
The Reactive Sites of Methane Activation: A Comparison of IrC3+ with PtC3+


Abstract
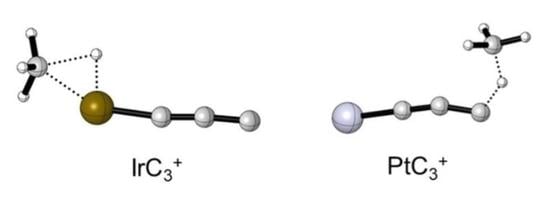
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental and Computational Methods
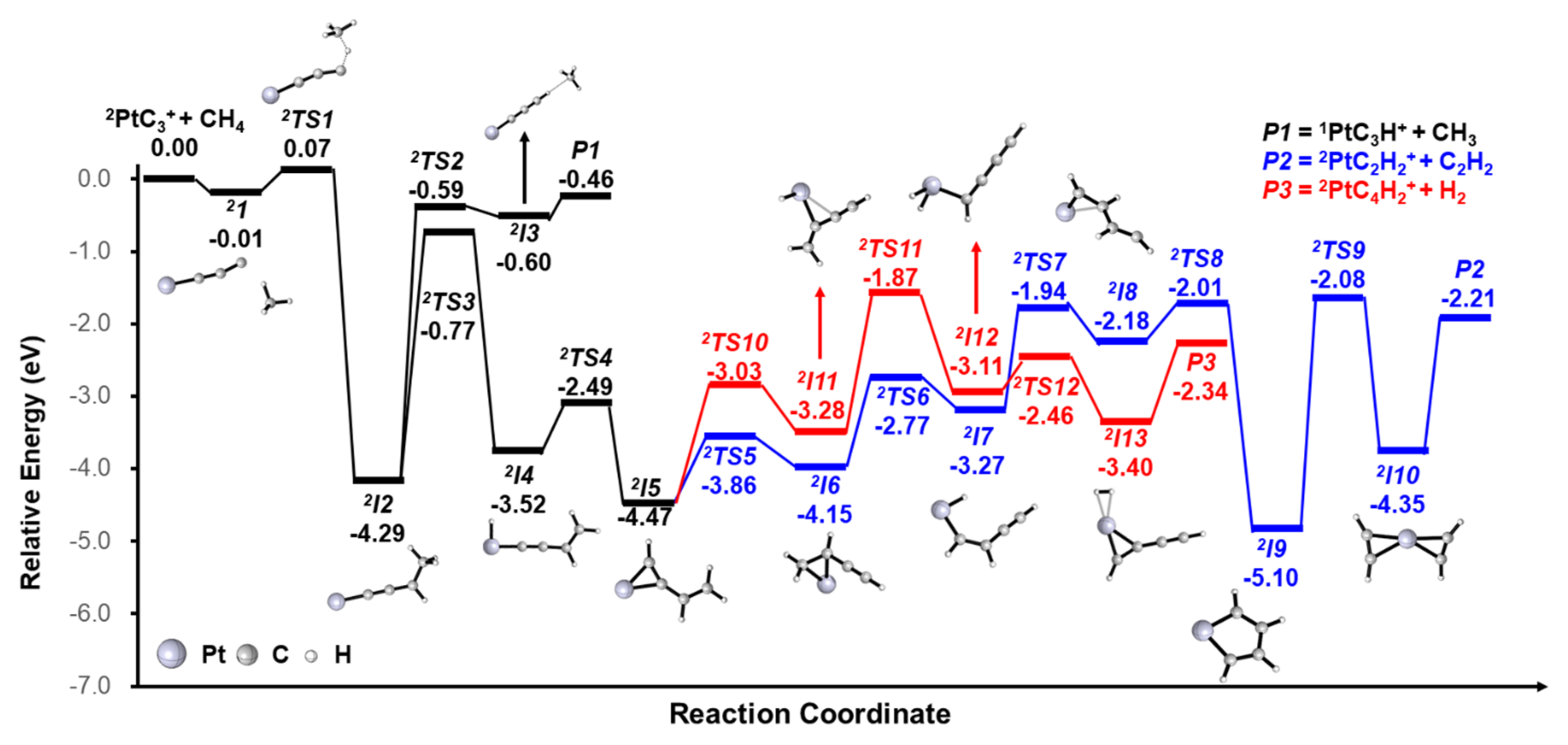
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Choudhary, T.; Aksoylu, E.; Goodman, D.W. Nonoxidative Activation of Methane. Catal. Rev. 2003, 45, 151–203. [Google Scholar] [CrossRef]
- Peter, M.; Marks, T.J. Platinum Metal-Free Catalysts for Selective Soft Oxidative Methane Ethylene Coupling. Scope and Mechanistic Observations. J. Am. Chem. Soc. 2015, 137, 15234–15240. [Google Scholar] [CrossRef]
- Schwarz, H. Chemistry with Methane: Concepts Rather than Recipes. Angew. Chem. Int. Ed. 2011, 50, 10096–10115. [Google Scholar] [CrossRef]
- Fokin, A.A.; Schreiner, P.R. Selective Alkane Transformations via Radicals and Radical Cations: Insights into the Activation Step from Experiment and Theory. Chem. Rev. 2002, 102, 1551–1594. [Google Scholar] [CrossRef]
- Schwarz, H.; Shaik, S.; Li, J. Electronic Effects on Room-Temperature, Gas-Phase C-H Bond Activations by Cluster Oxides and Metal Carbides: The Methane Challenge. J. Am. Chem. Soc. 2017, 139, 17201–17212. [Google Scholar] [CrossRef]
- Li, H.-F.; Jiang, L.-X.; Zhao, Y.-X.; Liu, Q.-Y.; Zhang, T.; He, S.-G. Formation of Acetylene in the Reaction of Methane with Iron Carbide Cluster Anions FeC3—Under High-Temperature Conditions. Angew. Chem. Int. Ed. 2018, 57, 2662–2666. [Google Scholar] [CrossRef]
- Geng, C.; Weiske, T.; Li, J.; Shaik, S.; Schwarz, H. Intrinsic Reactivity of Diatomic 3d Transition-Metal Carbides in the Thermal Activation of Methane: Striking Electronic Structure Effects. J. Am. Chem. Soc. 2018, 141, 599–610. [Google Scholar] [CrossRef]
- Ding, X.-L.; Wu, X.-N.; Zhao, Y.-X.; He, S.-G. C–H Bond Activation by Oxygen-Centered Radicals over Atomic Clusters. Accounts Chem. Res. 2011, 45, 382–390. [Google Scholar] [CrossRef]
- Gong, Y.; Zhou, M.; Andrews, L. Spectroscopic and Theoretical Studies of Transition Metal Oxides and Dioxygen Complexes. Chem. Rev. 2009, 109, 6765–6808. [Google Scholar] [CrossRef]
- Böhme, D.K.; Schwarz, H. Gas-Phase Catalysis by Atomic and Cluster Metal Ions: The Ultimate Single-Site Catalysts. Angew. Chem. Int. Ed. 2005, 44, 2336–2354. [Google Scholar] [CrossRef]
- Zhang, G.; Li, S.; Jiang, Y. Dehydrogenation of Methane by Gas-Phase Os+: A Density Functional Study. Organometallics 2003, 22, 3820–3830. [Google Scholar] [CrossRef]
- Wesendrup, R.; Schröder, D.; Schwarz, H. Catalytic Pt+-Mediated Oxidation of Methane by Molecular Oxygen in the Gas Phase. Angew. Chem. Int. Ed. 1994, 33, 1174–1176. [Google Scholar] [CrossRef]
- Irikura, K.K.; Beauchamp, J.L. Methane oligomerization in the gas phase by third-row transition-metal ions. J. Am. Chem. Soc. 1991, 113, 2769–2770. [Google Scholar] [CrossRef]
- Wang, G.; Chen, M.; Zhou, M. Activation of methane by Rh(0): Evidence for direct insertion of rhodium into the C–H bond at cryogenic temperatures. Chem. Phys. Lett. 2005, 412, 46–49. [Google Scholar] [CrossRef]
- Schröder, D.; Roithová, J. Low-Temperature Activation of Methane: It also Works Without a Transition Metal. Angew. Chem. Int. Ed. 2006, 45, 5705–5708. [Google Scholar] [CrossRef]
- Zhang, X.; Schwarz, H. Thermal Activation of Methane by Diatomic Metal Oxide Radical Cations: PbO+center dot as One of the Missing Pieces. Chemcatchem 2010, 2, 1391–1394. [Google Scholar] [CrossRef]
- Feyel, S.; Döbler, J.; Schröder, D.; Sauer, J.; Schwarz, H. Thermal Activation of Methane by Tetranuclear [V4O10]+. Angew. Chem. Int. Ed. 2006, 45, 4681–4685. [Google Scholar] [CrossRef]
- Zhao, Y.-X.; Wu, X.-N.; Wang, Z.-C.; He, S.-G.; Ding, X.-L. Hydrogen-atom abstraction from methane by stoichiometric early transition metal oxide cluster cations. Chem. Commun. 2010, 46, 1736–1738. [Google Scholar] [CrossRef]
- Li, H.-F.; Li, Z.-Y.; Liu, Q.-Y.; Li, X.-N.; Zhao, Y.-X.; He, S.-G. Methane Activation by Iron-Carbide Cluster Anions FeC6−. J. Phys. Chem. Lett. 2015, 6, 2287–2291. [Google Scholar] [CrossRef]
- Liu, Q.-Y.; Ma, J.-B.; Li, Z.-Y.; Zhao, C.; Ning, C.; Chen, H.; He, S. Activation of Methane Promoted by Adsorption of CO on Mo2C2−Cluster Anions. Angew. Chem. Int. Ed. 2016, 55, 5760–5764. [Google Scholar] [CrossRef]
- Li, H.-F.; Zhao, Y.-X.; Yuan, Z.; Liu, Q.-Y.; Li, Z.-Y.; Li, X.-N.; Ning, C.-G.; He, S.-G. Methane Activation by Tantalum Carbide Cluster Anions Ta2C4−. J. Phys. Chem. Lett. 2017, 8, 605–610. [Google Scholar] [CrossRef]
- Li, J.; Zhou, S.; Schlangen, M.; Weiske, T.; Schwarz, H. Hidden Hydride Transfer as a Decisive Mechanistic Step in the Reactions of the Unligated Gold Carbide [AuC]+ with Methane under Ambient Conditions. Angew. Chem. Int. Ed. 2016, 55, 13072–13075. [Google Scholar] [CrossRef]
- Geng, C.; Li, J.; Weiske, T.; Schwarz, H. A Reaction-Induced Localization of Spin Density Enables Thermal C–H Bond Activation of Methane by Pristine FeC4+. Chem.-A Eur. J. 2019, 25, 12940–12945. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.-N.; Liu, Z.; Wu, H.; Zhang, D.; Li, W.; Huang, Z.; Wang, G.; Xu, F.; Ding, C.-F.; Zhou, M. Reactions of Transition-Metal Carbyne Cations with Ethylene in the Gas Phase. J. Phys. Chem. A 2020, 124, 2628–2633. [Google Scholar] [CrossRef]
- Li, W.; Wu, X.-N.; Liu, Z.; Wu, H.; Zhang, D.; Ding, X.-L. The C/C Exchange in Activation/Coupling Reaction of Acetylene and Methane Mediated by Os+: A Comparison with Ir+, Pt+ and Au+. J. Phys. Chem. Lett. 2020, 11. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, G.; Petersson, A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Boese, A.D.; Martin, J.M.L. Development of density functionals for thermochemical kinetics. J. Chem. Phys. 2004, 121, 3405–3416. [Google Scholar] [CrossRef] [Green Version]
- Lu, L. Molclus program, Beijing Kein Research Center for Natural Science, China. 2016. Available online: http://www.keinsci.com/research/molclus.html (accessed on 1 September 2021).
- Peng, C.Y.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Fukui, K. The Path of Chemical-Reactions—The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Wu, H.; Li, W.; Wu, X. The Reactive Sites of Methane Activation: A Comparison of IrC3+ with PtC3+. Molecules 2021, 26, 6028. https://doi.org/10.3390/molecules26196028
Liu Z, Wu H, Li W, Wu X. The Reactive Sites of Methane Activation: A Comparison of IrC3+ with PtC3+. Molecules. 2021; 26(19):6028. https://doi.org/10.3390/molecules26196028
Chicago/Turabian StyleLiu, Zizhuang, Hechen Wu, Wei Li, and Xiaonan Wu. 2021. "The Reactive Sites of Methane Activation: A Comparison of IrC3+ with PtC3+" Molecules 26, no. 19: 6028. https://doi.org/10.3390/molecules26196028
APA StyleLiu, Z., Wu, H., Li, W., & Wu, X. (2021). The Reactive Sites of Methane Activation: A Comparison of IrC3+ with PtC3+. Molecules, 26(19), 6028. https://doi.org/10.3390/molecules26196028