula angustifoliaMill. | ||||||||
| Compounds | Itsexp | Itslit | Min | Max | Average | σ | Eur. Ph. | |
| 4 | 3-octanone | 989 | 986 | 0.4 | 1.3 | 0.9 | 0.3 | 0.1–5.0 |
| 9 | limonene | 1031 | 1031 | 0.4 | 1.1 | 0.7 | 0.2 | max 1.0 |
| 10 | 1,8-cineole | 1033 | 1033 | 1.3 | 2.6 | 2.4 | 1.2 | max 2.5 |
| 11 | cis-β-ocimene | 1041 | 1040 | 0.7 | 3.2 | 2.4 | 0.6 | |
| 12 | trans-β-ocimene | 1051 | 1050 | 0.2 | 2.2 | 1.8 | 0.5 | |
| 15 | linalool | 1100 | 1098 | 23.8 | 33.0 | 29.5 | 2.5 | 20.0–45.0 |
| 16 | 1-octen-3-yl acetate | 1116 | 1110 | 0.4 | 1.1 | 0.7 | 0.2 | |
| 17 | camphor | 1147 | 1143 | 0.6 | 1.2 | 1.0 | 0.2 | max 1.2 |
| 18 | lavandulol | 1171 | 1166 | 0.6 | 1.7 | 0.9 | 0.4 | min 0.1 |
| 19 | 4-terpineol | 1178 | 1177 | 2.6 | 6.0 | 3.6 | 1.0 | 0.1–8.0 |
| 20 | α-terpineol | 1191 | 1189 | 0.1 | 2.0 | 1.0 | 0.6 | max 2.0 |
| 22 | linalyl acetate | 1264 | 1257 | 25.1 | 40.7 | 35.4 | 4.0 | 25.0–47.0 |
| 24 | lavandulyl acetate | 1293 | 1289 | 2.7 | 5.3 | 3.5 | 0.7 | min 0.2 |
| 27 | trans-β-caryophyllene | 1419 | 1418 | 1.8 | 5.1 | 3.5 | 1.0 | |
| 29 | trans-β-farnesene | 1460 | 1458 | 1.2 | 5.2 | 2.5 | 1.0 | |
| Melaleuca alternifolia(Maiden & Betche) Cheel | ||||||||
| Compounds | Itsexp | Itslit | Min | Max | Average | σ | Eur. Ph. | |
| 1 | α-pinene | 936 | 939 | 1.8 | 6.0 | 3.8 | 1.8 | 1.0–6.0 |
| 3 | β-pinene | 978 | 980 | 0.1 | 1.1 | 0.6 | 0.3 | |
| 5 | β-myrcene | 992 | 991 | 0.1 | 1.1 | 0.5 | 0.4 | |
| 6 | α-phellandrene | 1004 | 1005 | 0.0 | 0.6 | 0.3 | 0.2 | |
| 7 | α-terpinene | 1018 | 1018 | 7.7 | 10.5 | 9.2 | 0.7 | 5.0–13.0 |
| 8 | p-cymene | 1028 | 1026 | 0.7 | 3.6 | 2.1 | 0.8 | 0.5–12.0 |
| 9 | limonene | 1031 | 1031 | 0.8 | 3.7 | 2.5 | 1.0 | 0.5–4.0 |
| 10 | 1,8-cineole | 1033 | 1033 | 1.9 | 7.1 | 3.9 | 1.5 | max 15.0 |
| 13 | γ-terpinene | 1061 | 1062 | 14.3 | 22.4 | 19.2 | 2.4 | 10.0–28.0 |
| 14 | α-terpinolene | 1089 | 1088 | 2.0 | 4.7 | 3.1 | 0.8 | 1.5–5.0 |
| 19 | 4-terpineol | 1178 | 1177 | 32.4 | 47.1 | 40.5 | 3.4 | min 30.0 |
| 20 | α-terpineol | 1191 | 1189 | 2.6 | 7.1 | 5.0 | 1.4 | 1.5–8.0 |
| 28 | aromadendrene | 1439 | 1439 | 0.1 | 5.8 | 1.3 | 1.4 | max 7.0 |
| Bergamot EO | Lavender EO | |||||||
| Itsexp | Itslit | Genuine | Adulterated CB-1 EO | Reference Values [15] | Genuine | Adulterated CL-1 EO | Reference Values [1] | |
| (R)-(−)-linalool | 1181 | 1174 | 99.6 | 76.0 | 99.4–99.7 | 93.8 | 66.4 | |
| (S)-(+)-linalool | 1196 | 1189 | 0.4 | 24.0 | 0.3–0.6 | 6.2 | 33.6 | max 12% |
| (R)-(−)-linalyl acetate | 1233 | 1231 | 99.7 | 89.1 | 99.7–99.9 | 99.4 | 80.4 | |
| (S)-(+)-linalyl acetate | 1243 | 1237 | 0.3 | 10.9 | 0.1–0.3 | 0.6 | 19.6 | max 1% |
| Chinese TTO | Australian TTO | |||||||
| Itsexp | Itslit | Genuine | Genuine | Adulterated CT-1 EO | Reference Values [16,17] | |||
| (R)-(−)-4-terpineol | 1258 | 1253 | 57.8 | 30.8 | 45.5 | |||
| (S)-(+)-4-terpineol | 1250 | 1248 | 42.2 | 69.2 | 54.5 | 67.4–69.6 | ||
| (R)-(+)-α-terpineol | 1317 | 1309 | 77.0 | 75.8 | 76.7 | 71.0–78.0 | ||
| (S)-(−)-α-terpineol | 1302 | 1296 | 23.0 | 24.2 | 23.3 | |||
| Citrus limonEO | Lavandula angustifoliaEO | ||||||||||||
| Genuine | Spiked Samples | Com. Sample CB-1 EO | Genuine | Spiked Samples | Com. Sample CL-1 EO | ||||||||
| Itsexp | Itslit | % | σ | % | σ | % | % | σ | % | σ | % | σ | |
| linalool | 1100 | 1098 | 11.4 | 0.1 | 15.2 | 0.1 | 15.0 | 30.0 | 0.2 | 38.8 | 0.1 | 39.2 | 0.3 |
| linalyl acetate | 1260 | 1259 | 27.5 | 0.2 | 32.6 | 0.4 | 33.1 | 35.0 | 0.4 | 42.8 | 0.1 | 42.9 | 0.5 |
| Melaleuca alternifoliaEO | |||||||||||||
| Australian | Mixed Origins | Com. Sample CT-1 EO | |||||||||||
| Itsexp | Itslit | % | σ | % | σ | % | σ | ||||||
| 4-terpineol | 1178 | 1177 | 44.0 | 0.6 | 42.6 | 0.1 | 41.4 | 0.6 | |||||
| α-terpineol | 1190 | 1189 | 3.0 | 0.1 | 4.8 | 0.04 | 5.0 | 0.7 | |||||
| Essential Oil | (Linalool) (g/100 g) | σ | (Linalyl Acetate) (g/100 g) | σ | (4-Terpineol) (g/100 g) | σ | (α-Terpineol) (g/100 g) | σ | |
|---|---|---|---|---|---|---|---|---|---|
| C. limon | genuine | 10.5 | 0.2 | 25.9 | 0.4 | ||||
| 5% spiked | 9.6 | 0.1 | 23.8 | 0.6 | |||||
| 20% spiked | 7.1 | 0.3 | 21.8 | 0.2 | |||||
| 50% spiked | 5.0 | 0.1 | 14.4 | 0.3 | |||||
| L. angustifolia | genuine | 23.4 | 1.2 | 27.0 | 0.9 | ||||
| 5% spiked | 20.1 | 0.5 | 23.4 | 0.2 | |||||
| 20% spiked | 18.4 | 0.3 | 21.2 | 0.6 | |||||
| 50% spiked | 13.4 | 0.2 | 15.3 | 0.4 | |||||
| CL-2 EO | 14.5 | 0.3 | 16.8 | 0.2 | |||||
| M. alternifolia | genuine | 44.2 | 2.7 | 8.5 | 1.5 | ||||
| 5% spiked | 42.0 | 2.2 | 6.9 | 0.9 | |||||
| 20% spiked | 33.5 | 1.8 | 3.5 | 0.2 | |||||
| 50% spiked | 26.7 | 2.1 | 2.4 | 0.1 |
| Essential Oil | EC% | ((R)-(−)-Linalool) (g/100 g) | σ | EC% | ((S)-(+)-Linalool) (g/100 g) | σ | |
|---|---|---|---|---|---|---|---|
| L. angustifolia | genuine | 99.6 | 22.1 | 0.2 | 0.4 | 1.5 | 0.1 |
| 5% spiked | 99.6 | 19.2 | 0.2 | 0.4 | 1.3 | 0.1 | |
| 20% spiked | 99.6 | 16.7 | 0.3 | 0.4 | 0.91 | 0.1 | |
| 50% spiked | 99.6 | 12.3 | 0.2 | 0.4 | 0.62 | 0.08 | |
| CL-2 EO | 99.6 | 14.5 | 0.2 | 0.4 | 0.74 | 0.09 | |
| EC% | ((R)-(−)-linalyl acetate) (g/100 g) | σ | EC% | ((S)-(+)-linalyl acetate) (g/100 g) | σ | ||
| genuine | 99.7 | 26.7 | 0.4 | 0.3 | 0.44 | 0.2 | |
| 5% spiked | 99.7 | 23.1 | 0.2 | 0.3 | 0.34 | 0.2 | |
| 20% spiked | 99.7 | 20.8 | 0.2 | 0.3 | 0.29 | 0.1 | |
| 50% spiked | 99.7 | 15.1 | 0.1 | 0.3 | 0.21 | 0.06 | |
| CL-2 EO | 99.7 | 16.8 | 0.2 | 0.3 | 0.26 | 0.07 |
| Essential Oil | Botanical Name | Plant Part Used | Selected Marker Compounds | Target Ion |
|---|---|---|---|---|
| Bergamot | Citrus limon (L.) Osbeck | Peel | Linalool, linalyl acetate | 71, 93 |
| Lavender | Lavandula angustifolia Mill. | Aerial part | Linalool, linalyl acetate | 71, 93 |
| Tea tree | Melaleuca alternifolia (Maiden & Betche) Cheel | Leaves | 4-terpineol, α-terpineol | 71, 59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capetti, F.; Marengo, A.; Cagliero, C.; Liberto, E.; Bicchi, C.; Rubiolo, P.; Sgorbini, B. Adulteration of Essential Oils: A Multitask Issue for Quality Control. Three Case Studies: Lavandula angustifolia Mill., Citrus limon (L.) Osbeck and Melaleuca alternifolia (Maiden & Betche) Cheel. Molecules 2021, 26, 5610. https://doi.org/10.3390/molecules26185610
Capetti F, Marengo A, Cagliero C, Liberto E, Bicchi C, Rubiolo P, Sgorbini B. Adulteration of Essential Oils: A Multitask Issue for Quality Control. Three Case Studies: Lavandula angustifolia Mill., Citrus limon (L.) Osbeck and Melaleuca alternifolia (Maiden & Betche) Cheel. Molecules. 2021; 26(18):5610. https://doi.org/10.3390/molecules26185610
Chicago/Turabian StyleCapetti, Francesca, Arianna Marengo, Cecilia Cagliero, Erica Liberto, Carlo Bicchi, Patrizia Rubiolo, and Barbara Sgorbini. 2021. "Adulteration of Essential Oils: A Multitask Issue for Quality Control. Three Case Studies: Lavandula angustifolia Mill., Citrus limon (L.) Osbeck and Melaleuca alternifolia (Maiden & Betche) Cheel" Molecules 26, no. 18: 5610. https://doi.org/10.3390/molecules26185610
APA StyleCapetti, F., Marengo, A., Cagliero, C., Liberto, E., Bicchi, C., Rubiolo, P., & Sgorbini, B. (2021). Adulteration of Essential Oils: A Multitask Issue for Quality Control. Three Case Studies: Lavandula angustifolia Mill., Citrus limon (L.) Osbeck and Melaleuca alternifolia (Maiden & Betche) Cheel. Molecules, 26(18), 5610. https://doi.org/10.3390/molecules26185610