
T3P-Promoted Synthesis of a Series of 2-Aryl-3-phenyl-2,3-dihydro-4H-pyrido[3,2-e][1,3]thiazin-4-ones and Their Activity against the Kinetoplastid Parasite Trypanosoma brucei

 , , ,
, , ,
Abstract
:1. Introduction
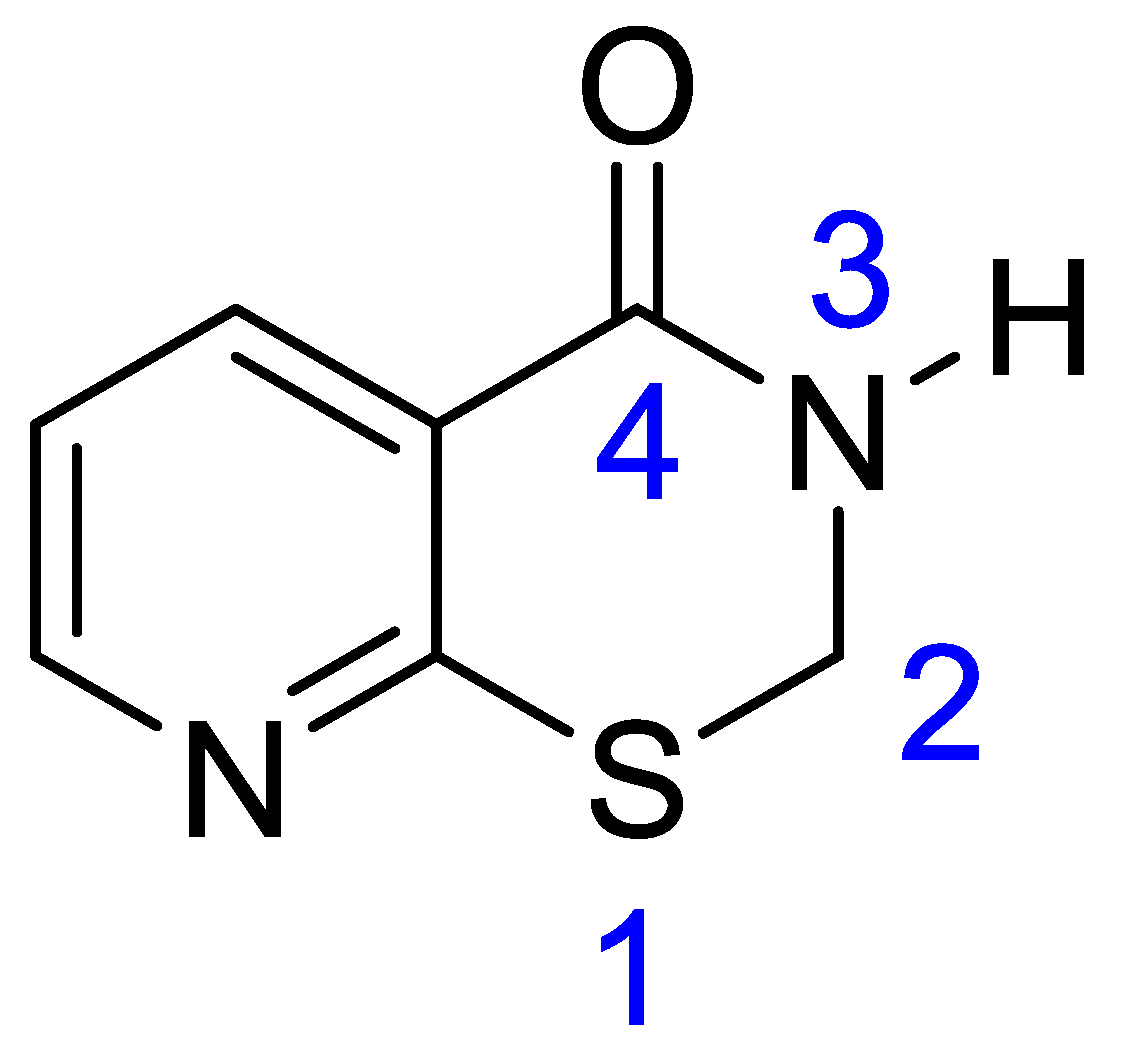
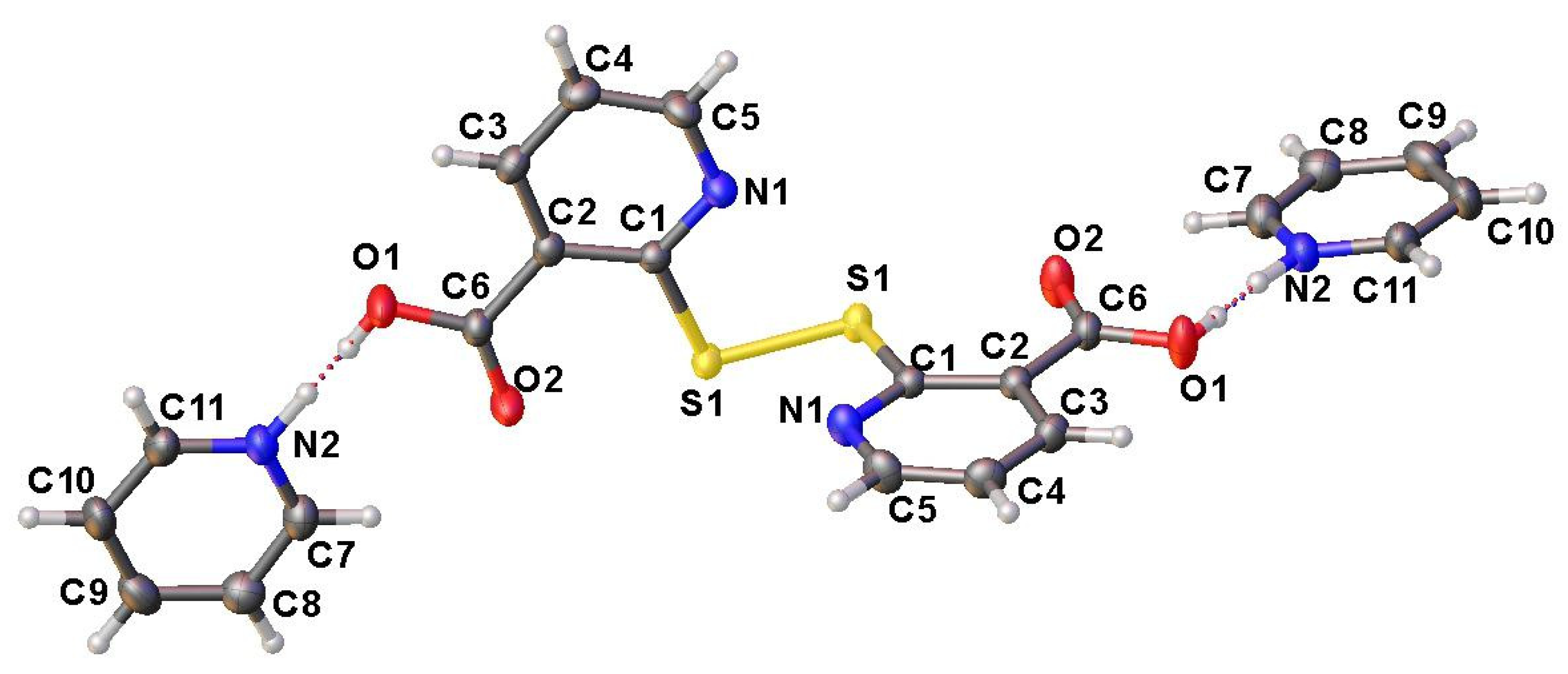
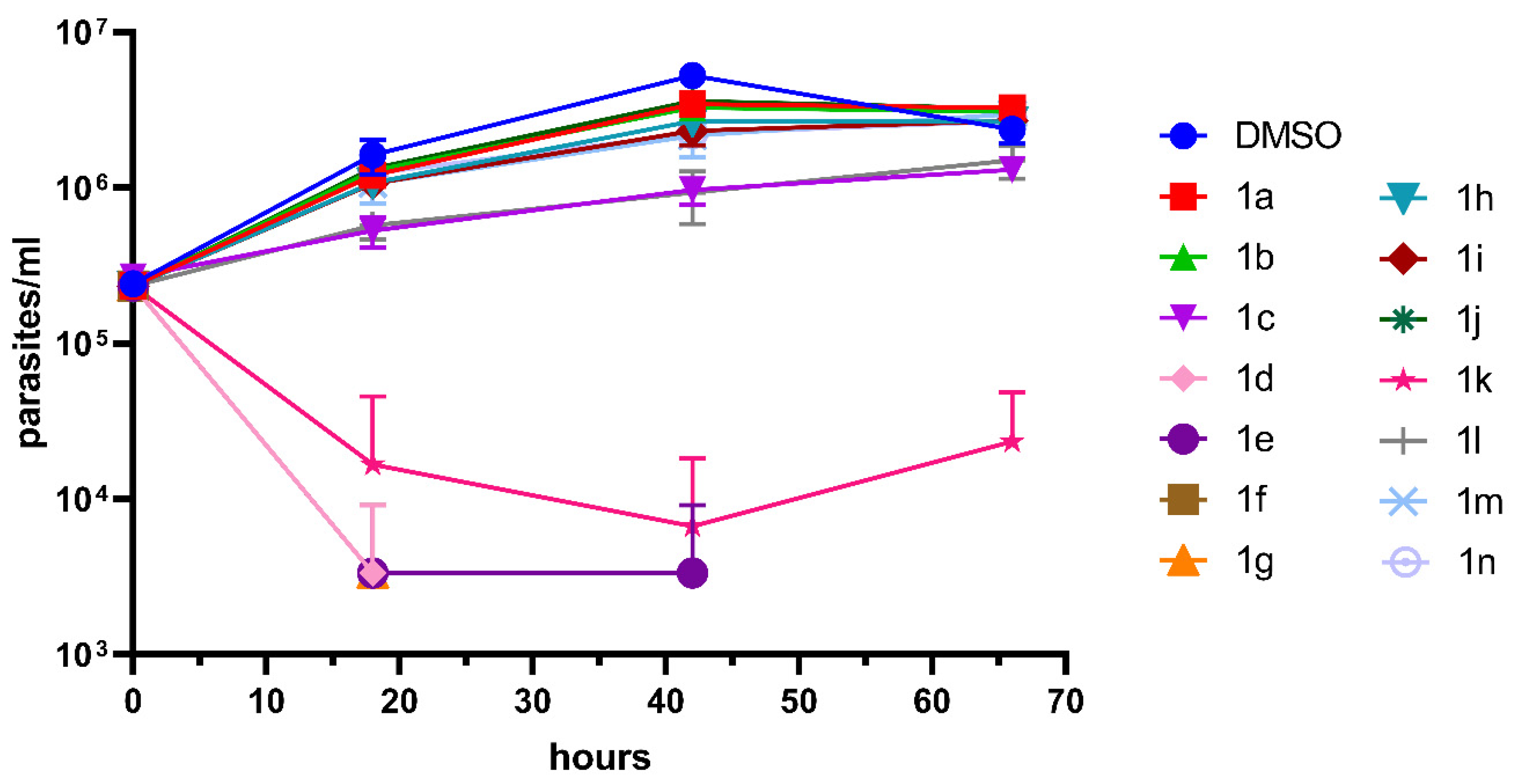
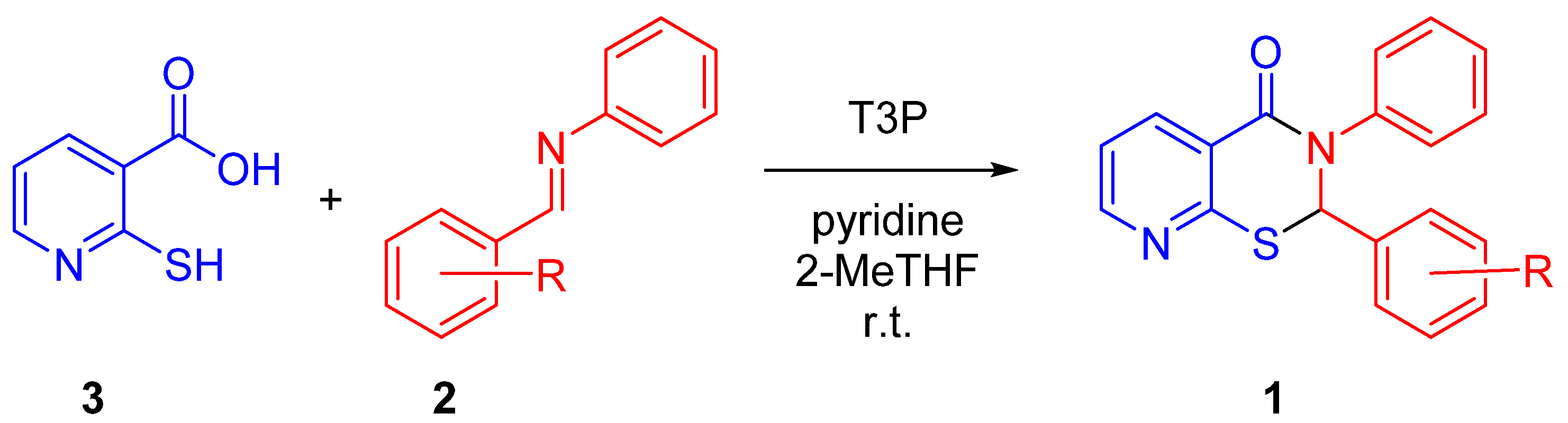
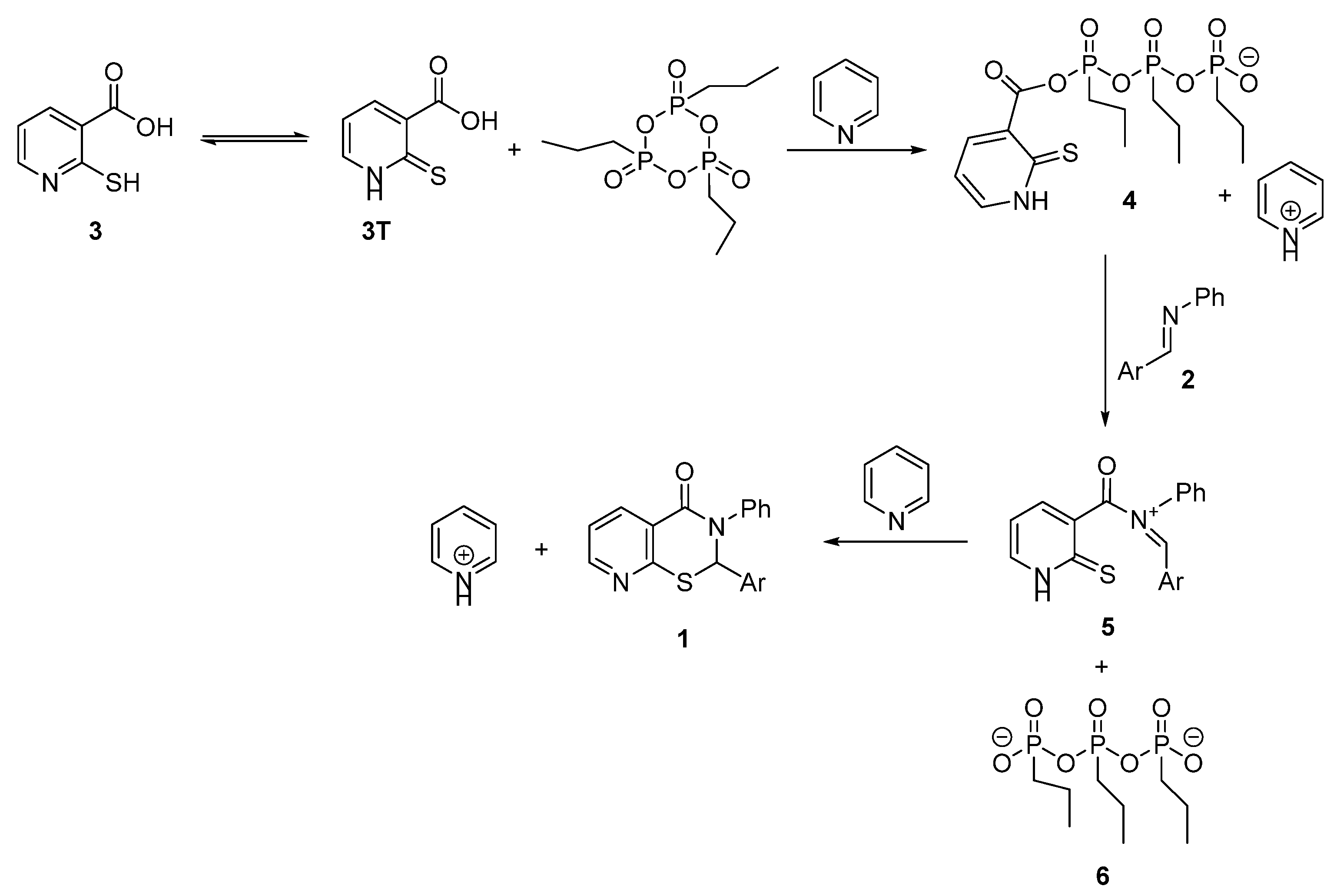
2. Results and Discussion
3. Materials and Methods
X-ray Crystallographic Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Arya, K.; Tomar, P.; Singh, J. Design, synthesis and biological evaluation of novel spiro[indole-pyridothiazine] analogs as antiproliferative agents. RSC Adv. 2014, 4, 3060–3064. [Google Scholar] [CrossRef]
- Shreedhara, S.H.; Vagdevi, H.M.; Jayanna, N.D.; Raghavendra, R. Synthesis, characterization and evaluation of cytotoxic, antibacterial and molecular docking studies of fused heterocyclic 6aH,13H benz[4’,5′]oxazole[2′,3′,:2,3][1,3]thiazino[6,5-b]quinolin-13-one derivatives. Int. J. Pharma. Res. Health Sci. 2017, 5, 2055–2063. [Google Scholar] [CrossRef]
- Li, X.; Qin, Z.; Yang, T.; Zhang, H.; Wei, S.; Li, C.; Chen, H.; Meng, M. Synthesis and biological activity of bi/tricyclic azasugars fused thiazolidin-4-one and thiazinan-4-one by microwave-assisted tandem Staudinger/aza-Wittig/cyclization. Bioorg. Med. Chem. Lett. 2012, 22, 2712–2716. [Google Scholar] [CrossRef]
- Yennawar, H.P.; Singh, H.; Silverberg, L.J. 2,3-Diphenyl-2,3-dihydro-4H-pyrido[3,2-e][1,3]thiazin-4-one. Acta Cryst. 2014, E70, o638. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, L.J.; Pacheco, C.N.; Lagalante, A.; Cannon, K.C.; Bachert, J.T.; Xie, Y.; Baker, L.; Bayliff, J.A. Synthesis and Spectroscopic Properties of 2,3-Diphenyl-1,3-thiaza-4-one Heterocycles. Int. J. Chem. 2015, 7, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Malfara, M.F.; Silverberg, L.J.; DiMaio, J.; Iatsenko, K.; Povelones, M.L. 2,3-Diphenyl-2,3-dihydro-4H-1,3-thiaza-4-one heterocycles inhibit growth and block completion of cytokinesis in kinetoplastid parasites. Mol. Biochem. Parasitol. 2021, 245, 111396. [Google Scholar] [CrossRef] [PubMed]
- Liporagi-Lopes, L.; Sobhi, H.F.; Silverberg, L.J.; Cordero, R.J.B.; Casadevall, A. Antifungal activity of 2,3-diphenyl-2,3-dihydro-1,3-thiaza-4-ones against two human pathogenic fungi. BioRxiv 2020. [Google Scholar] [CrossRef]
- Arya, K.; Diwan Rawata, S.; Sasaib, H. Zeolite supported Brønsted-acid ionic liquids: An eco approach for synthesis of spiro[indole-pyrido[3,2-e]thiazine] in water under ultrasonication. Green Chem. 2012, 14, 1956–1963. [Google Scholar] [CrossRef]
- Dandia, A.; Arya, K.; Sati, M.; Gautam, S. Microwave assisted green chemical synthesis of novel spiro[indole-pyrido thiazines]: A system reluctant to be formed under thermal conditions. Tetrahedron 2004, 60, 5253–5258. [Google Scholar] [CrossRef]
- Chakravarty, S.; Pham, S.M.; Kankanala, J.; Agarwal, A.K.; Pujala, B.; Soni, S.; Arya, S.K.; Palve, D.; Kumar, V. Preparation of Heterocyclic Compounds, Especially Substituted 2H-pyrimido[5,4-e][1,3]oxazin-4(3H)-ones and 2H-pyrimido[5,4-e][1,3]thiazin-4(3H)-ones and Their Analogs, as Wee1 Inhibitors. U.S. Patent 20190106436 A1, 11 April 2019. [Google Scholar]
- Silverberg, L.J.; Moyer, Q.J. Chemistry of 1,3-thiazin-4-ones and their derivatives, 1995-mid 2018. Arkivoc 2019, 1, 139–227. [Google Scholar] [CrossRef] [Green Version]
- Silverberg, L.J.; Pacheco, C.; Sahu, D.; Scholl, P.; Sobhi, H.F.; Bradley, H.G.; Cardenas, O.A.; Gonzalez, K.M.; Islam, J.M.; Kimmel, E.G.; et al. T3P-Promoted Synthesis of a Series of Novel 2-Aryl-3-phenyl-2,3,5,6-tetrahydro-4H-1,3-thiazin-4-ones. Tetrahedron Lett. 2020, 61, 151836. [Google Scholar] [CrossRef]
- Silverberg, L.J.; Pacheco, C.; Sahu, D.; Scholl, P.; Sobhi, H.F.; Bachert, J.T.; Bandholz, K.; Bendinsky, R.V.; Bradley, H.G.; Colburn, B.K.; et al. T3P-promoted synthesis of a series of novel 3-aryl-2-phenyl-2,3-dihydro-4H-1,3-benzothiazin-4-ones. J. Heterocycl. Chem. 2020, 57, 1797–1805. [Google Scholar] [CrossRef]
- Silverberg, L.J.; Tierney, J.; Pacheco, C.; Lagalante, A.; Bachert, J.T.; Bayliff, J.A.; Bendinsky, R.V.; Cali, A.S.; Chen, L.; Cooper, A.D.; et al. Synthesis and spectroscopic properties of a series of novel 2-aryl-3-phenyl-2,3-dihydro-4H-1,3-benzothiazin-4-ones. Arkivoc 2016, 6, 122–143. [Google Scholar] [CrossRef] [Green Version]
- Yennawar, H.P.; Li, J.; Thompson, E.N.; Silverberg, L.J. Crystal structures of two solvated 2-aryl-3-phenyl-2,3-dihydro-4H-pyrido[3,2-e][1,3]thiazin-4-ones. Acta Cryst. 2019, E75, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Dunetz, J.R.; Xiang, Y.; Baldwin, A.; Ringling, J. General and Scalable Amide Bond Formation with Epimerization-Prone Substrates Using T3P and Pyridine. Org. Lett. 2011, 13, 5048–5051. [Google Scholar] [CrossRef]
- Unsworth, W.P.; Kitsiou, C.; Taylor, R.J.K. Direct Imine Acylation: Rapid Access to Diverse Heterocyclic Scaffolds. Org. Lett. 2013, 15, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Unsworth, W.P.; Coulthard, G.; Kitsiou, C.; Taylor, R.J.K. Direct imine acylation for molecular diversity in heterocyclic synthesis. J. Org. Chem. 2014, 79, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Kitsiou, C.; Unsworth, W.P.; Coulthard, G.; Taylor, R.J.K. Substrate scope in the direct imine acylation of ortho-substituted benzoic acid derivatives: The total synthesis (+/-)-cavidine. Tetrahedron 2014, 70, 7172–7180. [Google Scholar] [CrossRef]
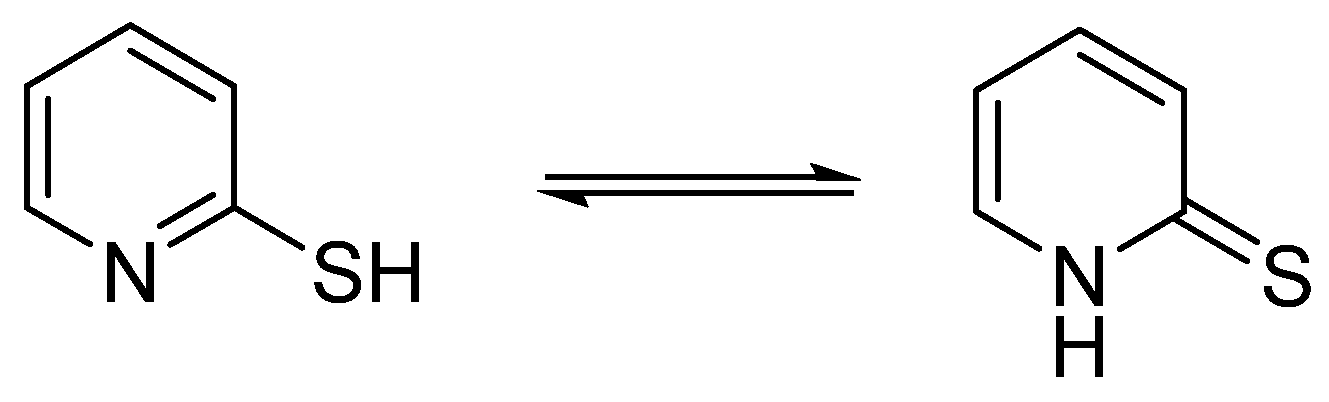
- Moran, D.; Sukcharoenphon, K.; Puchta, R.; Schaefer, H.F., III; Schleyer, P.V.R.; Hoff, C.D. 2-Pyridinethiol/2-Pyridinethione Tautomeric Equilibrium. A Comparative Experimental and Computational Study. J. Org. Chem. 2002, 67, 9061–9069. [Google Scholar] [CrossRef]
- Saleh, M.S.; Idriss, K.A.; Au-Bakr, M.S.; Hashem, E.Y. Acid Dissociation and Solution Equilibria of Some Pyridinecarboxylic Acids. Analyst 1992, 117, 1003–1007. [Google Scholar] [CrossRef]
- Smith, G.; Sagatys, D.S. Ammonium 2-mercaptopyridine-3-carboxylate hydrate. Acta. Cryst. 2003, E59, o540–o541. [Google Scholar] [CrossRef]
- Corban, G.J.; Antoniadis, C.D.; Hadjikakou, S.K.; Kourkoumelis, N.; Tyurin, V.Y.; Dolgano, A.; Milaeva, E.R.; Kubicki, M.; Bernhardt, P.V.; Tiekink, E.R.T.; et al. Reactivity of di-iodine toward thiol: Desulfuration reaction of 5-nitro-2-mercapto-benzimidazole upon reaction with di-iodine. Heteroatom Chem. 2012, 23, 498–511. [Google Scholar] [CrossRef]
- Zhu, J.; Zhao, Y.; Hong, M.; Sun, D.; Shi, Q.; Cao, R. A novel 2D Mn(II)-disulphide complex [Mn(2,20-DTDN)(H2O)2]n•2nH2O. Chem. Lett. 2002, 31, 484–485. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Maginnity, P.M.; Eisenmann, J.L. Derivatives of o-, m-, and p-aminobenzotrifluoride. II. azomethines containing the trifluoromethyl group. J. Am. Chem. Soc. 1952, 74, 6119–6121. [Google Scholar] [CrossRef]
- Hirumi, H.; Hirumi, K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
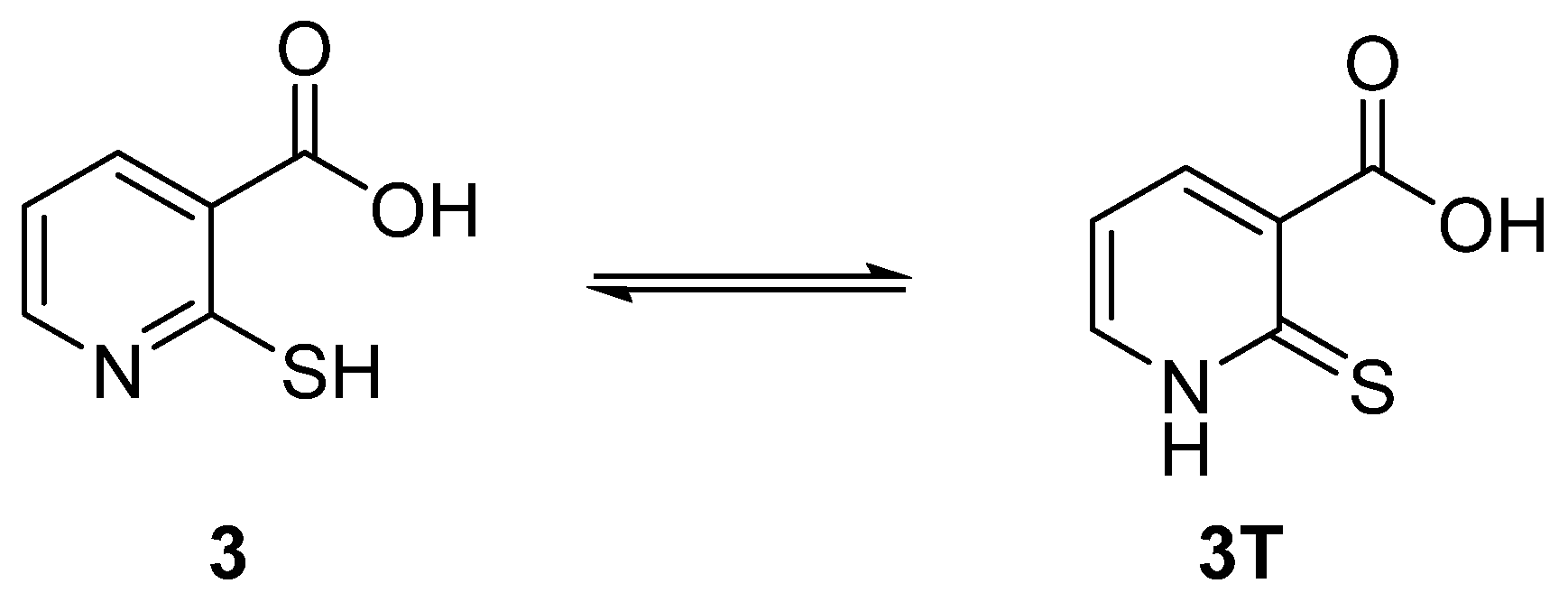{kind=link}
{kind=link}
| Compound | R | Yield of 1 from 2 (%) |
|---|---|---|
| 1a [15] | p-NO2 | 45% a |
| 1b | m-NO2 | 52% a |
| 1c | o-NO2 | 22% a |
| 1d | p-CF3 | 63% a |
| 1e | m-CF3 | 54% a |
| 1f | p-Br | 40% a |
| 1g | m-Br | 42% a,b |
| 1h [15] | p-F | 50% a |
| 1i | m-F | 43% a |
| 1j [4,5] | H | 48% a |
| 1k | p-Me | 53% c |
| 1l | m-Me | 35% a,b |
| 1m | p-OMe | 55% a |
| 1n | m-OMe | 44% a,b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silverberg, L.J.; Mal, T.K.; Pacheco, C.N.; Povelones, M.L.; Malfara, M.F.; Lagalante, A.F.; Olsen, M.A.; Yennawar, H.P.; Sobhi, H.F.; Baney, K.R.; et al. T3P-Promoted Synthesis of a Series of 2-Aryl-3-phenyl-2,3-dihydro-4H-pyrido[3,2-e][1,3]thiazin-4-ones and Their Activity against the Kinetoplastid Parasite Trypanosoma brucei. Molecules 2021, 26, 6099. https://doi.org/10.3390/molecules26206099
Silverberg LJ, Mal TK, Pacheco CN, Povelones ML, Malfara MF, Lagalante AF, Olsen MA, Yennawar HP, Sobhi HF, Baney KR, et al. T3P-Promoted Synthesis of a Series of 2-Aryl-3-phenyl-2,3-dihydro-4H-pyrido[3,2-e][1,3]thiazin-4-ones and Their Activity against the Kinetoplastid Parasite Trypanosoma brucei. Molecules. 2021; 26(20):6099. https://doi.org/10.3390/molecules26206099
Chicago/Turabian StyleSilverberg, Lee J., Tapas K. Mal, Carlos N. Pacheco, Megan L. Povelones, Madeline F. Malfara, Anthony F. Lagalante, Mark A. Olsen, Hemant P. Yennawar, Hany F. Sobhi, Kayla R. Baney, and et al. 2021. "T3P-Promoted Synthesis of a Series of 2-Aryl-3-phenyl-2,3-dihydro-4H-pyrido[3,2-e][1,3]thiazin-4-ones and Their Activity against the Kinetoplastid Parasite Trypanosoma brucei" Molecules 26, no. 20: 6099. https://doi.org/10.3390/molecules26206099
APA StyleSilverberg, L. J., Mal, T. K., Pacheco, C. N., Povelones, M. L., Malfara, M. F., Lagalante, A. F., Olsen, M. A., Yennawar, H. P., Sobhi, H. F., Baney, K. R., Bozeman, R. L., Eroh, C. S., Fleming, M. J., Garcia, T. L., Gregory, C. L., Hahn, J. E., Hatter, A. M., Johns, L. L., Klinger, T. L., ... Yazdani, S. F. (2021). T3P-Promoted Synthesis of a Series of 2-Aryl-3-phenyl-2,3-dihydro-4H-pyrido[3,2-e][1,3]thiazin-4-ones and Their Activity against the Kinetoplastid Parasite Trypanosoma brucei. Molecules, 26(20), 6099. https://doi.org/10.3390/molecules26206099