
Probing the Suitability of Different Ca2+ Parameters for Long Simulations of Diisopropyl Fluorophosphatase



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Equilibrium Simulations of DFPase under Different Treatment
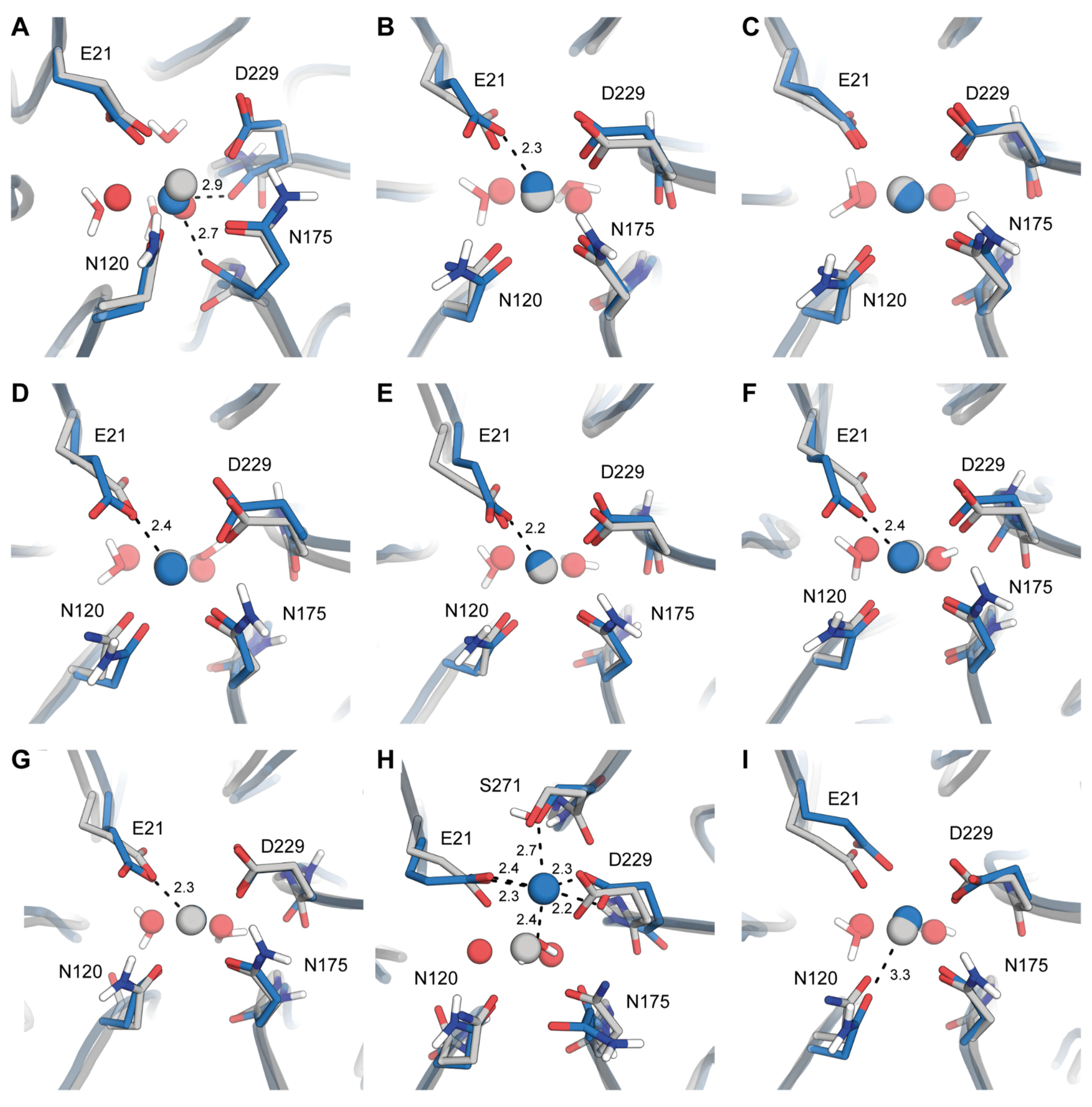
2.2. Origins of Structural Disturbances in the Catalytic Ca2+ Site
2.3. Origins of a Switched Conformation of E21
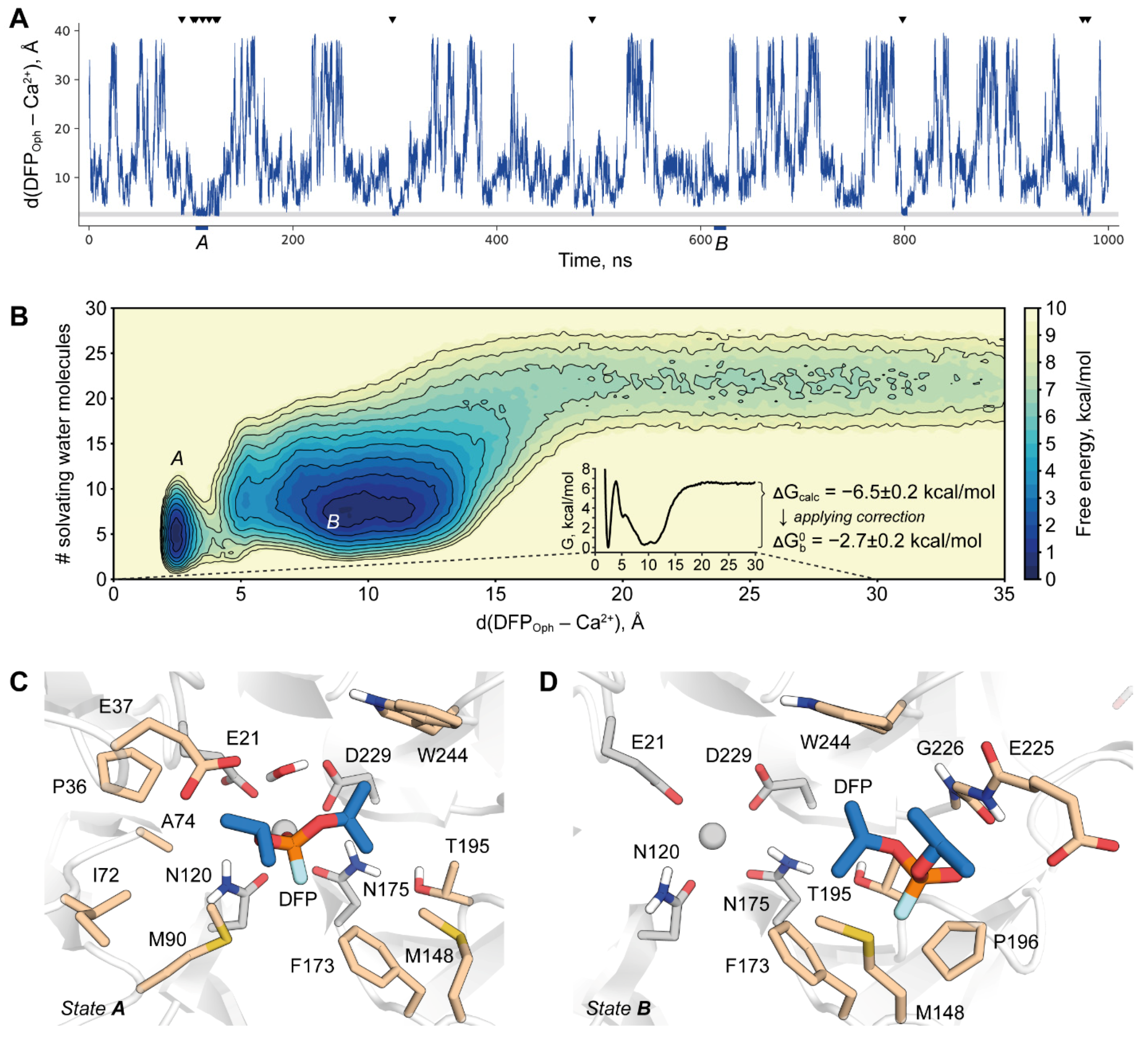
2.4. DFPase-DFP Binding/Unbinding Process
3. Materials and Methods
3.1. Parameter Sets
3.2. System Preparation
3.3. Molecular Dynamics
3.4. Analysis of Equilibration Dynamics
3.5. QM/MM Simulations
3.6. MM Simulations of E21 Conformation
3.7. Funnel Metadynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hoskin, F.C.; Long, R.J. Purification of a DFP-Hydrolyzing Enzyme from Squid Head Ganglion. Arch. Biochem. Biophys. 1972, 150, 548–555. [Google Scholar] [CrossRef]
- Hoskin, F.C.; Roush, A.H. Hydrolysis of Nerve Gas by Squid-Type Diisopropyl Phosphorofluoridate Hydrolyzing Enzyme on Agarose Resin. Science 1982, 215, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Matula, M.; Kucera, T.; Soukup, O.; Pejchal, J. Enzymatic Degradation of Organophosphorus Pesticides and Nerve Agents by EC: 3.1.8.2. Catalysts 2020, 10, 1365. [Google Scholar] [CrossRef]
- Blum, M.M.; Mustyakimov, M.; Rüterjans, H.; Kehe, K.; Schoenborn, B.P.; Langan, P.; Chen, J.C.-H. Rapid Determination of Hydrogen Positions and Protonation States of Diisopropyl Fluorophosphatase by Joint Neutron and X-Ray Diffraction Refinement. Proc. Natl. Acad. Sci. USA 2009, 106, 713–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, M.M.; Löhr, F.; Richardt, A.; Rüterjans, H.; Chen, J.C.H. Binding of a Designed Substrate Analogue to Diisopropyl Fluorophosphatase: Implications for the Phosphotriesterase Mechanism. J. Am. Chem. Soc. 2006, 128, 12750–12757. [Google Scholar] [CrossRef]
- Koepke, J.; Scharff, E.I.; Lücke, C.; Rüterjans, H.; Fritzsch, G. Atomic Resolution Crystal Structure of Squid Ganglion DFPase. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1757–1759. [Google Scholar] [CrossRef] [PubMed]
- Scharff, E.I.; Koepke, J.; Fritzsch, G.; Lücke, C.; Rüterjans, H. Crystal Structure of Diisopropylfluorophosphatase from Loligo Vulgaris. Structure 2001, 9, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Blum, M.M.; Chen, J.C.H. Structural Characterization of the Catalytic Calcium-Binding Site in Diisopropyl Fluorophosphatase (DFPase)--Comparison with Related Beta-Propeller Enzymes. Chem. Biol. Interact. 2010, 187, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Wymore, T.; Field, M.J.; Langan, P.; Smith, J.C.; Parks, J.M. Hydrolysis of DFP and the Nerve Agent (S)-Sarin by DFPase Proceeds along Two Different Reaction Pathways: Implications for Engineering Bioscavengers. J. Phys. Chem. B 2014, 118, 4479–4489. [Google Scholar] [CrossRef]
- Xu, C.; Yang, L.; Yu, J.G.; Liao, R.Z. What Roles Do the Residue Asp229 and the Coordination Variation of Calcium Play of the Reaction Mechanism of the Diisopropyl-Fluorophosphatase? A DFT Investigation. Theor. Chem. Acc. 2016, 135, 138. [Google Scholar] [CrossRef]
- Purg, M.; Elias, M.; Kamerlin, S.C.L. Similar Active Sites and Mechanisms Do Not Lead to Cross-Promiscuity in Organophosphate Hydrolysis: Implications for Biotherapeutic Engineering. J. Am. Chem. Soc. 2017, 139, 17533–17546. [Google Scholar] [CrossRef]
- Melzer, M.; Heidenreich, A.; Dorandeu, F.; Gäb, J.; Kehe, K.; Thiermann, H.; Letzel, T.; Blum, M.-M. In Vitro and in Vivo Efficacy of PEGylated Diisopropyl Fluorophosphatase (DFPase). Drug Test. Anal. 2012, 4, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.; Chen, J.C.-H.; Heidenreich, A.; Gäb, J.; Koller, M.; Kehe, K.; Blum, M.-M. Reversed Enantioselectivity of Diisopropyl Fluorophosphatase against Organophosphorus Nerve Agents by Rational Design. J. Am. Chem. Soc. 2009, 131, 17226–17232. [Google Scholar] [CrossRef] [PubMed]
- Raniolo, S.; Limongelli, V. Ligand Binding Free-Energy Calculations with Funnel Metadynamics. Nat. Protoc. 2020, 15, 2837–2866. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, V.; Bonomi, M.; Parrinello, M. Funnel Metadynamics as Accurate Binding Free-Energy Method. Proc. Natl. Acad. Sci. USA 2013, 110, 6358–6363. [Google Scholar] [CrossRef] [Green Version]
- Souza, P.C.T.; Thallmair, S.; Conflitti, P.; Ramírez-Palacios, C.; Alessandri, R.; Raniolo, S.; Limongelli, V.; Marrink, S.J. Protein-Ligand Binding with the Coarse-Grained Martini Model. Nat. Commun. 2020, 11, 3714. [Google Scholar] [CrossRef]
- Ribeiro, J.M.L.; Tsai, S.T.; Pramanik, D.; Wang, Y.; Tiwary, P. Kinetics of Ligand-Protein Dissociation from All-Atom Simulations: Are We There Yet? Biochemistry 2019, 58, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Bhattarai, A.; Wang, J. Ligand Gaussian Accelerated Molecular Dynamics (LiGaMD): Characterization of Ligand Binding Thermodynamics and Kinetics. J. Chem. Theory Comput. 2020, 16, 5526–5547. [Google Scholar] [CrossRef]
- Araki, M.; Matsumoto, S.; Bekker, G.J.; Isaka, Y.; Sagae, Y.; Kamiya, N.; Okuno, Y. Exploring Ligand Binding Pathways on Proteins Using Hypersound-Accelerated Molecular Dynamics. Nat. Commun. 2021, 12, 2793. [Google Scholar] [CrossRef]
- Wolf, S.; Lickert, B.; Bray, S.; Stock, G. Multisecond Ligand Dissociation Dynamics from Atomistic Simulations. Nat. Commun. 2020, 11, 2918. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M., Jr. Metal Ion Modeling Using Classical Mechanics. Chem. Rev. 2017, 117, 1564–1686. [Google Scholar] [CrossRef]
- Feenstra, K.A.; Hess, B.; Berendsen, H.J.C. Improving Efficiency of Large Time-Scale Molecular Dynamics Simulations of Hydrogen-Rich Systems. J. Comput. Chem. 1999, 20, 786–798. [Google Scholar] [CrossRef]
- Jung, J.; Sugita, Y. Group-Based Evaluation of Temperature and Pressure for Molecular Dynamics Simulation with a Large Time Step. J. Chem. Phys. 2020, 153, 234115. [Google Scholar] [CrossRef]
- Duarte, F.; Bauer, P.; Barrozo, A.; Amrein, B.A.; Purg, M.; Aqvist, J.; Kamerlin, S.C.L. Force Field Independent Metal Parameters Using a Nonbonded Dummy Model. J. Phys. Chem. B 2014, 118, 4351–4362. [Google Scholar] [CrossRef]
- Li, Z.; Song, L.F.; Li, P.; Merz, K.M., Jr. Systematic Parametrization of Divalent Metal Ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB Water Models. J. Chem. Theory Comput. 2020, 16, 4429–4442. [Google Scholar] [CrossRef] [PubMed]
- Babu, C.S.; Lim, C. Empirical Force Fields for Biologically Active Divalent Metal Cations in Water. J. Phys. Chem. A 2006, 110, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Wilson, J.; Aksimentiev, A. Improved Model of Hydrated Calcium Ion for Molecular Dynamics Simulations Using Classical Biomolecular Force Fields. Biopolymers 2016, 105, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.; Aksimentiev, A. New Tricks for Old Dogs: Improving the Accuracy of Biomolecular Force Fields by Pair-Specific Corrections to Non-Bonded Interactions. Phys. Chem. Chem. Phys. 2018, 20, 8432–8449. [Google Scholar] [CrossRef] [PubMed]
- Terekhov, S.S.; Mokrushina, Y.A.; Nazarov, A.S.; Zlobin, A.; Zalevsky, A.; Bourenkov, G.; Golovin, A.; Belogurov, A., Jr.; Osterman, I.A.; Kulikova, A.A.; et al. A Kinase Bioscavenger Provides Antibiotic Resistance by Extremely Tight Substrate Binding. Sci. Adv. 2020, 6, eaaz9861. [Google Scholar] [CrossRef]
- Rizzi, V.; Bonati, L.; Ansari, N.; Parrinello, M. The Role of Water in Host-Guest Interaction. Nat. Commun. 2021, 12, 93. [Google Scholar] [CrossRef]
- Capelli, R.; Lyu, W.; Bolnykh, V.; Meloni, S.; Olsen, J.M.H.; Rothlisberger, U.; Parrinello, M.; Carloni, P. Accuracy of Molecular Simulation-Based Predictions of Values: A Metadynamics Study. J. Phys. Chem. Lett. 2020, 11, 6373–6381. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ramezanghorbani, F.; Isayev, O.; Smith, J.S.; Roitberg, A.E. TorchANI: A Free and Open Source PyTorch-Based Deep Learning Implementation of the ANI Neural Network Potentials. J. Chem. Inf. Model. 2020, 60, 3408–3415. [Google Scholar] [CrossRef] [PubMed]
- Lahey, S.L.J.; Rowley, C.N. Simulating Protein-Ligand Binding with Neural Network Potentials. Chem. Sci. 2020, 11, 2362–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubatyuk, R.; Smith, J.S.; Nebgen, B.T.; Tretiak, S.; Isayev, O. Teaching a Neural Network to Attach and Detach Electrons from Molecules. Nat. Commun. 2021, 12, 4870. [Google Scholar] [CrossRef]
- Jindal, G.; Slanska, K.; Kolev, V.; Damborsky, J.; Prokop, Z.; Warshel, A. Exploring the Challenges of Computational Enzyme Design by Rebuilding the Active Site of a Dehalogenase. Proc. Natl. Acad. Sci. USA 2019, 116, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, F.; Kamerlin, S.C.L. (Eds.) Theory and Applications of the Empirical Valence Bond Approach: From Physical Chemistry to Chemical Biology; John Wiley & Sons: Nashville, TN, USA, 2017; ISBN 9781119245391. [Google Scholar]
- Repič, M.; Vianello, R.; Purg, M.; Duarte, F.; Bauer, P.; Kamerlin, S.C.L.; Mavri, J. Empirical Valence Bond Simulations of the Hydride Transfer Step in the Monoamine Oxidase B Catalyzed Metabolism of Dopamine. Proteins 2014, 82, 3347–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warshel, A. Computer Modeling of Chemical Reactions in Enzymes and Solutions; Wiley-Interscience: Hoboken, NJ, USA, 1991. [Google Scholar]
- Bauer, P.; Barrozo, A.; Purg, M.; Amrein, B.A.; Esguerra, M.; Wilson, P.B.; Major, D.T.; Åqvist, J.; Kamerlin, S.C.L. Q6: A Comprehensive Toolkit for Empirical Valence Bond and Related Free Energy Calculations. SoftwareX 2018, 7, 388–395. [Google Scholar] [CrossRef]
- Senftle, T.P.; Hong, S.; Islam, M.M.; Kylasa, S.B.; Zheng, Y.; Shin, Y.K.; Junkermeier, C.; Engel-Herbert, R.; Janik, M.J.; Aktulga, H.M.; et al. The ReaxFF Reactive Force-Field: Development, Applications and Future Directions. npj Comput. Mater. 2016, 2. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hourahine, B.; Aradi, B.; Blum, V.; Bonafé, F.; Buccheri, A.; Camacho, C.; Cevallos, C.; Deshaye, M.Y.; Dumitrică, T.; Dominguez, A.; et al. DFTB+, a Software Package for Efficient Approximate Density Functional Theory Based Atomistic Simulations. J. Chem. Phys. 2020, 152, 124101. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.; Elstner, M.; Kubař, T. Coupled-Perturbed DFTB-QM/MM Metadynamics: Application to Proton-Coupled Electron Transfer. J. Chem. Phys. 2018, 149, 072328. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Tribello, G.A. Analyzing and Biasing Simulations with PLUMED. Methods Mol. Biol. 2019, 2022, 529–578. [Google Scholar] [PubMed] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLSAA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef]
- Dodda, L.S.; de Cabeza Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen Web Server: An Automatic OPLS-AA Parameter Generator for Organic Ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [Green Version]
- Izadi, S.; Onufriev, A.V. Accuracy Limit of Rigid 3-Point Water Models. J. Chem. Phys. 2016, 145, 074501. [Google Scholar] [CrossRef] [Green Version]
- Elias, M.; Liebschner, D.; Koepke, J.; Lecomte, C.; Guillot, B.; Jelsch, C.; Chabriere, E. Hydrogen Atoms in Protein Structures: High-Resolution X-Ray Diffraction Structure of the DFPase. BMC Res. Notes 2013, 6, 308. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernetti, M.; Bussi, G. Pressure Control Using Stochastic Cell Rescaling. J. Chem. Phys. 2020, 153, 114107. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [Green Version]
- Tiwary, P.; Parrinello, M. A Time-Independent Free Energy Estimator for Metadynamics. J. Phys. Chem. B 2015, 119, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.-Z.; Thiel, W. Convergence in the QM-Only and QM/MM Modeling of Enzymatic Reactions: A Case Study for Acetylene Hydratase. J. Comput. Chem. 2013, 34, 2389–2397. [Google Scholar] [CrossRef] [Green Version]
- Jindal, G.; Warshel, A. Exploring the Dependence of QM/MM Calculations of Enzyme Catalysis on the Size of the QM Region. J. Phys. Chem. B 2016, 120, 9913–9921. [Google Scholar] [CrossRef]
- Gaus, M.; Cui, Q.; Elstner, M. DFTB3: Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB). J. Chem. Theory Comput. 2012, 7, 931–948. [Google Scholar] [CrossRef] [Green Version]
- Gaus, M.; Goez, A.; Elstner, M. Parametrization and Benchmark of DFTB3 for Organic Molecules. J. Chem. Theory Comput. 2013, 9, 338–354. [Google Scholar] [CrossRef]
- Mokrushina, Y.A.; Golovin, A.V.; Smirnov, I.V.; Chatziefthimiou, S.D.; Stepanova, A.V.; Bobik, T.V.; Zalevsky, A.O.; Zlobin, A.S.; Konovalov, K.A.; Terekhov, S.S.; et al. Multiscale Computation Delivers Organophosphorus Reactivity and Stereoselectivity to Immunoglobulin Scavengers. Proc. Natl. Acad. Sci. USA 2020, 117, 22841–22848. [Google Scholar] [CrossRef] [PubMed]
- Zlobin, A.S.; Zalevsky, A.O.; Mokrushina, Y.A.; Kartseva, O.V.; Golovin, A.V.; Smirnov, I.V. The Preferable Binding Pose of Canonical Butyrylcholinesterase Substrates Is Unproductive for Echothiophate. Acta Nat. 2018, 10, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Zlobin, A.; Mokrushina, Y.; Terekhov, S.; Zalevsky, A.; Bobik, T.; Stepanova, A.; Aliseychik, M.; Kartseva, O.; Panteleev, S.; Golovin, A.; et al. QM/MM Description of Newly Selected Catalytic Bioscavengers Against Organophosphorus Compounds Revealed Reactivation Stimulus Mediated by Histidine Residue in the Acyl-Binding Loop. Front. Pharmacol. 2018, 9, 834. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Gaus, M.; Elstner, M.; Cui, Q. Parametrization of DFTB3/3OB for Magnesium and Zinc for Chemical and Biological Applications. J. Phys. Chem. B 2015, 119, 1062–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vujović, M.; Huynh, M.; Steiner, S.; Garcia-Fernandez, P.; Elstner, M.; Cui, Q.; Gruden, M. Exploring the Applicability of Density Functional Tight Binding to Transition Metal Ions. Parameterization for Nickel with the Spin-Polarized DFTB3 Model. J. Comput. Chem. 2019, 40, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Roston, D.; Demapan, D.; Cui, Q. Extensive Free-Energy Simulations Identify Water as the Base in Nucleotide Addition by DNA Polymerase. Proc. Natl. Acad. Sci. USA 2019, 116, 25048–25056. [Google Scholar] [CrossRef]
- Kubillus, M.; Kubař, T.; Gaus, M.; Řezáč, J.; Elstner, M. Parameterization of the DFTB3 Method for Br, Ca, Cl, F, I, K, and Na in Organic and Biological Systems. J. Chem. Theory Comput. 2015, 11, 332–342. [Google Scholar] [CrossRef]
- Polynski, M.V.; Sapova, M.D.; Ananikov, V.P. Understanding the Solubilization of Ca Acetylide with a New Computational Model for Ionic Pairs. Chem. Sci. 2020, 11, 13102–13112. [Google Scholar] [CrossRef]
- Supercomputer Lomonosov-2: Large Scale, Deep Monitoring and Fine Analytics for the User Community. Supercomput. Front. Innov. 2019, 6. [CrossRef] [Green Version]
- Bhakat, S.; Söderhjelm, P. Resolving the Problem of Trapped Water in Binding Cavities: Prediction of Host-Guest Binding Free Energies in the SAMPL5 Challenge by Funnel Metadynamics. J. Comput. Aided Mol. Des. 2017, 31, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Galvelis, R.; Doerr, S.; Damas, J.M.; Harvey, M.J.; De Fabritiis, G. A Scalable Molecular Force Field Parameterization Method Based on Density Functional Theory and Quantum-Level Machine Learning. J. Chem. Inf. Model. 2019, 59, 3485–3493. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zlobin, A.; Diankin, I.; Pushkarev, S.; Golovin, A. Probing the Suitability of Different Ca2+ Parameters for Long Simulations of Diisopropyl Fluorophosphatase. Molecules 2021, 26, 5839. https://doi.org/10.3390/molecules26195839
Zlobin A, Diankin I, Pushkarev S, Golovin A. Probing the Suitability of Different Ca2+ Parameters for Long Simulations of Diisopropyl Fluorophosphatase. Molecules. 2021; 26(19):5839. https://doi.org/10.3390/molecules26195839
Chicago/Turabian StyleZlobin, Alexander, Igor Diankin, Sergey Pushkarev, and Andrey Golovin. 2021. "Probing the Suitability of Different Ca2+ Parameters for Long Simulations of Diisopropyl Fluorophosphatase" Molecules 26, no. 19: 5839. https://doi.org/10.3390/molecules26195839
APA StyleZlobin, A., Diankin, I., Pushkarev, S., & Golovin, A. (2021). Probing the Suitability of Different Ca2+ Parameters for Long Simulations of Diisopropyl Fluorophosphatase. Molecules, 26(19), 5839. https://doi.org/10.3390/molecules26195839