
Stereoelectronic Features of a Complex Ketene Dimerization Reaction



Abstract
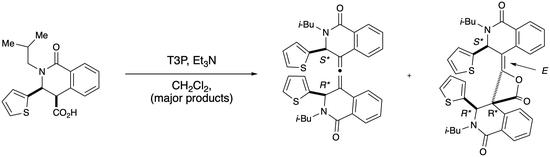
:
1. Introduction
2. Results and Discussion
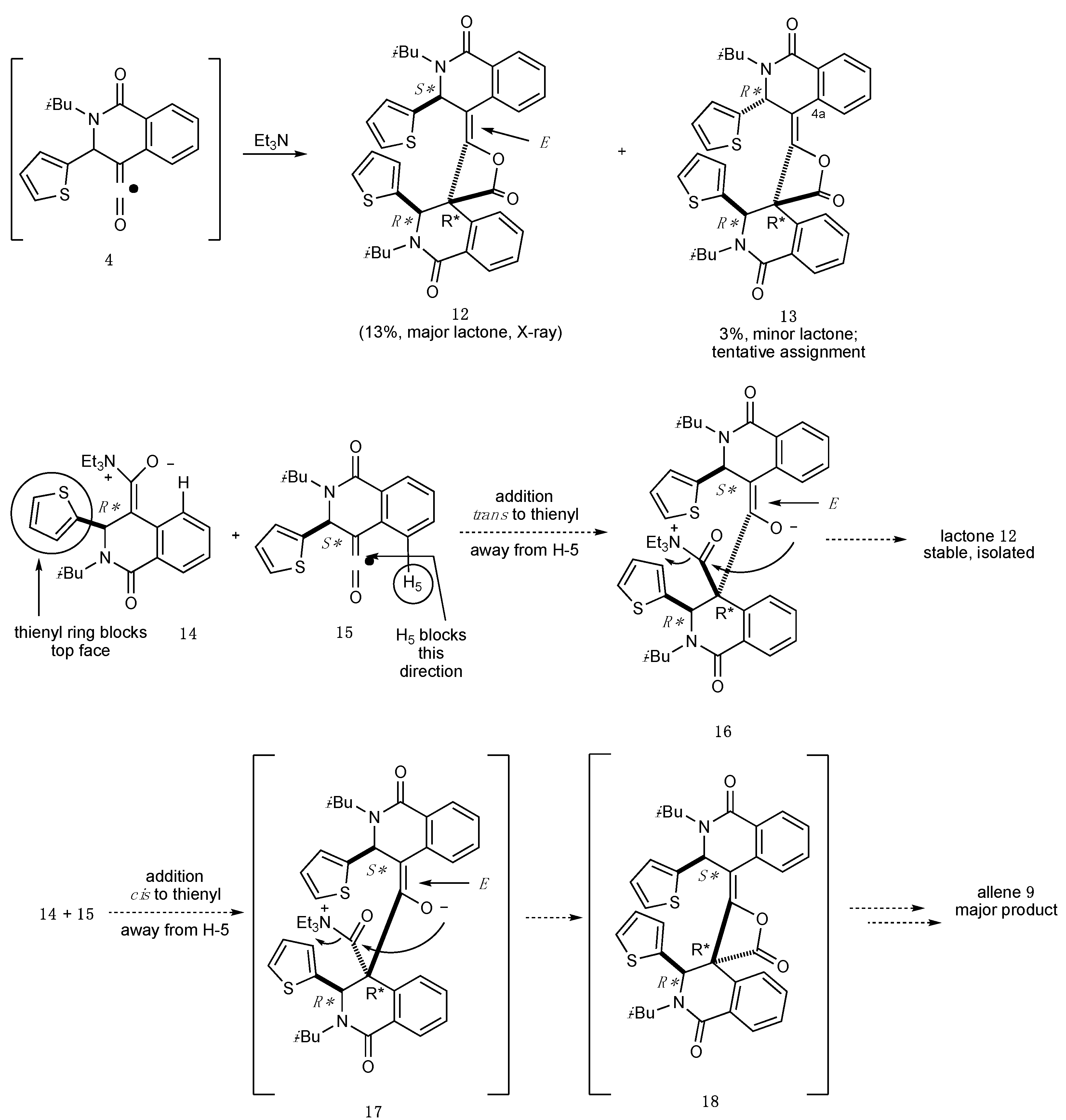
2.1. Dimeric Allene Formation from THIQ Carboxylates
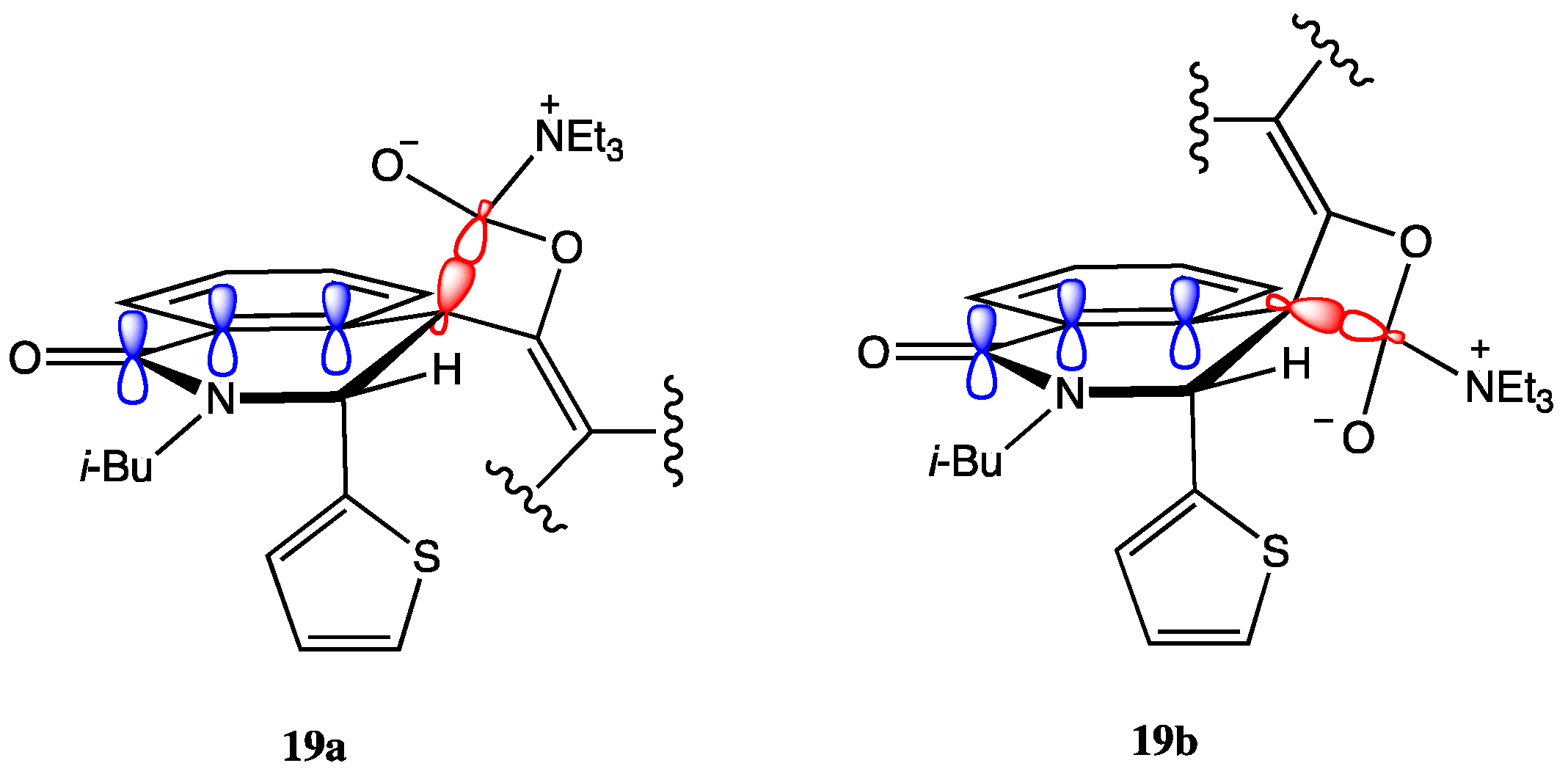
2.2. Determination of the Dimeric Lactones
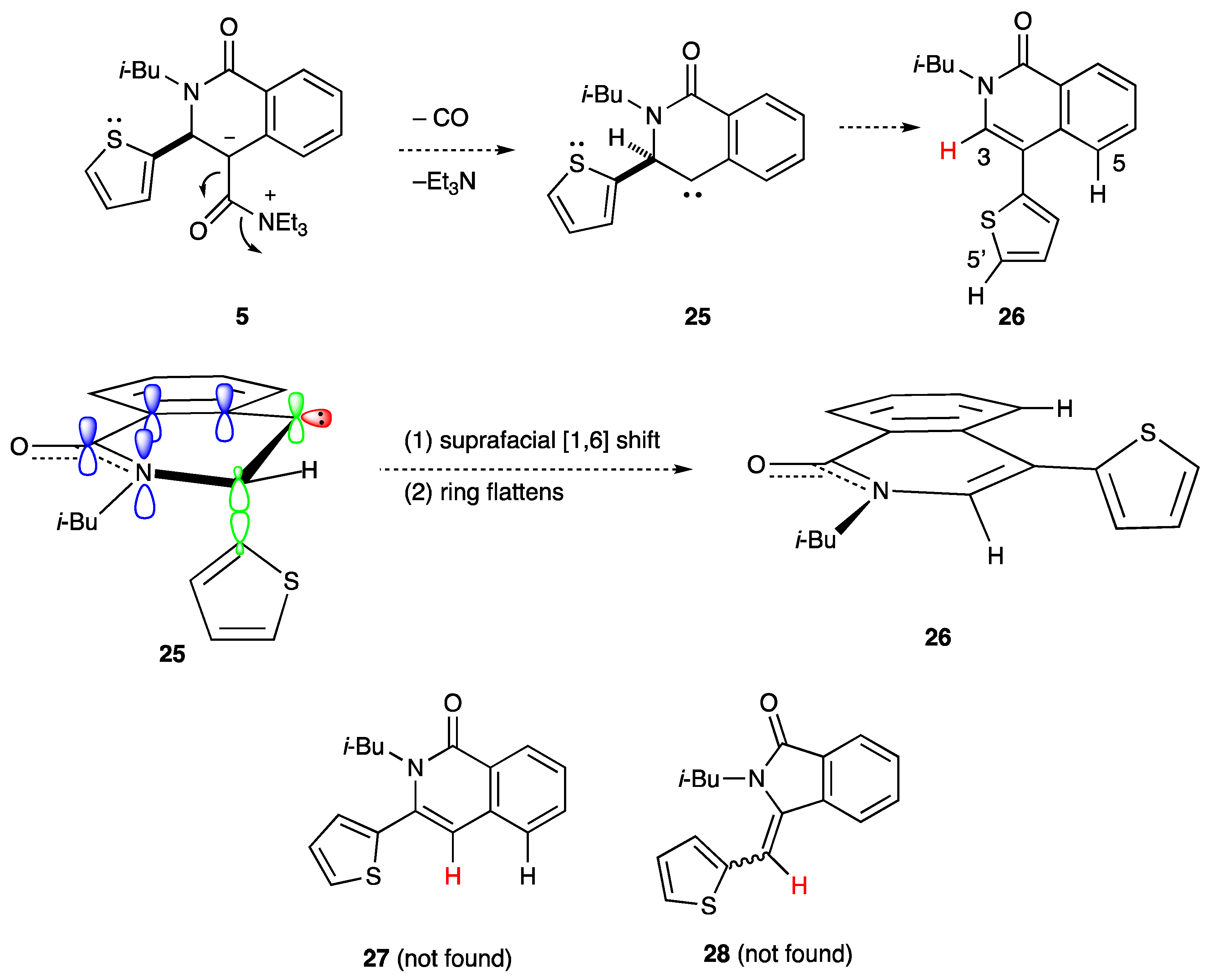
2.3. Determination of the Other Products
3. Conclusions
4. Materials and Methods
4.1. General
4.2. Dehydration/Dimerization of (3S*,4R*)-2-Isobutyl-1-oxo-3-(thiophen-3-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (4)
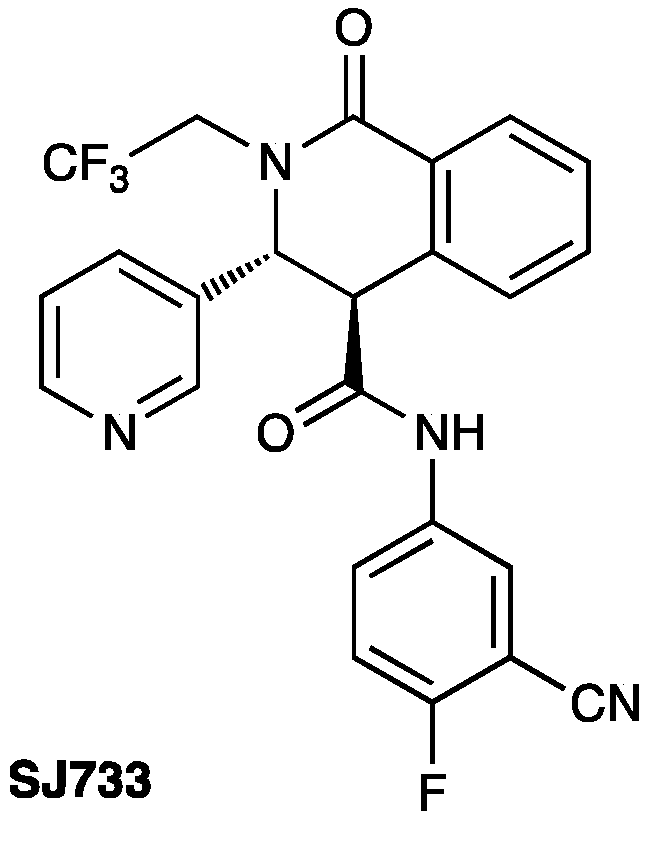
4.3. (3R*,4S*)-2-Isobutyl-2’-((S*,E)-2-isobutyl-1-oxo-3-(thiophen-2-yl)-2,3-dihydroisoquinolin-4(1H)-ylidene)-3-(thiophen-2-yl)-2,3-dihydro-1H-spiro[isoquinoline-4,3’-oxetane]-1,4’-dione (Major Lactone 12)
4.4. (3R*,4S*)-2-Isobutyl-2’-((R*,E)-2-isobutyl-1-oxo-3-(thiophen-2-yl)-2,3-dihydroisoquinolin-4(1H)-ylidene)-3-(thiophen-2-yl)-2,3-dihydro-1H-spiro[isoquinoline-4,3’-oxetane]-1,4’-dione (Assigned to Minor Lactone 13)
4.5. 6-Isobutyl-5H-thieno[3’,2’:4,5]cyclopenta[1,2-c]isoquinoline-5,10(6H)-dione (Purple Ketone 21)
4.6. 2-Isobutyl-4-(thiophen-2-yl)isoquinolin-1(2H)-one (Decarbonylated Product 26, Partially Purified)
4.7. Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- WHO World Malaria Report 2020; CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2020.
- Phyo, A.P.; Nkhoma, S.; Stepniewska, K.; Ashley, E.A.; Nair, S.; McGready, R.; Moo, C.l.; Al-Saai, S.; Dondorp, A.M.; Lwin, K.M.; et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: A longitudinal study. Lancet 2012, 379, 1960–1966. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.D.; Devine, S.M.; Möhrle, J.J.; Laleu, B.; Burrows, J.N.; Charman, S.A.; Creek, D.J.; Sleebs, B.E. The development process for discovery and clinical advancement of modern antimalarials. J. Med. Chem. 2019, 62, 10526–10562. [Google Scholar] [CrossRef] [PubMed]
- Floyd, D.M.; Stein, P.; Wang, Z.; Liu, J.; Castro, S.; Clark, J.A.; Connelly, M.; Zhu, F.; Holbrook, G.; Matheny, A.; et al. Hit-to-lead studies for the antimalarial tetrahydroisoquinolone carboxanilides. J. Med. Chem. 2016, 59, 7950–7962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Díaz, M.-B.; Ebert, D.; Salinas, Y.; Pradhan, A.; Lehane, A.M.; Myrand-Lapierre, M.-E.; O’Loughlin, K.G.; Shackleford, D.M.; Lage de Almeida, M.J.; Clark, J.; et al. A clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of plasmodium. Proc. Nat. Acad. Sci. USA 2014, 111, E5455–E5462. [Google Scholar] [CrossRef] [Green Version]
- Guy, R.K. An Open Label Phase 2a Study to Assess the Efficacy, Safety, Tolerability, and Pharmacokinetics of (+)-SJ000557733 (SJ733) with or without Cobicistat in Adult Patients with Acute, Uncomplicated Malaria over a 42-Day Period. Available online: https://clinicaltrials.gov/ct2/show/NCT04709692 (accessed on 9 November 2021).
- Liu, J.; Wang, Z.; Levin, A.; Emge, T.J.; Rablen, P.R.; Floyd, D.M.; Knapp, S. N-methylimidazole promotes the reaction of homophthalic anhydride with imines. J. Org. Chem. 2014, 79, 7593–7599. [Google Scholar] [CrossRef]
- Jarvis, C.L.; Jemal, N.M.; Knapp, S.; Seidel, D. Formal [4+2] cycloaddition of imines with alkoxyisocoumarins. Org. Biomol. Chem. 2018, 16, 4231–4235. [Google Scholar] [CrossRef]
- Polyak, D.; Phung, N.; Barrows, R.; Emge, T.J.; Knapp, S. Stereochemistry and reactivity of the HPA-imine Mannich intermediate. Tetrahedron Lett. 2017, 58, 3879–3883. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Wang, Z.; Levin, A.; Emge, T.J.; Floyd, D.M.; Knapp, S. Dimerization and comments on the reactivity of homophthalic anhydride. Tetrahedron Lett. 2015, 56, 3001–3004. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Barrows, R.D.; Emge, T.J.; Knapp, S. Stereochemical aspects of T3P amidations. Org. Process Res. Dev. 2017, 21, 399–407. [Google Scholar] [CrossRef]
- Moore, H.W.; Duncan, W.G. Reaction of tert-butylcyanoketene with tertiary amines. Synthesis of 1,3-di-tert-butyl-1,3-dicyanoallene. J. Org. Chem. 1973, 38, 156–158. [Google Scholar] [CrossRef]
- Zarei, Z. An efficient and green method for the synthesis of 2-azetidinones mediated by propylphosphonic anhydride (T3P). Monatsch. Chem. 2014, 145, 1495–1499. [Google Scholar] [CrossRef]
- Crichfield, K.S.; Hart, J.E.; Lampert, J.T.; Vaid, R.K. Propane phosphonic acid anhydride: A mild reagent for beta-lactam synthesis. Synth. Commun. 2000, 30, 3737–3744. [Google Scholar] [CrossRef]
- Coulthard, G.; Unsworth, W.P.; Taylor, R.J.K. Propylphosphonic anhydride (T3P) mediated synthesis of beta-lactams from imines and aryl-substituted acetic acids. Tetrahedron Lett. 2015, 56, 3113–3116. [Google Scholar] [CrossRef]
- Dunetz, J.R.; Xiang, Y.; Baldwin, A.; Ringling, J. General and scalable amide bond formation with epimerization-prone substrates using T3P and pyridine. Org. Lett. 2011, 13, 5048–5051. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.D.; Andraos, J.; Tidwell, T.T.; Vukovic, S. Ketene reactions with tertiary amines. J. Org. Chem. 2014, 79, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Yamataka, H.; Aleksiuk, O.; Biali, S.E.; Rappoport, Z. Stereoelectronic effects in the nucleophilic addition to the sp-hybridized carbon of a ketene and vinyl cation: When is a mesityl effectively smaller than a phenyl ring? J. Am. Chem. Soc. 1996, 118, 12580–12587. [Google Scholar] [CrossRef]
- Gong, L.; Leung-Toung, R.; Tidwell, T.T. Nucleophilic additions to ketenes by (trimethylsilyl)lithium and by enolates. J. Org. Chem. 1990, 55, 3634–3639. [Google Scholar] [CrossRef]
- Couture, A.; Deniau, E.; Grandclaudon, P.; Woisel, P. A new synthetic route to 2-alkyl-4-aryl-1(2H)-isoquinolones and 2-alkyl-4-aryl-1,2,3,4-tetrahydroisoquinolines. Tetrahedron 1996, 52, 4433–4448. [Google Scholar] [CrossRef]
- Couture, A.; Cornet, H.; Grandclaudon, P. A new synthetic route to 2-methyl-3-(aryl or alkyl)-1-oxo-1,2-dihydroisoquinolines via an intramolecular Wittig reaction. Tetrahedron 1992, 48, 3857–3866. [Google Scholar] [CrossRef]
- Munoz, S.B.; Aloia, A.N.; Moore, A.K.; Papp, A.; Mathew, T.; Fustero, S.; Olah, G.A.; Prakash, G.K.S. Synthesis of 3-substituted isoindolin-1-ones via a tandem desilylation, cross-coupling, hydroamidation sequence under aqueous phase-transfer conditions. Org. Biomol. Chem. 2016, 14, 85–92. [Google Scholar] [CrossRef]
- Pathare, R.S.; Sharma, S.; Elagandhula, S.; Saini, V.; Sawant, D.M.; Yadav, M.; Sharon, A.; Khan, S.; Pardasani, R.T. Application of isocyanides as amine surrogates in the synthesis of diverse isoindolin-1-one derivatives by a palladium-catalyzed tandem carboxamidation/hydroamidation reaction. Eur. J. Org. Chem. 2016, 33, 5579–5587. [Google Scholar] [CrossRef]
- Chapman, O.L.; Chang, C.-C.; Kolc, J.; Rosenquist, N.R.; Tomioka, H. A photochemical method for the introduction of strained multiple bonds. J. Am. Chem. Soc. 1975, 97, 6586–6588. [Google Scholar] [CrossRef]
- Mitsudo, T.-A.; Kadokura, M.; Watanabe, Y. Palladium-catalysed reactions of ketenes with allyl acetates or allyl carbonates: Novel syntheses of 1,3-dienes and allylated esters. J. Chem. Soc. Chem. Commun. 1986, 1539–1541. [Google Scholar] [CrossRef]
- Allen, A.D.; Tidwell, T.T. New directions in ketene chemistry: The land of opportunity. Eur. J. Org. Chem. 2012, 1081–1096. [Google Scholar] [CrossRef]
- Dale, H.J.A.; Nottingham, C.; Poree, C.; Lloyd-Jones, G.C. Systematic evaluation of 1,2-migratory aptitude in alkylidene carbenes. J. Am. Chem. Soc. 2021, 143, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Wang, T.; Zhang, X.; Wang, P.; Zhang, B.; Wei, J.; Zhang, Z. One-pot synthesis of 4-heteroaryl-substituted pyrazoles: A gold-catalyzed oxidation/1,2-heteroaryl migration cascade constitutes the key step. Adv. Synth. Cat. 2016, 358, 1534–1539. [Google Scholar] [CrossRef]
- Tidwell, T.T. Ketenes, 2nd ed.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | NMR Yield | Structural Evidence |
|---|---|---|
| Allene isomers 9, 10, and 11 | 58% combined | Previously isolated and characterized; X-ray structure of major isomer 9 |
| Major lactone 12 | 13% | NMR, LC-MS, X-ray structure |
| Minor lactone 13 | 3% | Stereochem tentative; NMR signals in mixture, LC-MS |
| Purple ketone 21 | 6% | X-ray structure, NMR, MS |
| Decarbonylated DHIQ product 26 | 4% | NMR, MS, comparison to structurally related compounds |
| Trans acid 1 | 10% | Comparison to the known compound |
| Recovered 2 (sm) | <1% | Comparison to the known compound |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrows, R.D.; Dresel, M.J.; Emge, T.J.; Rablen, P.R.; Knapp, S. Stereoelectronic Features of a Complex Ketene Dimerization Reaction. Molecules 2022, 27, 66. https://doi.org/10.3390/molecules27010066
Barrows RD, Dresel MJ, Emge TJ, Rablen PR, Knapp S. Stereoelectronic Features of a Complex Ketene Dimerization Reaction. Molecules. 2022; 27(1):66. https://doi.org/10.3390/molecules27010066
Chicago/Turabian StyleBarrows, Robert D., Mark J. Dresel, Thomas J. Emge, Paul R. Rablen, and Spencer Knapp. 2022. "Stereoelectronic Features of a Complex Ketene Dimerization Reaction" Molecules 27, no. 1: 66. https://doi.org/10.3390/molecules27010066
APA StyleBarrows, R. D., Dresel, M. J., Emge, T. J., Rablen, P. R., & Knapp, S. (2022). Stereoelectronic Features of a Complex Ketene Dimerization Reaction. Molecules, 27(1), 66. https://doi.org/10.3390/molecules27010066