
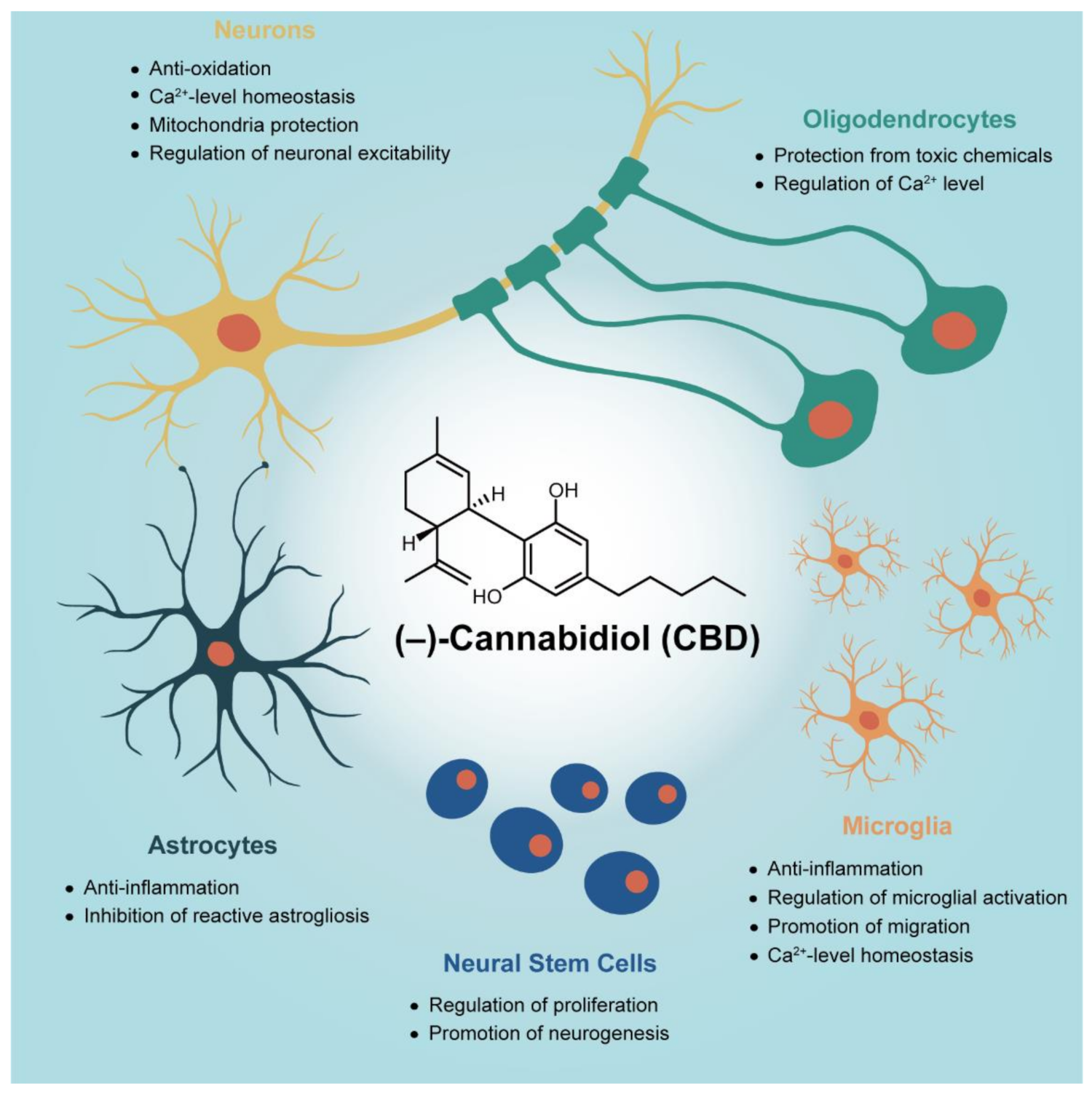
In Vitro Studies on Therapeutic Effects of Cannabidiol in Neural Cells: Neurons, Glia, and Neural Stem Cells


Abstract
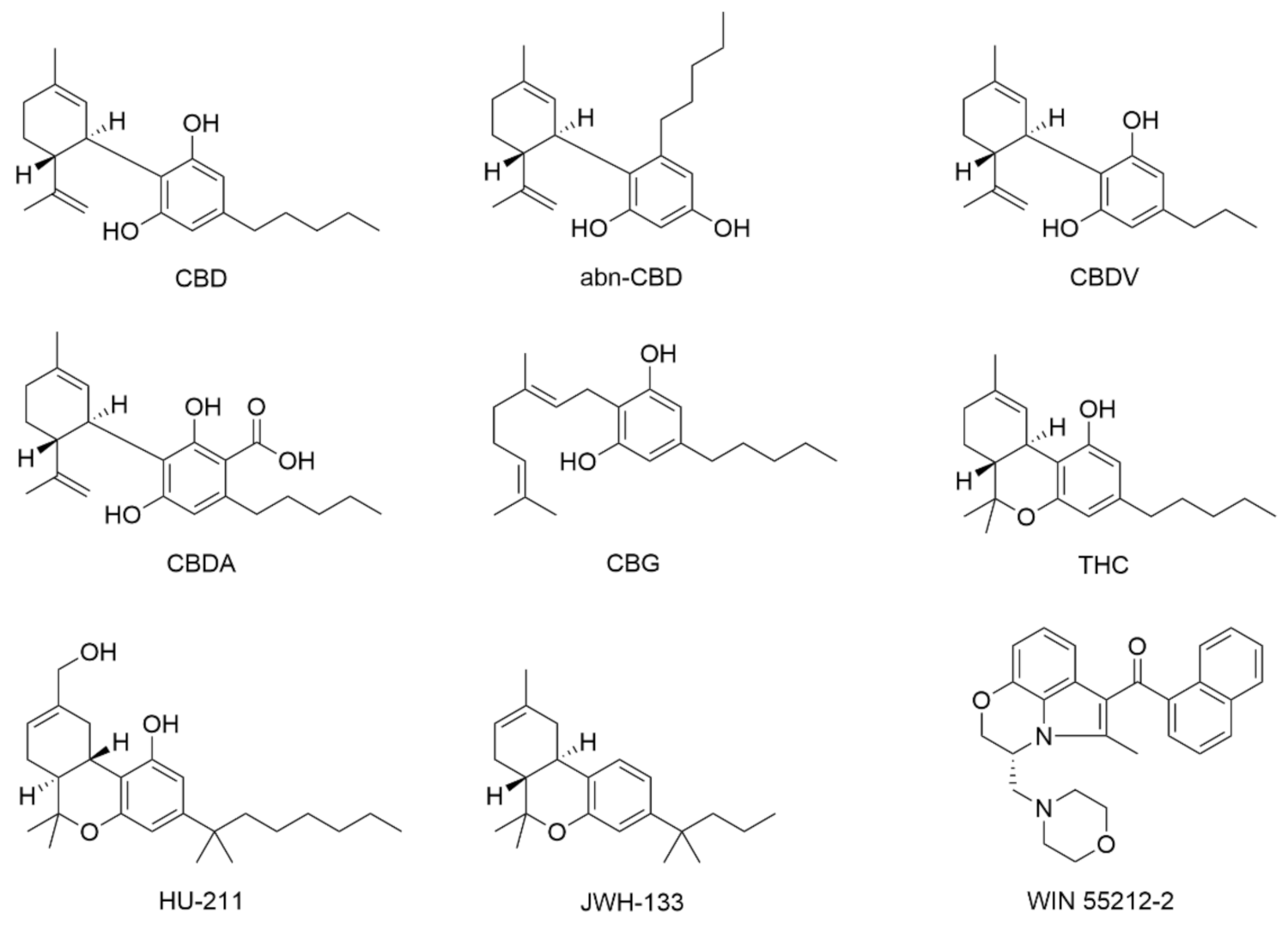
:1. Introduction
2. Neuroprotection of CBD on Primary Neurons
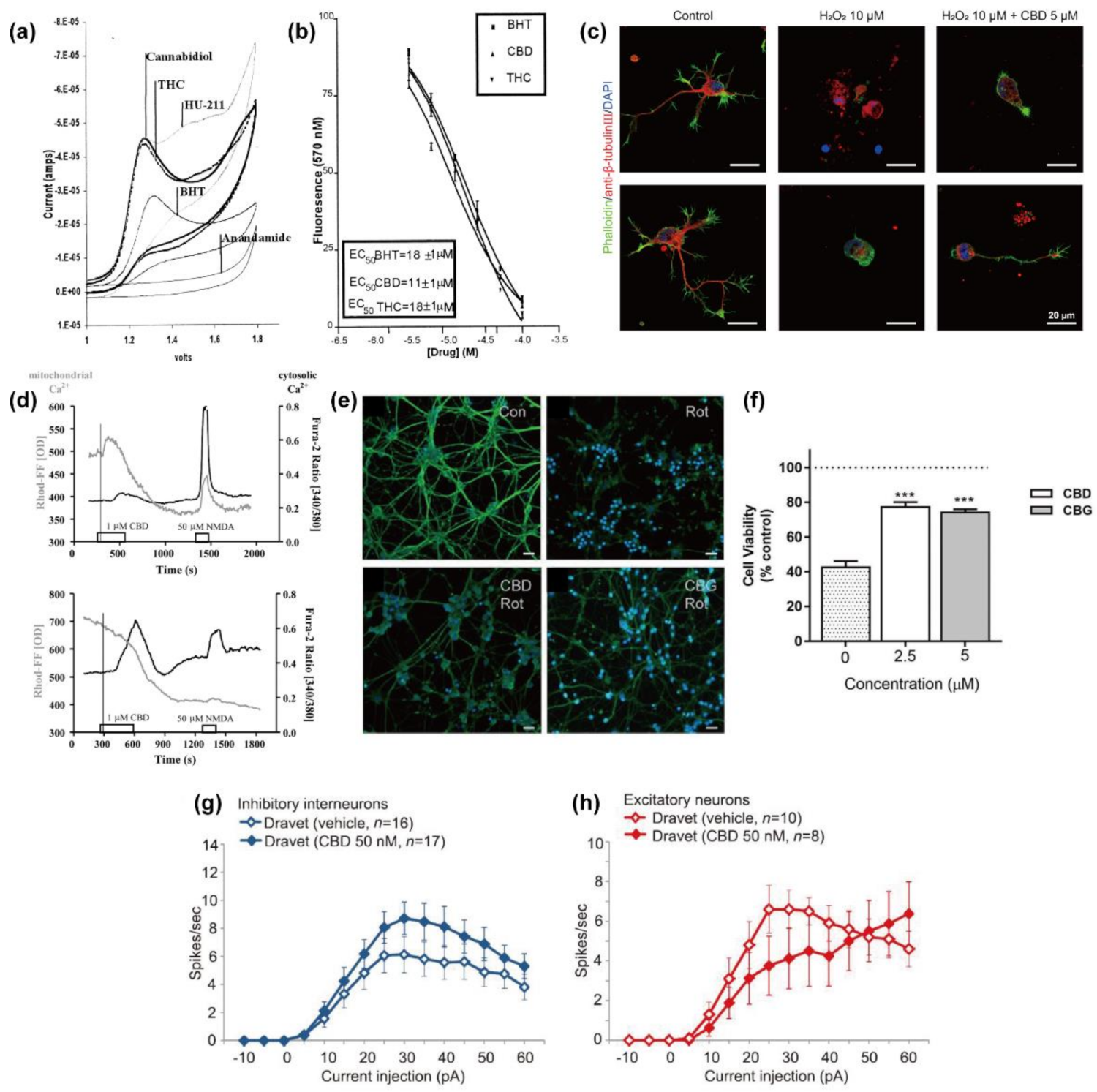
2.1. Anti-Oxidation
2.2. Homeostasis of Intracellular Ca2+ Level
2.2.1. Mitochondria
2.2.2. Endoplasmic Reticulum (ER)
2.2.3. Endogenous Cannabinoid Systems
2.3. Protection against Mitochondrial Dysfunction
2.4. Regulatory Actions on Network Activity and Neuronal Excitability
3. Anti-Gliosis and Anti-Inflammatory Effects of CBD on Primary Glial Cells
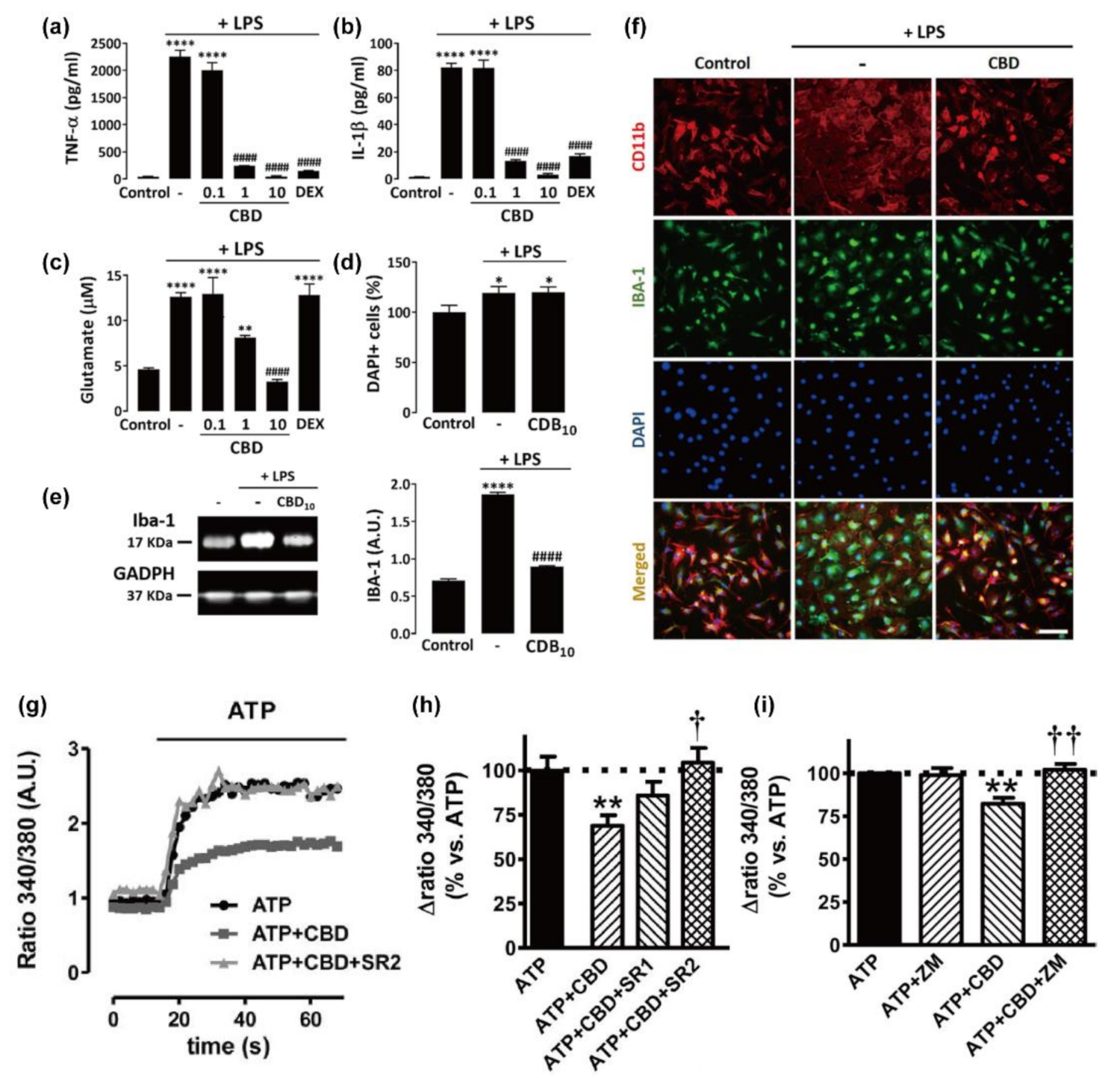
3.1. Microglia
3.2. Astrocytes
3.3. Oligodendrocytes
4. Regeneration from Neural Stem Cells
5. Perspective and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| 2-APB | 2-aminoethoxydiphenyl borate |
| 4-AP | 4-aminopyridine |
| 5-HT1A receptor | serotonin 1A receptor |
| A2A receptor | adenosine A2A receptor |
| AIF | apoptosis-induced factor |
| Akt | protein kinase B |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| ATP | adenosine triphosphate |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell lymphoma 2 protein |
| bFGF | basic fibroblast growth factor |
| BrdU | 5-bromo-2′-deoxyuridine |
| CaCl2 | calcium chloride |
| CB1 | cannabinoid receptor type 1 |
| CB2 | cannabinoid receptor type 2 |
| CCL | chemokine (C-C motif) ligand |
| CHOP | C/EBP homologous protein |
| CNS | central nervous system |
| CoCl2 | cobalt(II) chloride |
| CPZ | capsazepine |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| CSPG | chondroitin sulfate proteoglycan |
| CUS | chronic unpredictable stress |
| DCX | double cortin |
| DMSO | dimethyl sulfoxide |
| EC50 | half maximal effective concentration |
| EGTA | ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid |
| eiF2α | eukaryotic translation initiation factor 2α |
| ER | endoplasmic reticulum |
| EtOH | ethanol |
| FAAH | fatty acid amide hydrolase |
| FDA | U.S. Food and Drug Administration |
| GABA | γ-aminobutyric acid |
| GAP43 | Growth-associated protein 43 |
| GCV | ganciclovir |
| GFAP | glial fibrillary acidic protein |
| GFP | green fluorescent protein |
| GPCR | G protein-coupled receptor |
| GPR18 | G protein-coupled receptor 18 |
| GPR55 | G protein-coupled receptor 55 |
| HEPES | hydroxyethyl piperazine ethane sulfonic acid |
| hGMSC | human gingival mesenchymal stem cell |
| HO | heme oxygenase |
| hPLMSC | human periodontal ligament-derived mesenchymal stem cell |
| Iba-1 | ionized calcium-binding adapter molecule 1 |
| IC50 | half maximal inhibitory concentration |
| IFNγ | interferon γ |
| ihPSC | induced human pluripotent stem cell |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| iPSC | induced pluripotent stem cell |
| IP3 | inositol trisphosphate |
| KCl | potassium chloride |
| LDL | low-density lipoprotein |
| LPS | lipopolysaccharide |
| MAGL | monoacylglycerol lipase |
| MAP2 | microtubule-associated protein 2 |
| MARK1 | mitogen-activated protein kinase 1 |
| MEA | multi-electrode array |
| MgCl2 | magnesium dichloride |
| mNCX | mitochondrial Na+/Ca2+ exchanger |
| mPTP | mitochondrial permeability transition pore |
| mTOR | mammalian target of rapamycin |
| MSC | mesenchymal stem cells |
| NaCl | sodium chloride |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NAGly | N-Arachidonylglycine |
| NF-κB | nuclear factor-κB |
| NMDAR | N-methyl-D-aspartic acid receptor |
| PAPR-1 | poly(ADP-ribose) polymerase 1 |
| PAX6 | paired box protein |
| PI3K | phosphatidylinositol3-kinase |
| PKR | protein kinase R |
| PLC | phospholipase C |
| PPARγ | peroxisome proliferator-activated receptor γ |
| PrPres | protease-resistant prion protein |
| PSC | pluripotent stem cells |
| PTZ | pentylenetetrazole |
| S100B | S100 calcium binding protein B |
| SCN1A | sodium voltage-gated channel alpha subunit 1 |
| SOCC | store-operated calcium channel |
| SOCE | store-operated calcium entry |
| TGF-β1 | transforming growth factor β1 |
| TH | tyrosine hydroxylase |
| TMEV | Theiler’s murine encephalomyelitis virus |
| TNF-α | tumor necrosis factor-α |
| TRPC | transient receptor potential cation channel subfamily C |
| TRPV | transient receptor potential cation channel subfamily V |
| VCAM-1 | vascular cell adhesion protein 1 |
| VGCC | voltage-gated calcium channel |
| Xbp | X-box protein |
| XT-1 | xylosyltransferase 1 |
References
- Reekie, T.; Scott, M.; Kassiou, M. The evolving science of phytocannabinoids. Nat. Rev. Chem. 2018, 2, 0101. [Google Scholar] [CrossRef]
- Campos, A.C.; Fogaça, M.V.; Sonego, A.B.; Guimarães, F.S. Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol. Res. 2016, 112, 119–127. [Google Scholar] [CrossRef]
- Scuderi, C.; Filippis, D.D.; Iuvone, T.; Blasio, A.; Steardo, A.; Esposito, G. Cannabidiol in medicine: A review of its therapeutic potential in CNS disorders. Phytother. Res. 2009, 23, 597–602. [Google Scholar] [CrossRef]
- Iuvone, T.; Esposito, G.; Filippis, D.D.; Scuderi, C.; Steardo, L. Cannabidiol: A promising drug for neurodegenerative disorders? CNS Neurosci. Ther. 2009, 15, 65–75. [Google Scholar] [CrossRef]
- Elsaid, S.; Le Foll, B. The complexity of pharmacology of cannabidiol (CBD) and its implications in the treatment of brain disorders. Neuropsychopharmacology 2020, 45, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, M.M.; Queiroz, R.H.; Zuardi, A.W.; Crippa, J.A. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr. Drug Saf. 2011, 6, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Bih, C.I.; Chen, T.; Nunn, A.V.; Bazelot, M.; Dallas, M.; Whalley, B.J. Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics 2015, 12, 699–730. [Google Scholar]
- McGuire, P.; Robson, P.; Cubala, W.J.; Vasile, D.; Morrison, P.D.; Barron, R.; Taylor, A.; Wright, S. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: A multicenter randomized controlled trial. Am. J. Psychiatry 2018, 175, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenstein, M. From menace to medicine. Nature 2019, 572, S2–S4. [Google Scholar] [CrossRef] [Green Version]
- Whiting, P.F.; Wolff, R.F.; Deshpande, S.; Nisio, M.D.; Duffy, S.; Hernandez, A.V.; Keurentjes, J.C.; Lang, S.; Misso, K.; Ryder, S.; et al. Cannabinoids for medical use: A systematic review and meta-analysis. J. Am. Med. Assoc. 2015, 313, 2456–2473. [Google Scholar] [CrossRef]
- Huestis, M.A.; Solimini, R.; Pichini, S.; Pacifici, R.; Carlier, J.; Busardò, F.P. Cannabidiol adverse effects and toxicity. Curr. Neuropharmacol. 2019, 17, 974–989. [Google Scholar] [CrossRef]
- Chesney, E.; Oliver, D.; Green, A.; Sovi, S.; Wilson, J.; Englund, A.; Freeman, T.P.; McGuire, P. Adverse effects of cannabidiol: A systematic review and meta-analysis of randomized clinical trials. Neuropsychopharmacol 2020, 45, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Millar, S.A.; Stone, N.L.; Bellman, Z.D.; Yates, A.S.; England, T.J.; O’Sullivan, S.E. A systematic review of cannabidiol dosing in clinical populations. Br. J. Clin. Pharmacol. 2019, 85, 1888–1900. [Google Scholar] [CrossRef]
- Gould, J. The cannabis crop. Nature 2015, 525, S2–S3. [Google Scholar] [CrossRef] [Green Version]
- Vitale, R.M.; Iannotti, F.A.; Amodeo, P. The (poly)pharmacology of cannabidiol in neurological and neuropsychiatric disorders: Molecular mechanisms and targets. Int. J. Mol. Sci. 2021, 22, 4876. [Google Scholar] [CrossRef] [PubMed]
- Sonego, A.B.; Prado, D.S.; Vale, G.T.; Sepulveda-Diaz, J.E.; Cunha, T.M.; Tirapelli, C.R.; Del Bel, E.A.; Raisman-Vozari, R.; Guimarães, F.S. Cannabidiol prevents haloperidol-induced vacuos chewing movements and inflammatory changes in mice via PPARγ receptors. Brain Behav. Immun. 2018, 74, 241–251. [Google Scholar] [CrossRef]
- Mecha, M.; Feliú, A.; Iñigo, P.M.; Mestre, L.; Carrillo-Salinas, F.J.; Guaza, C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: A role for A2A receptors. Neurobiol. Dis. 2013, 59, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Banker, G.A.; Goslin, K. Types of nerve cell cultures, their advantages and limitations. In Culturing Nerve Cells, 2nd ed.; Banker, G.A., Goslin, K., Eds.; The MIT Press: Cambridge, MA, USA, 1998; pp. 11–35. [Google Scholar]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef]
- Howlett, A.C.; Blume, L.C.; Dalton, G.D. CB1 cannabinoid receptors and their associated proteins. Curr. Med. Chem. 2010, 17, 1382–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsicano, G.; Moosmann, B.; Hermann, H.; Lutz, B.; Behl, C. Neuroprotective properties of cannabinoids against oxidative stress: Role of the cannabinoid receptor CB1. J. Neurochem. 2002, 80, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Kriegstein, A.R.; Dichter, M.A. Morphological classification of rat cortical neurons in cell culture. J. Neurosci. 1983, 3, 1634–1647. [Google Scholar] [CrossRef] [PubMed]
- Hampson, A.J.; Grimaldi, M.; Axelrod, J.; Wink, D. Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc. Natl. Acad. Sci. USA 1998, 95, 8268–8273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Choi, J.Y.; Seo, J.; Choi, I.S. Neuroprotective effect of cannabidiol against hydrogen peroxide in hippocampal neuron culture. Cannabis Cannabinoid Res. 2021, 6, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.; Drysdale, A.J.; Lafourcade, C.; Pertwee, R.G.; Platt, B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J. Neurosci. 2009, 29, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Echeverry, C.; Prunell, G.; Narbondo, C.; de Medina, V.S.; Nadal, X.; Reyes-Parada, M.; Scorza, C. A comparative in vitro study of the neuroprotective effect induced by cannabidiol, cannabigerol, and their respective acid forms: Relevance of the 5-HT1A receptors. Neurotox. Res. 2021, 39, 335–348. [Google Scholar] [CrossRef]
- Sun, Y.; Dolmetsch, R.E. Investigating the therapeutic mechanism of cannabidiol in a human induced pluripotent stem cell (iPSC)-based model of Dravet syndrome. Cold Spring Harb. Symp. Quant. Biol. 2018, 83, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drysdale, A.J.; Ryan, D.; Pertwee, R.G.; Platt, B. Cannabidiol-induced intracellular Ca2+ elevations in hippocampal cells. Neuropharmacology 2006, 50, 621–631. [Google Scholar] [CrossRef]
- Ryan, D.; Drysdale, A.J.; Pertwee, R.G.; Platt, B. Differential effects of cannabis extracts and pure plant cannabinoids on hippocampal neurones and glia. Neurosci. Lett. 2006, 408, 236–241. [Google Scholar] [CrossRef]
- Ryan, D.; Drysdale, A.J.; Pertwee, R.G.; Platt, B. Interactions of cannabidiol with endocannabinoid signalling in hippocampal tissue. Eur. J. Neurosci. 2007, 25, 2093–2102. [Google Scholar] [CrossRef]
- Bouron, A. Phyto and endocannabinoids exert complex actions on calcium and zinc signaling in mouse cortical neurons. Biochem. Pharmacol. 2018, 152, 244–251. [Google Scholar] [CrossRef]
- Moldzio, R.; Pacher, T.; Krewenka, C.; Kranner, B.; Novak, J.; Duvigneau, J.C.; Rausch, W.D. Effects of cannabinoids Δ(9)-tetrahydrocannabinol, Δ(9)-tetrahydrocannabinolic acid and cannabidiol in MPP+ affected murine mesencephalic cultures. Phytomedicine 2012, 19, 819–824. [Google Scholar] [CrossRef]
- Duvigneau, J.C.; Trovato, A.; Müllebner, A.; Miller, I.; Krewenka, C.; Krenn, K.; Zich, W.; Moldzio, R. Cannabidiol protects dopaminergic neurons in mesencephalic cultures against the complex I inhibitor rotenone via modulation of heme oxygenase activity and bilirubin. Antioxidants 2020, 9, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, C.J.; Finch, P.; Müller-Taubenberger, A.; Leung, K.Y.; Warren, E.C.; Damstra-Oddy, J.; Sharma, D.; Patra, P.H.; Glyn, S.; Boberska, J.; et al. A new mechanism for cannabidiol in regulating the one-carbon cycle and methionine levels in Dictyostelium and in mammalian epilepsy models. Br. J. Pharmacol. 2020, 177, 912–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, I.V.A.; Bellozi, P.M.Q.; Batista, E.M.; Vilela, L.R.; Brandão, I.L.; Ribeiro, F.M.; Moraes, M.F.D.; Moreira, F.A.; de Oliveira, A.C.P. Cannabidiol anticonvulsant effect is mediated by the PI3Kγ pathway. Neuropharmacology 2020, 176, 108156. [Google Scholar] [CrossRef]
- Ledgerwood, C.J.; Greenwood, S.M.; Brett, R.R.; Pratt, J.A.; Bushell, T.J. Cannabidiol inhibits synaptic transmission in rat hippocampal cultures and slices via multiple receptor pathways. Br. J. Pharmacol. 2011, 162, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Paşca, S.P.; Portmann, T.; Goold, C.; Worringer, K.A.; Guan, W.; Chan, K.C.; Gai, H.; Vogt, D.; Chen, Y.J.; et al. A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet Syndrome patients. Elife 2016, 5, e13073. [Google Scholar] [CrossRef]
- Hill, A.J.; Jones, N.A.; Smith, I.; Hill, C.L.; Williams, C.M.; Stephens, G.J.; Whalley, B.J. Voltage-gated sodium (NaV) channel blockade by plant cannabinoids does not confer anticonvulsant effects per se. Neurosci. Lett. 2014, 566, 269–274. [Google Scholar] [CrossRef]
- Mendis, G.D.C.; Berecki, G.; Morrisroe, E.; Pachernegg, S.; Li, M.; Varney, M.; Osborne, P.B.; Reid, C.A.; Halgamuge, S.; Petrou, S. Discovering the pharmacodynamics of conolidine and cannabidiol using a cultured neuronal network based workflow. Sci. Rep. 2019, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Rosenthaler, S.; Pöhn, B.; Kolmanz, C.; Huu, C.N.; Krewenka, C.; Huber, A.; Kranner, B.; Rausch, W.D.; Moldzio, R. Differences in receptor binding affinity of several phytocannabinoids do not explain their effects on neural cell cultures. Neurotoxicol. Teratol. 2014, 46, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Dirikoc, S.; Priola, S.A.; Marella, M.; Zsürger, N.; Chabry, J. Nonpsychoactive cannabidiol prevents prion accumulation and protects neurons against prion toxicity. J. Neurosci. 2007, 27, 9537–9544. [Google Scholar] [CrossRef] [Green Version]
- Mato, S.; Victoria Sánchez-Gómez, M.; Matute, C. Cannabidiol induces intracellular calcium elevation and cytotoxicity in oligodendrocytes. Glia 2010, 58, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Valentão, P.; Pereira, J.A.; Andrade, P.B. Phenolics: From chemistry to biology. Molecules 2009, 14, 2202–2211. [Google Scholar] [CrossRef]
- Song, J.H.; Shin, S.H.; Wang, W.; Ross, G.M. Involvement of oxidative stress in ascorbate-induced proapoptotic death of PC12 cells. Exp. Neurol. 2001, 169, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, B.; Behl, C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc. Natl. Acad. Sci. USA 1999, 96, 8867–8872. [Google Scholar] [CrossRef] [Green Version]
- Gleichmann, M.; Mattson, M.P. Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal. 2011, 14, 1261–1273. [Google Scholar] [CrossRef] [Green Version]
- Zündorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Grienberger, C.; Konnerth, A. Imaging calcium in neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Mattson, M.P.; Chan, S.J. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 2003, 34, 385–397. [Google Scholar] [CrossRef]
- Szewczyk, B. Zinc homeostasis and neurodegenerative disorders. Front. Aging Neurosci. 2013, 5, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisogno, T.; Hanus, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Scott, R.H.; Ross, R.A. Multiple actions of anandamide on neonatal rat cultured sensory neurones. Br. J. Pharmacol. 2004, 141, 1223–1233. [Google Scholar] [CrossRef] [Green Version]
- Movsesyan, V.A.; Stoica, B.A.; Yakovlev, A.G.; Knoblach, S.M.; Lea IV, P.M.; Cernak, I.; Vink, R.; Faden, A.I. Anandamide-induced cell death in primary neuronal cultures: Role of calpain and caspase pathways. Cell Death Differ. 2004, 11, 1121–1132. [Google Scholar] [CrossRef]
- Sugiura, T.; Kodaka, T.; Kondo, S.; Tonegawa, T.; Nakane, S.; Kishimoto, S.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol, a putative endogenous cannabinoid receptor ligand, induces rapid, transient elevation of intracellular free Ca2+ in neuroblastoma x glioma hybrid NG108-15 cells. Biochem. Biophys. Res. Commun. 1996, 229, 58–64. [Google Scholar] [CrossRef]
- Brady, C.M.; DasGupta, R.; Dalton, C.; Wiseman, O.J.; Berkley, K.J.; Fowler, C.J. An open-label pilot study of cannabis-based extracts for bladdery function in advanced multiple sclerosis. Mult. Scler. 2004, 10, 425–433. [Google Scholar] [CrossRef]
- Whalley, B.J.; Wilkinson, J.D.; Williamson, E.M.; Constanti, A. A novel component of cannabis extract potentiates excitatory synaptic transmission in rat olfactory cortex in vitro. Neurosci. Lett. 2004, 365, 58–63. [Google Scholar] [CrossRef]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Rad, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Kitamura, Y.; Kakimura, J.; Shibagaki, K.; Taniguchi, T.; Gebicke-Haerter, P.J.; Smith, M.A.; Perry, G.; Shimohama, S. Possible protective mechanisms of heme oxygenase-1 in the brain. Ann. N. Y. Acad. Sci. 2002, 977, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Colín-González, A.L.; Orozco-Ibarra, M.; Chánez-Cárdenas, M.E.; Rangel-López, E.; Santamaría, A.; Pedraza-Chaverri, J.; Barrera-Oviedo, D.; Maldonado, P.D. Heme oxygenase-1 (HO-1) upregulation delays morphological and oxidative damage induced in an excitotoxic/pro-oxidant model in the rat striatum. Neuroscience 2013, 231, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Perucca, E. Cannabinoids in the treatment of epilepsy: Hard evidence at last? J. Epilepsy Res. 2017, 7, 61–76. [Google Scholar] [CrossRef]
- GW Pharmaceuticals plc and it’s U.S. subsidiary Greenwich Biosciences announce FDA approval of EPIDIOLEX® (cannabidiol) oral solution—the first plant-derived cannabinoid prescription medicine. Available online: https://ir.gwpharm.com/news-releases/news-release-details/gw-pharmaceuticals-plc-and-its-us-subsidiary-greenwich (accessed on 25 June 2018).
- Köhling, R. Voltage-gated sodium channels in epilepsy. Epilepsia 2002, 43, 1278–1295. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.T.; Pathmanathan, J.; Cash, S.S. From ion channels to complex networks: Magic bullet versus magic shotgun approaches to anticonvulsant pharmacotherapy. Med. Hypotheses 2009, 72, 297–305. [Google Scholar] [CrossRef]
- Schmitz, D.; Empson, R.M.; Heinemann, U. Serotonin reduces inhibition via 5-HT1A receptors in area CA1 of rat hippocampal slices in vitro. J. Neurosci. 1995, 15, 7217–7225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, M.J.; Wiley, J.L.; Martin, B.R.; DeLorenzo, R.J. Assessment of the role of CB1 receptors in cannabinoid anticonvulsant effects. Eur. J. Pharmacol. 2001, 428, 51–57. [Google Scholar] [CrossRef]
- Rakhshan, F.; Day, T.A.; Blakely, R.D.; Barker, E.L. Carrier-mediated uptake of the endogenous cannabinoid anandamide in RBL-2H3 cells. J. Pharmacol. Exp. Ther. 2000, 292, 960–967. [Google Scholar]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A.; Kalume, F.; Oakley, J.C. NaV1.1 channels and epilepsy. J. Physiol. 2010, 588, 1849–1859. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Rodríguez, J.J.; Verkhratsky, A. Neuroglia in neurodegeneration. Brain Res. Rev. 2010, 63, 189–211. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef] [PubMed]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte crosstalk in CNS inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Martín-Moreno, A.M.; Reigada, D.; Ramírez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to Alzheimer’s disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos-Santos-Pereira, M.; Guimarães, F.S.; Del-Bel, E.; Raisman-Vozari, R.; Michel, P.P. Cannabidiol prevents LPS-induced microglial inflammation by inhibiting ROS/NF-κB-dependent signaling and glucose consumption. Glia 2020, 68, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Eldeeb, K.; Millns, P.J.; Bennett, A.J.; Alexander, S.P.; Kendall, D.A. Cannabidiol enhances microglial phagocytosis via transient receptor potential (TRP) channel activation. Br. J. Pharmacol. 2014, 171, 2426–2439. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.Y.; Goble, K.; Mecha, M.; Wang, C.C.; Huang, C.H.; Guaza, C.; Jan, T.R. Cannabidiol-induced apoptosis in murine microglial cells through lipid raft. Glia 2012, 60, 1182–1190. [Google Scholar] [CrossRef] [Green Version]
- Feliú, A.; Moreno-Martet, M.; Mecha, M.; Carrillo-Salinas, F.J.; de Lago, E.; Fernández-Ruiz, J.; Guaza, C. A Sativex(®) -like combination of phytocannabinoids as a disease-modifying therapy in a viral model of multiple sclerosis. Br. J. Pharmacol. 2015, 172, 3579–3595. [Google Scholar] [CrossRef] [Green Version]
- von Widdern, J.C.; Hohmann, T.; Dehghani, F. Abnormal cannabidiol affects production of pro-inflammatory mediators and astrocyte wound closure in primary astrocytic-microglial cocultures. Molecules 2020, 25, 496. [Google Scholar] [CrossRef] [Green Version]
- Mecha, M.; Torrao, A.S.; Mestre, L.; Carrillo-Salinas, F.J.; Mechoulam, R.; Guaza, C. Cannabidiol protects oligodendrocyte progenitor cells from inflammation-induced apoptosis by attenuating endoplasmic reticulum stress. Cell Death Dis. 2012, 3, e331. [Google Scholar] [CrossRef] [Green Version]
- Auzmendi, J.; Palestro, P.; Blachman, A.; Gavernet, L.; Merelli, A.; Talevi, A.; Calabrese, G.C.; Ramos, A.J.; Lazarowski, A. Cannabidiol (CBD) inhibited Rhodamine-123 efflux in cultured vascular endothelial cells and astrocytes under hypoxic conditions. Front. Behav. Neurosci. 2020, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Microglia in steady state. J. Clin. Investig. 2017, 127, 3201–3209. [Google Scholar] [CrossRef] [Green Version]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic immuneffector cell of the brain. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Perry, V.; Nicoll, J.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef]
- Diz-Chaves, Y.; Pernía, O.; Carrero, P.; Garcia-Segura, L.M. Prenatal stress causes alterations in the morphology of microglia and the inflammatory response of the hippocampus of adult female mice. J. Neuroinflammation 2012, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichhoff, G.; Brawek, B.; Garaschuk, O. Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochim. Biophys. Acta 2011, 1813, 1014–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.Y.; Chang, A.C.; Wang, C.C.; Kuo, F.H.; Lee, C.Y.; Liu, D.Z.; Jan, T.R. Cannabidiol induced a contrasting pro-apoptotic effect of between freshly isolated and precultured human monocytes. Toxicol. Appl. Pharmacol. 2010, 246, 141–147. [Google Scholar] [CrossRef]
- Wu, H.Y.; Chu, R.M.; Wang, C.C.; Lee, C.Y.; Lin, S.H.; Jan, T.R. Cannabidiol-induced apoptosis in primary lymphocytes is associated with oxidative stress-dependent activation of caspase-8. Toxicol. Appl. Pharmacol. 2008, 226, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Boatright, K.M.; Salvesen, G.S. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 2003, 15, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Nashar, T.O.; Williams, N.A.; Hirst, T.R. Cross-linking of cell surface ganglioside GM1 induces the selective apoptosis of mature CD8+ T lymphocytes. Int. Immunol. 1996, 8, 731–736. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Shao, H.; Cox, N.R.; Baker, H.J.; Ewald, S.J. Gangliosides enhance apoptosis of thymocytes. Cell. Immunol. 1998, 183, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Gargalovic, P.; Dory, L. Cellular apoptosis is associated with increased caveolin-1 expression in macrophages. J. Lipid Res. 2003, 44, 1622–1632. [Google Scholar] [CrossRef] [Green Version]
- Banker, G.A. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science 1980, 209, 809–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamby, M.E.; Sofroniew, M.V. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 2010, 7, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef]
- Robb, J.L.; Hammad, N.A.; Weightman, P.P.G.; Chilton, J.K.; Beall, C.; Ellacott, K.L.J. The metabolic response to inflammation in astrocytes is regulated by nuclear factor-kappa B signaling. Glia 2020, 68, 2246–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebert, J.R.; Conta Steencken, A.; Osterhout, D.J. Chondroitin sulfate proteoglycans in the nervous system: Inhibitors to repair. Biomed. Res. Int. 2014, 2014, 845323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tognatta, R.; Miller, R.H. Contribution of the oligodendrocyte lineage to CNS repair and neurodegenerative pathologies. Neuropharmacology 2016, 110, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Ettle, B.; Schlachetzki, J.C.M.; Winkler, J. Oligodendroglia and myelin in neurodegenerative diseases: More than just bystanders? Mol. Neurobiol. 2016, 53, 3046–3062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhou, J. Oligodendrocytes in neurodegenerative diseases. Front. Biol. 2013, 8, 127–133. [Google Scholar] [CrossRef]
- Ogata, T. Therapeutic strategies for oligodendrocyte-mediated remyelination. Adv. Exp. Med. Biol. 2019, 1190, 265–279. [Google Scholar]
- Hong, S.J.; Dawson, T.M.; Dawson, V.L. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol. Sci. 2004, 25, 259–264. [Google Scholar] [CrossRef]
- Proud, C.G. eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 2005, 16, 3–12. [Google Scholar] [CrossRef]
- Rossi, F.; Cattaneo, E. Neural stem cell therapy for neurological diseases: Dreams and reality. Nat. Rev. Neurosci. 2002, 3, 401–409. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, L.; Binda, E. Concise review: Self-renewal in the central nervous system: Neural stem cells from embryo to adult. Stem Cells Transl. Med. 2012, 1, 298–308. [Google Scholar] [CrossRef]
- Rodrigues, R.S.; Lourenço, D.M.; Paulo, S.L.; Mateus, J.M.; Ferreira, M.F.; Mouro, F.M.; Moreira, J.B.; Ribeiro, F.F.; Sebastião, A.M.; Xapelli, S. Cannabinoid actions on neural stem cells: Implications for pathophysiology. Molecules 2019, 24, 1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luján, M.Á.; Valverde, O. The pro-neurogenic effects of cannabidiol and its potential therapeutic implications in psychiatric disorders. Front. Behav. Neurosci. 2020, 14, 109. [Google Scholar] [CrossRef] [PubMed]
- Rajan, T.S.; Giacoppo, S.; Scionti, D.; Diomede, F.; Grassi, G.; Pollastro, F.; Piattelli, A.; Bramanti, P.; Mazzon, E.; Trubiani, O. Cannabidiol activates neuronal precursor genes in human gingival mesenchymal stromal cells. J. Cell. Biochem. 2017, 118, 1531–1546. [Google Scholar] [CrossRef]
- Lanza Cariccio, V.; Scionti, D.; Raffa, A.; Iori, R.; Pollastro, F.; Diomede, F.; Bramanti, P.; Trubiani, O.; Mazzon, E. Treatment of periodontal ligament stem cells with MOR and CBD promotes cell survival and neuronal differentiation via the PI3K/Akt/mTOR pathway. Int. J. Mol. Sci. 2018, 19, 2341. [Google Scholar] [CrossRef] [Green Version]
- Campos, A.C.; Ortega, Z.; Palazuelos, J.; Fogaça, M.V.; Aguiar, D.C.; Díaz-Alonso, J.; Ortega-Gutiérrez, S.; Vázquez-Villa, H.; Moreira, F.A.; Guzmán, M.; et al. The anxiolytic effect of cannabidiol on chronically stressed mice depends on hippocampal neurogenesis: Involvement of the endocannabinoid system. Int. J. Neuropsychopharmacol. 2013, 16, 1407–1419. [Google Scholar] [CrossRef] [Green Version]
- Miranda, C.C.; Barata, T.; Vaz, S.H.; Ferreira, C.; Quintas, A.; Bekman, E.P. hiPSC-based model of prenatal exposure to cannabinoids: Effect on neuronal differentiation. Front. Mol. Neurosci. 2020, 13, 119. [Google Scholar] [CrossRef]
- Schiavon, A.P.; Bonato, J.M.; Milani, H.; Guimarães, F.S.; Weffort de Oliveira, R.M. Influence of single and repeated cannabidiol administration on emotional behavior and markers of cell proliferation and neurogenesis in non-stressed mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Bick-Sander, A.; Fabel, K.; Leal-Galicia, P.; Tauber, S.; Ramirez-Rodriguez, G.; Müller, A.; Melnik, A.; Waltinger, T.P.; Ullrich, O.; et al. Cannabinoid receptor CB1 mediates baseline and activity-induced survival of new neurons in adult hippocampal neurogenesis. Cell Commun. Signal. 2010, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogaça, M.V.; Campos, A.C.; Coelho, L.D.; Duman, R.S.; Guimarães, F.S. The anxiolytic effects of cannabidiol in chronically stressed mice are mediated by the endocannabinoid system: Role of neurogenesis and dendritic remodeling. Neuropharmacology 2018, 135, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jia, D.; Li, A.; Chau, J.; He, D.; Ruan, X.; Liu, F.; Li, J.; He, L.; Li, B. p53 regulates neural stem cell proliferation and differentiation via BMP-Smad1 signaling and Id1. Stem Cells Dev. 2013, 22, 913–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, E.G.; An, J.J.; Orefice, L.L.; Baydyuk, M.; Liao, G.-Y.; Zheng, K.; Lu, B.; Xu, B. BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J. Neurosci. 2012, 32, 14318–14330. [Google Scholar] [CrossRef]
- Brunet, A.; Datta, S.R.; Greenberg, M.E. Transcription-dependent and -independent control of neuronal survival by the PI3K–Akt signaling pathway. Cur. Opin. Neurobiol. 2001, 11, 297–305. [Google Scholar] [CrossRef]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef]
- Abranches, E.; Silva, M.; Pradier, L.; Schulz, H.; Hummel, O.; Henrique, D.; Bekman, E. Neural differentiation of embryonic stem cells in vitro: A road map to neurogenesis in the embryo. PLoS ONE 2009, 4, e6286. [Google Scholar] [CrossRef] [PubMed]
- Macías, W.; Carlson, R.; Rajadhyaksha, A.; Barczak, A.; Konradi, C. Potassium chloride depolarization mediates CREB phosphorylation in striatal neurons in an NMDA receptor-dependent manner. Brain Res. 2001, 890, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Wade, D.T.; Collin, C.; Stott, C.; Duncombe, P. Meta-analysis of the efficacy and safety of Sativex (nabiximols), on spasticity in people with multiple sclerosis. Mult. Scler. 2010, 16, 707–714. [Google Scholar] [CrossRef]
- Mitelpunkt, A.; Kramer, U.; Hausman, K.M.; Zilbershot, F.E.; Orbach, R.; Chernuha, V.; Fattal-Valevski, A.; Deutsch, L.; Heffetz, D.; Sacks, H. The safety, tolerability, and effectiveness of PTL-101, an oral cannabidiol formulation, in pediatric intractable epilepsy: A phase II, open-label, single-center study. Epilepsy Behav. 2019, 98, 233–237. [Google Scholar] [CrossRef]
- Atsmon, J.; Heffetz, D.; Deutsch, L.; Deutsch, F.; Sacks, H. Single-dose pharmacokinetics of oral cannabidiol following administration of PTL101: A new formulation based on gelatin matrix pellets technology. Clin. Pharmacol. Drug Dev. 2018, 7, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Heussler, H.; Cohen, J.; Silove, N.; Tich, N.; Bonn-Miller, M.O.; Du, W.; O’Neill, C.; Sebree, T. A phase 1/2, open-label assessment of the safety, tolerability, and efficacy of transdermal cannabidiol (ZYN002) for the treatment of pediatric fragile X syndrome. J. Neurodev. Disord. 2019, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.; Hulihan, J.; Messenheimer, J.; Ali, S.; Davis, S.; Gutterman, D.; Sebree, T.; Sadleir, L. Cannabidiol transdermal gel in children and adolescents with developmental and epileptic encephalopathies: An open-label clinical trial (1631). Neurology 2020, 94, 1631. [Google Scholar]
- Jung, B.; Lee, J.K.; Kim, J.; Kang, E.K.; Han, S.Y.; Lee, H.-Y.; Choi, I.S. Synthetic strategies for (-)-cannabidiol and its structural analogs. Chem. Asian J. 2019, 14, 3749–3762. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Reiter, M.A.; d’Espaux, L.; Wong, J.; Denby, C.M.; Lechner, A.; Zhang, Y.; Grzybowski, A.T.; Harth, S.; Lin, W.; et al. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 2019, 567, 123–126. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Simitzi, C.; Ranella, A.; Stratakis, E. Controlling the morphology and outgrowth of nerve and neuroglial cells: The effect of surface topography. Acta Biomater. 2017, 51, 21–52. [Google Scholar] [CrossRef] [Green Version]
- Marcus, M.; Baranes, K.; Park, M.; Choi, I.S.; Kang, K.; Shefi, O. Interactions of neurons with physical environments. Adv. Healthcare Mater. 2017, 6, 1700267. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
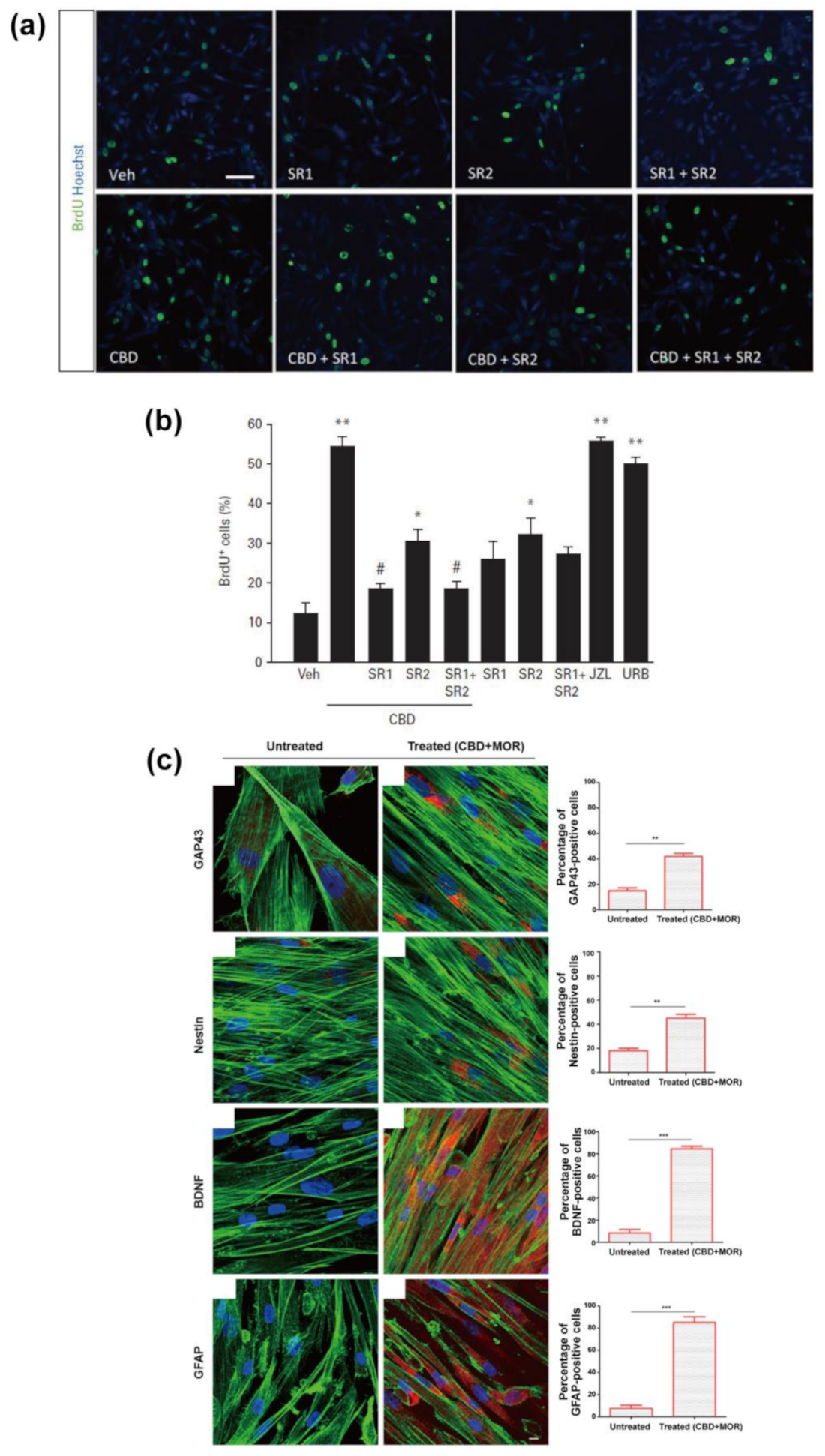{kind=link}
| Cell Type/Source | DIV | Toxicant | CBD Concentration [μM] | CBD Exposure | CBD Effects | Related Molecular Targets | Ref. |
|---|---|---|---|---|---|---|---|
| cortical neurons (Wistar rat pups, E17) | 7–18 | glutamate; tert-butyl hydroperoxide | 1/3.16/10/31.6 | 18–20 h | neuroprotection against NMDAR-mediated and AMPA/kainate receptor-mediated glutamate toxicity, ROS-induced toxicity, and the glutamate-induced toxicity | (not CB1/2) | [23] |
| cerebellar granular neurons (Wistar rats, 6–8 days) | 6–7 | H2O2; rotenone | 2.5/5/10 | 24/48 h | neuroprotection against ROS-induced toxicity and mitochondrial dysfunction | (not CB1/2 and 5-HT1A receptor) | [26] |
| hippocampal neurons (Sprague–Dawley rat pups, E18) | 1 | H2O2 | 0.1/1/3/5/10/15/30/50/100 | 24 h | neuroprotection against H2O2-induced toxicity | [24] | |
| hippocampal neurons (Sprague–Dawley rats, 1–3 days) | 5–10 | 1/10 | 0 | regulation of [Ca2+]i homeostasis | Ca2+ channels (including L-type VGCC), CB1, and TRPV1 | [28] | |
| hippocampal neurons (Lister-hooded rats, 1–3 days) | 5–10 | 1 | 0 | regulation of [Ca2+]i homeostasis | [29] | ||
| hippocampal neurons (Lister-hooded rats, 1-3 days) | 5–10 | 1 | 0 | regulation of [Ca2+]i homeostasis | CB1, TRPV1, and PLC; (not FAAH, MAGL, and Gi/o-linked GPCR) | [30] | |
| hippocampal neurons (Lister-hooded rats, 1–3 days) | 5–10 | 4-AP; oligomycin; FCCP | 0.1/1 | 0/overnight | regulation of [Ca2+]i homeostasis; neuroprotection against oligomycin and FCCP | mNCX; (not mPTP, and ryanodine and IP3 receptors) | [25] |
| cortical neurons (C57BL/6J mouse pups, E13) | 2–3 | 1/10/20 | 0 | regulation of [Ca2+]i homeostasis | (not Gi/o-linked GPCRs and CB1/2) | [31] | |
| mesencephalic neurons (OF1/SPF mouse pups, E14) | 11 | MPP | 0.01/0.1/1/10 | 48 h | neuroprotection against MPP-induced toxicity | [32] | |
| mesencephalic neurons (OF1/SPF mouse pups, E14) | 12 | rotenone | 10 | 48 h | neuroprotection against rotenone-induced toxicity | [33] | |
| hippocampal neurons (Sprague–Dawley rat pups, E18) | 21 | PTZ | 1.89 | 1 h | reduction of PTZ-induced increase in methionine level | [34] | |
| hippocampal neurons (C57BL/6 mice, 1 day) | 10–12 | glutamate | 0.1/1/10 | 4 h | neuroprotection against glutamate-induced toxicity | PI3Kγ | [35] |
| hippocampal neurons (Sprague–Dawley rats, 1–2 days) | 13–16 | 0.1/1/10 | 5 min | regulation of synaptic activity | Gi/o-linked GPCR and CB1; (not 5-HT1A receptor) | [36] | |
| cortical neurons (Sprague–Dawley rat pups, E18) | 13–14 | 0.005/0.015/0.05/0.15/0.5/1.5/5/15 | 5–6 min/48 h | regulation of neuronal activities ([Ca2+]i and action potential) | [37] | ||
| telencephalic inhibitory and excitatory neurons (Dravet syndrome patients-derived iPSCs) | 0.05 | 5–6 min | regulation of action potential activity | [37] | |||
| cortical neurons (NIHS mouse pups, E13–15) | 4–11 | 10 | 20 min | regulation of Nav channel | [38] | ||
| cortical neurons (C57BL/6 mice, 0–2 days) | 14–21 | 10 | 10 min | regulation of network activity | Cav2.2 channel | [39] | |
| mesencephalic neurons (OF1/SPF mouse pups, E14) | 12 | 0.1/1/10 | 48 h | neurotoxicity | [40] | ||
| cortical neurons (C57BL/6 mouse pups, E14) | 6–8 | PrPres | 1/5 | 48 h | neuroprotection against PrPres | [41] | |
| cortical neurons (Sprague–Dawley rat pups, E18) | 8 | 0.1/1/10 | 20–30 min | neurotoxicity | [42] |
| Cell Type/Source | DIV | Toxicant | CBD Concentration [μM] | CBD Exposure | CBD Effects | Related Molecular Targets | Ref. |
|---|---|---|---|---|---|---|---|
| microglia (cortices of neonatal rats) | 1 | ATP; LPS | 0.001–1 | 0/24 h | anti-inflammatory effect; promotion of migration; [Ca2+]i regulation | CB1/2 and A2A receptor | [75] |
| microglia (brains of C57BL/6J mice) | LPS | 0.1/1/10 | 24 h/30 min | anti-inflammatory effect; astrogliosis inhibition | CB2; (not CB1, GPR55, and PPARγ) | [76] | |
| microglia (brains of C57BL/6J mice) | 14–16 | LPS | 10 | 4 h | anti-inflammatory effect; inhibition of microglial activation | PPARγ | [16] |
| microglia (brains and spinal cords of C57BL/6 mice, 8–10 weeks) | 4 | 10 | 24 h | promotion of phagocytosis | TRP channels (TRPV1, TRPV2, and TRPC); (not Gi/o-linked GPCR and CB1/2) | [77] | |
| primary microglial cells (forebrain of newborn BALB/c mice) | 1/4/8/16/24 | 1-24 h | induction of microglial apoptosis | (not CB1/2, TPRV1, and GPR55) | [78] | ||
| astrocytes (cortices of Sprague–Dawley rats, 2 days) | Aβ1-42 | 0.001/0.01/0.1 | 24 h | anti-inflammatory effect; astrogliosis inhibition | PPARγ; (not PPARα) | [73] | |
| astrocytes (Wistar rats, 0–2 days) | IL-1β + TNF-α | 1/5 | 6 h | immunosuppression | A2A receptor | [17] | |
| astrocytes (Wistar rats, 0-2 days) | 4 | TGF-1β + bFGF | Sativex® (0.1/0.5/1) | 24/48/72 h | astrogliosis inhibition | [79] | |
| astrocytes (brains of C57BL/6 mice, 0–1 day) | 1 | LPS; scratch | 1/10 | 30 min/72 h | anti-inflammatory effect; astrogliosis inhibition | (not CB2) | [80] |
| oligodendrocytes (optic nerves of Sprague–Dawley rats, 12 days) | 1 | 0.1/1/10 | 0/20–30 min | oligodendro-toxicity; [Ca2+]i regulation | (not CB1/2, TPRV1, PPARγ, A2A, ryanodine and IP3 receptors, and L-type VGCC) | [42] | |
| OPCs (brains of Wistar rats, 0–2 days) | 3 | H2O2; tunicamycin; LPS/IFNγ | 0.1/1/2.5/5 | 2/24/48 h | oligodendro-protection | (not CB1/2, TPRV1, and PPARγ) | [81] |
| astrocytes and microglia (brains of C57BL/6 mice, 0–1 day) | 1 | LPS; scratch | 1/10 | 30 min, 72 h | anti-inflammatory effect; astrogliosis inhibition | (not CB2) | [80] |
| astrocytes and microglia (cortices of Wistar rats, 3–5 days) | CoCl2 | 5/50/100 | 30 min | regulation of hemodynamics | P-gp | [82] | |
| mixed glial cells (hippocampi of Lister-hooded rats, 1–3 days) | 5–10 | 1 | 0 | [Ca2+]i regulation | [29] | ||
| mixed glial cells (hippocampi of Lister-hooded rats, 1–3 days) | 5–10 | 1 | 0 | [Ca2+]i regulation | CB1, TRPV1, and PLC; (not FAAH, MAGL, and Gi/o-linked GPCR) | [30] | |
| mixed glial cells (hippocampi of Sprague–Dawley rats, 1–3 days) | 5–10 | 1/10 | 0 | [Ca2+]i regulation | Ca2+ channels (including L-type VGCC), CB1, and TRPV1 | [28] | |
| mixed glial cells (hippocampi of Lister-hooded rats, 1–3 days) | 5–10 | 4-AP; H2O2; oligomycin | 1 | 0 | [Ca2+]i regulation | mNCX; (not mPTP, and ryanodine and IP3 receptors) | [25] |
| Cell Type | Cell Source | DIV | CBD Concentration [μM] | CBD Exposure | CBD Effects | Related Molecular Targets | Ref. |
|---|---|---|---|---|---|---|---|
| MSCs | gingival tissues of healthy adult patients | 2nd passage | 5/10/25 | 24/48/96 h | promotion of differentiation toward neural progenitor cells | [111] | |
| MSCs | periodontal ligament tissues of healthy adult patients | 1st passage | CBD + MOR (0.5 each) | 24/48/72 h | promotion of cell survival and neuronal differentiation | [112] | |
| NPCs | HiB5 hippocampal progenitor cell line | 50/100/250/500 | 16/48 h | promotion of cell survival and proliferation; and modulation of cell cycle | CB1/2; (not 5-HT1A receptor) | [113] | |
| PSCs | human-induced PSC lines (iPSC6.2 and F002.1A.13) | 19 | 1/10 | 11/37 days | effect on differentiation/maturation of neural progenitor cells. | [114] |
| Cell Type | Biochemical Actions | Ref. | |
|---|---|---|---|
| neurons | anti-oxidation | [21,23,24,25] | |
| [Ca2+]i homeostasis | [25,28,29,30,31] | ||
| protection against mitochondrial dysfunction | [26,32,33] | ||
| regulation of neural-network activity | [36,37,38,39] | ||
| glia | microglia | anti-inflammation | [16,17,35,75,76,79,80] |
| regulation of microglial activation | [16,17,35,76,79,80] | ||
| promotion of phagocytosis | [77] | ||
| [Ca2+]i homeostasis | [25,28,29,30,75,77] | ||
| microglial migration | [41,75,89] | ||
| induction of microglial apoptosis | [78] | ||
| regulation of haemodynamics | [82] | ||
| astrocytes | anti-inflammation | [73,79,80] | |
| regulation of astroglial activation | [35,73,79,80] | ||
| immunosuppression | [17] | ||
| regulation of haemodynamics | [82] | ||
| [Ca2+]i homeostasis | [25,28,29,30] | ||
| oligodendrocytes | oligodendro-protection | [81] | |
| oligodendro-toxicity | [42] | ||
| [Ca2+]i homeostasis | [25,28,29,30,42,81] | ||
| neural stem cells | promotion of proliferation | [111,113,115,116,117] | |
| support of survival | [112,113,115,116,117] | ||
| promotion of neurogenesis | [111,112,113,115,116,117] | ||
| protection against impairment of neurogenesis | [73,113,117] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Choi, H.; Kang, E.K.; Ji, G.Y.; Kim, Y.; Choi, I.S. In Vitro Studies on Therapeutic Effects of Cannabidiol in Neural Cells: Neurons, Glia, and Neural Stem Cells. Molecules 2021, 26, 6077. https://doi.org/10.3390/molecules26196077
Kim J, Choi H, Kang EK, Ji GY, Kim Y, Choi IS. In Vitro Studies on Therapeutic Effects of Cannabidiol in Neural Cells: Neurons, Glia, and Neural Stem Cells. Molecules. 2021; 26(19):6077. https://doi.org/10.3390/molecules26196077
Chicago/Turabian StyleKim, Jungnam, Hyunwoo Choi, Eunhye K. Kang, Gil Yong Ji, Youjeong Kim, and Insung S. Choi. 2021. "In Vitro Studies on Therapeutic Effects of Cannabidiol in Neural Cells: Neurons, Glia, and Neural Stem Cells" Molecules 26, no. 19: 6077. https://doi.org/10.3390/molecules26196077
APA StyleKim, J., Choi, H., Kang, E. K., Ji, G. Y., Kim, Y., & Choi, I. S. (2021). In Vitro Studies on Therapeutic Effects of Cannabidiol in Neural Cells: Neurons, Glia, and Neural Stem Cells. Molecules, 26(19), 6077. https://doi.org/10.3390/molecules26196077