
Design, Cytotoxicity and Antiproliferative Activity of 4-Amino-5-methyl-thieno[2,3-d]pyrimidine-6-carboxylates against MFC-7 and MDA-MB-231 Breast Cancer Cell Lines

 ,
,  , ,
, ,  and
and 
Abstract
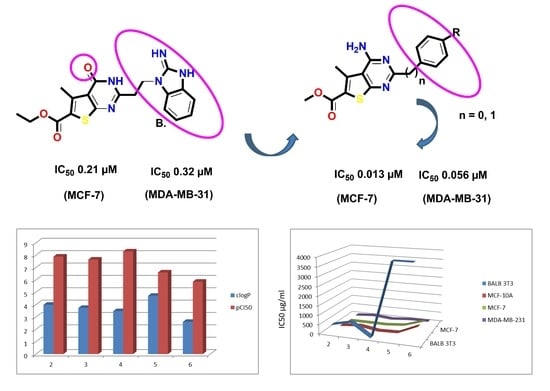
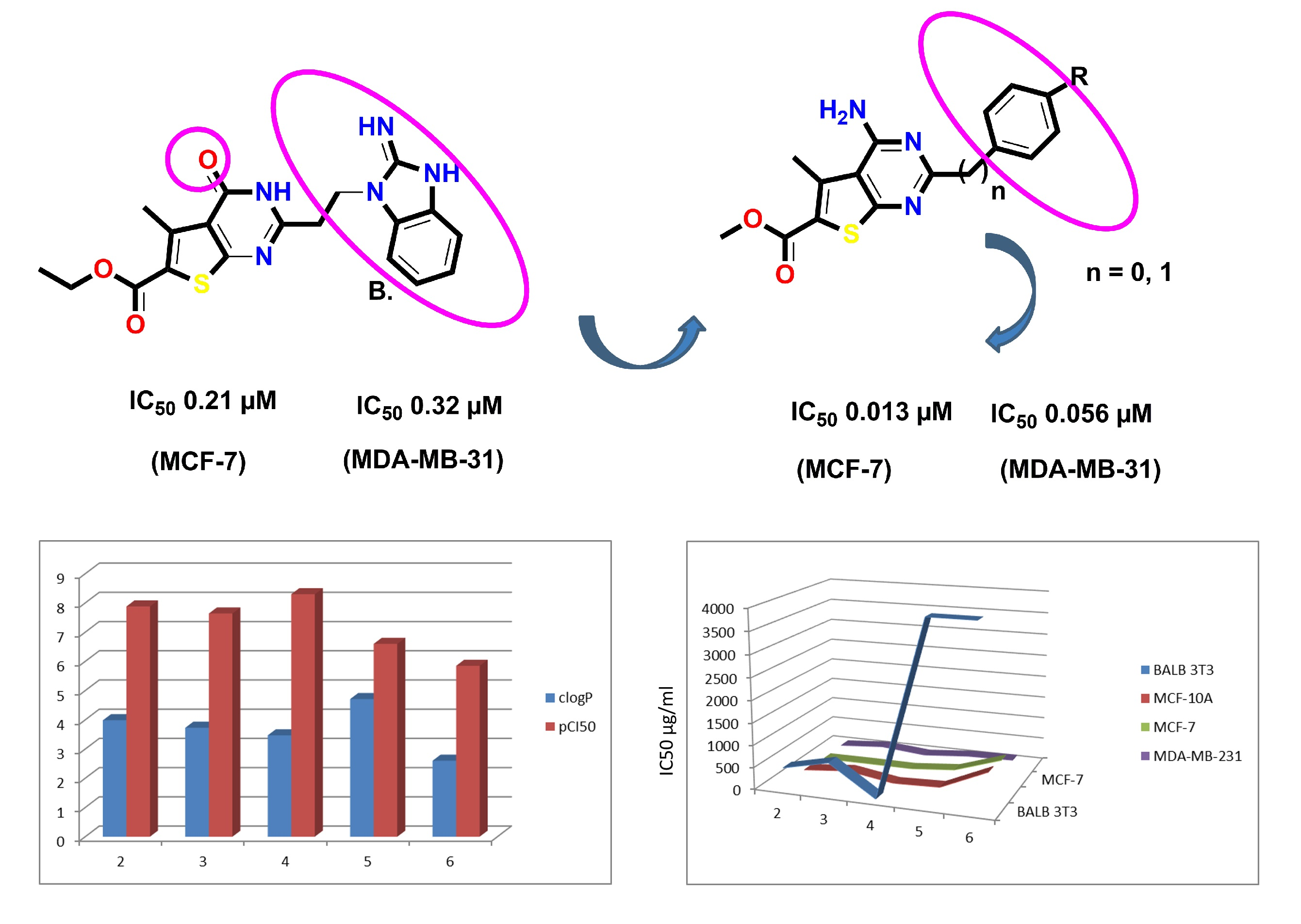
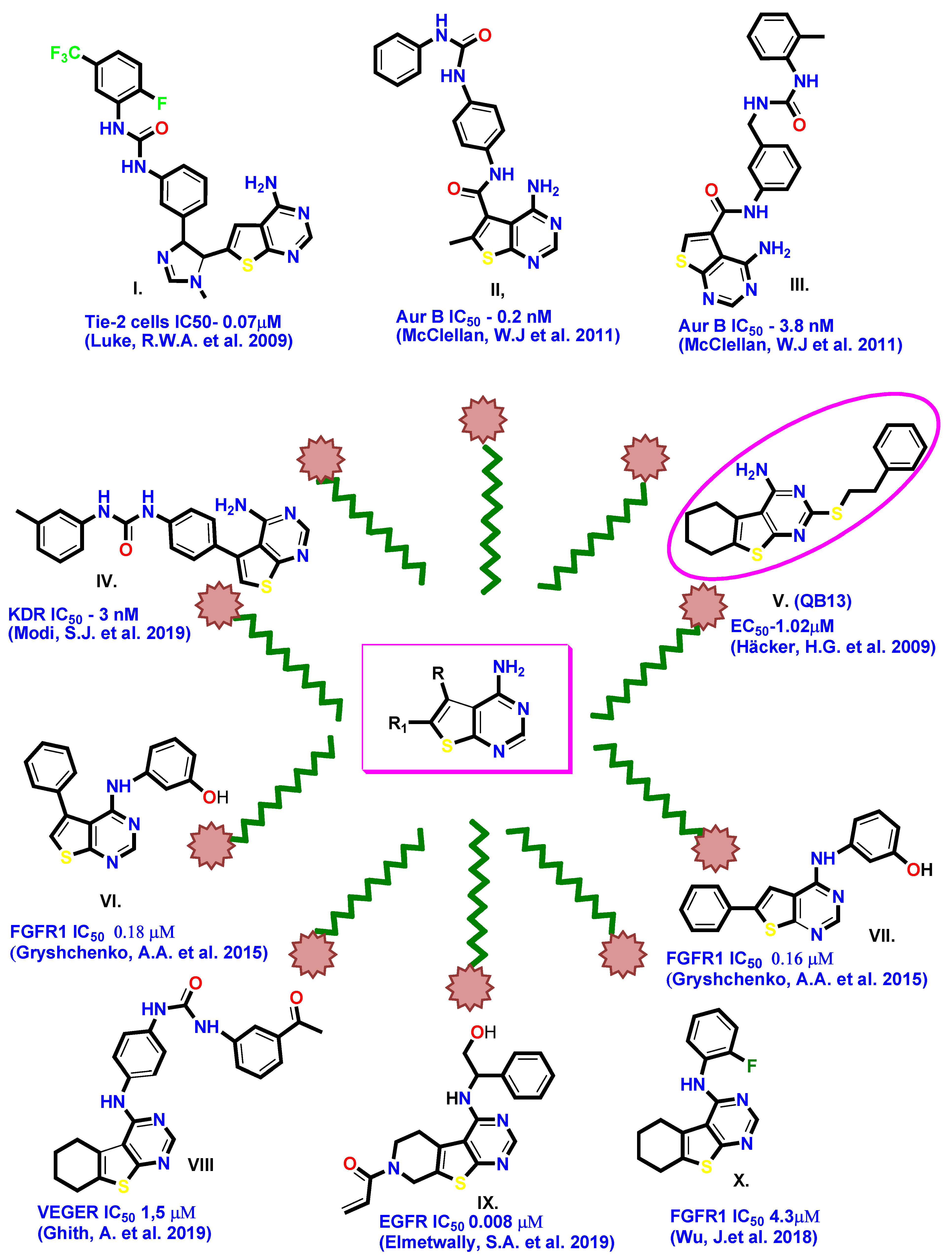
:
1. Introduction
2. Results and Discussion
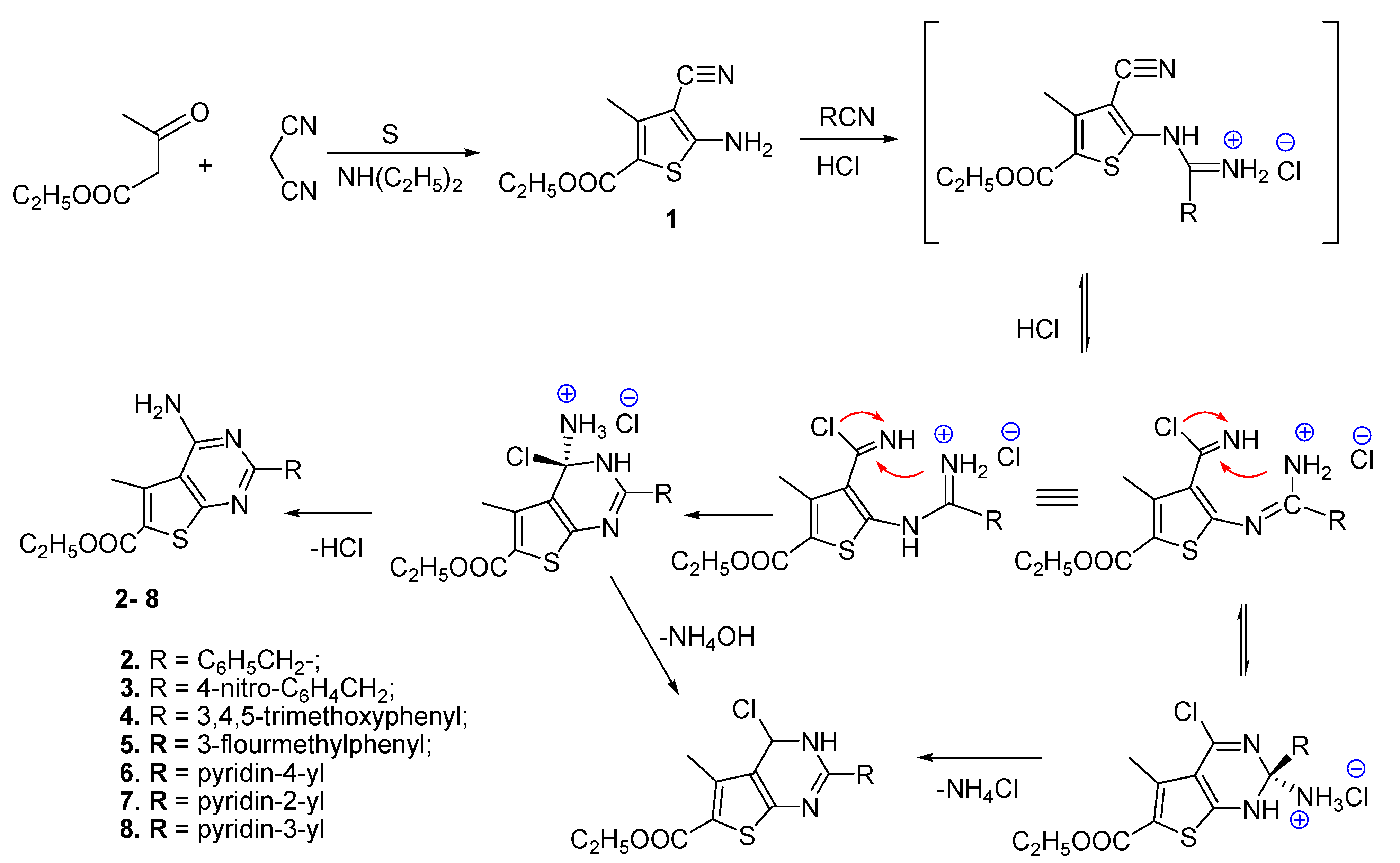
2.1. Chemistry
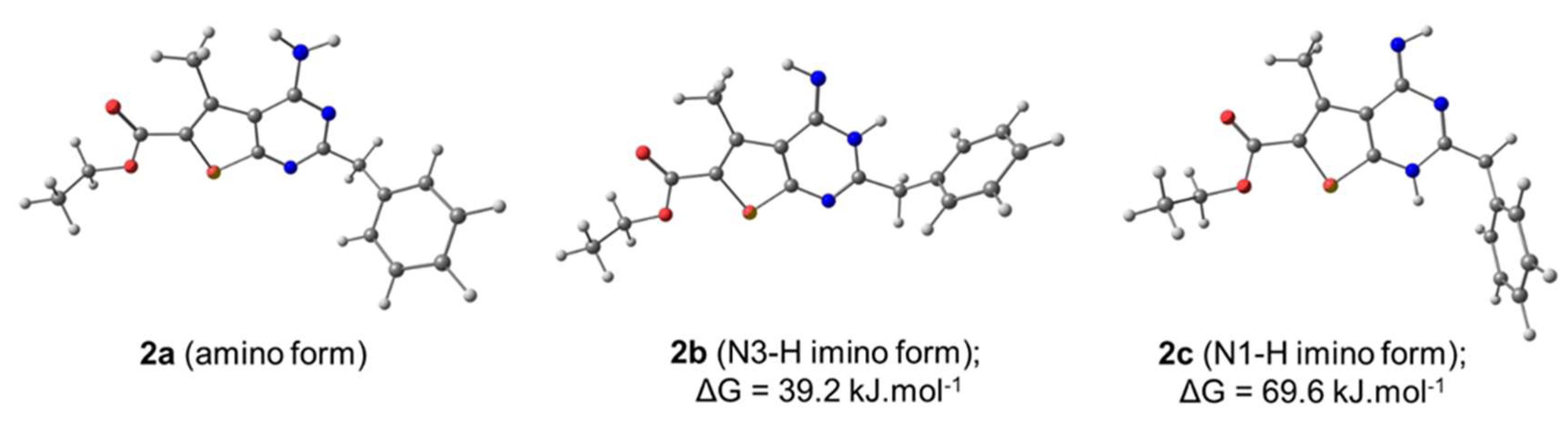
2.2. Molecular Structure Characterization
2.3. Biological Evaluation
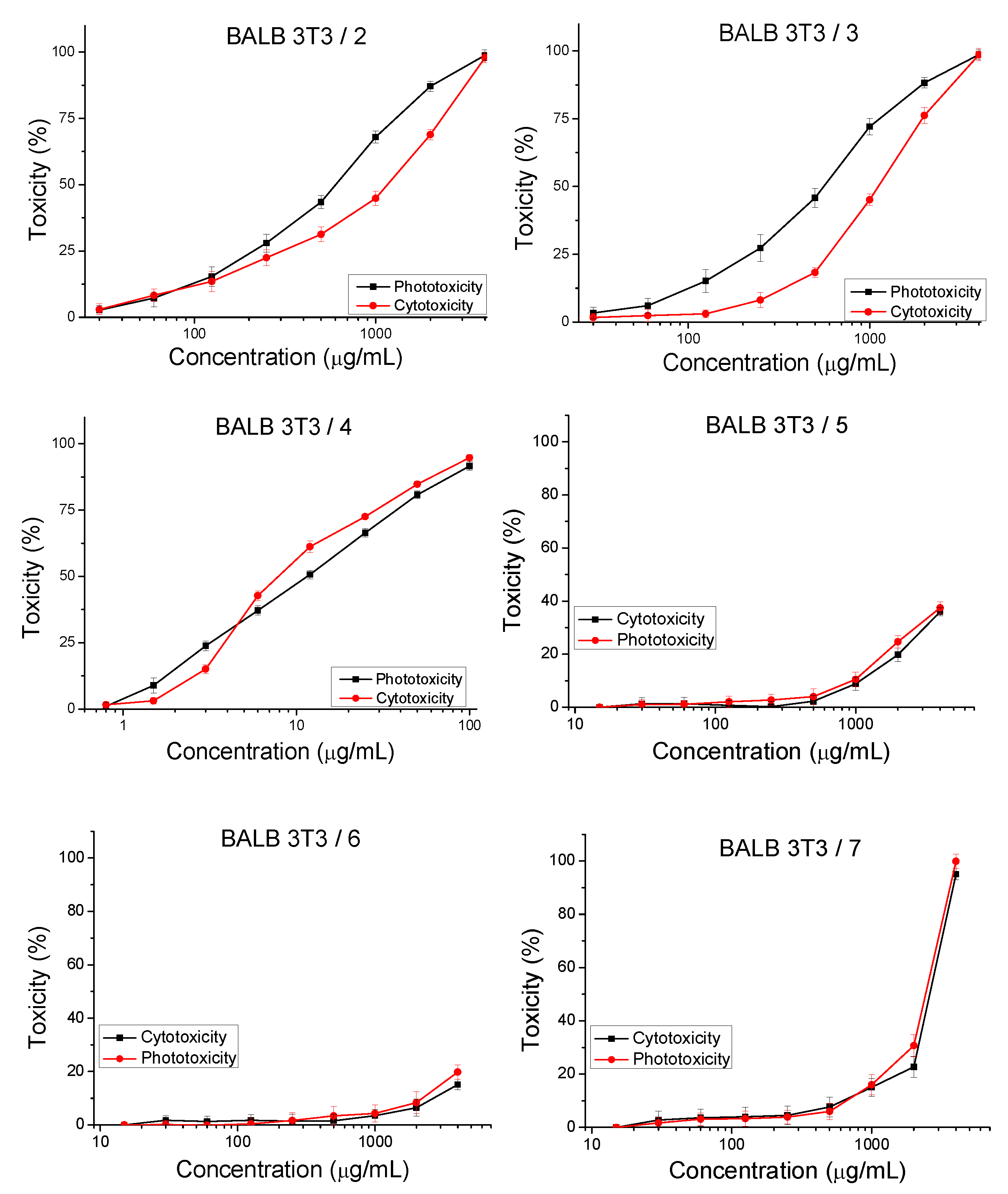
2.3.1. Cyto- and Phototoxicity
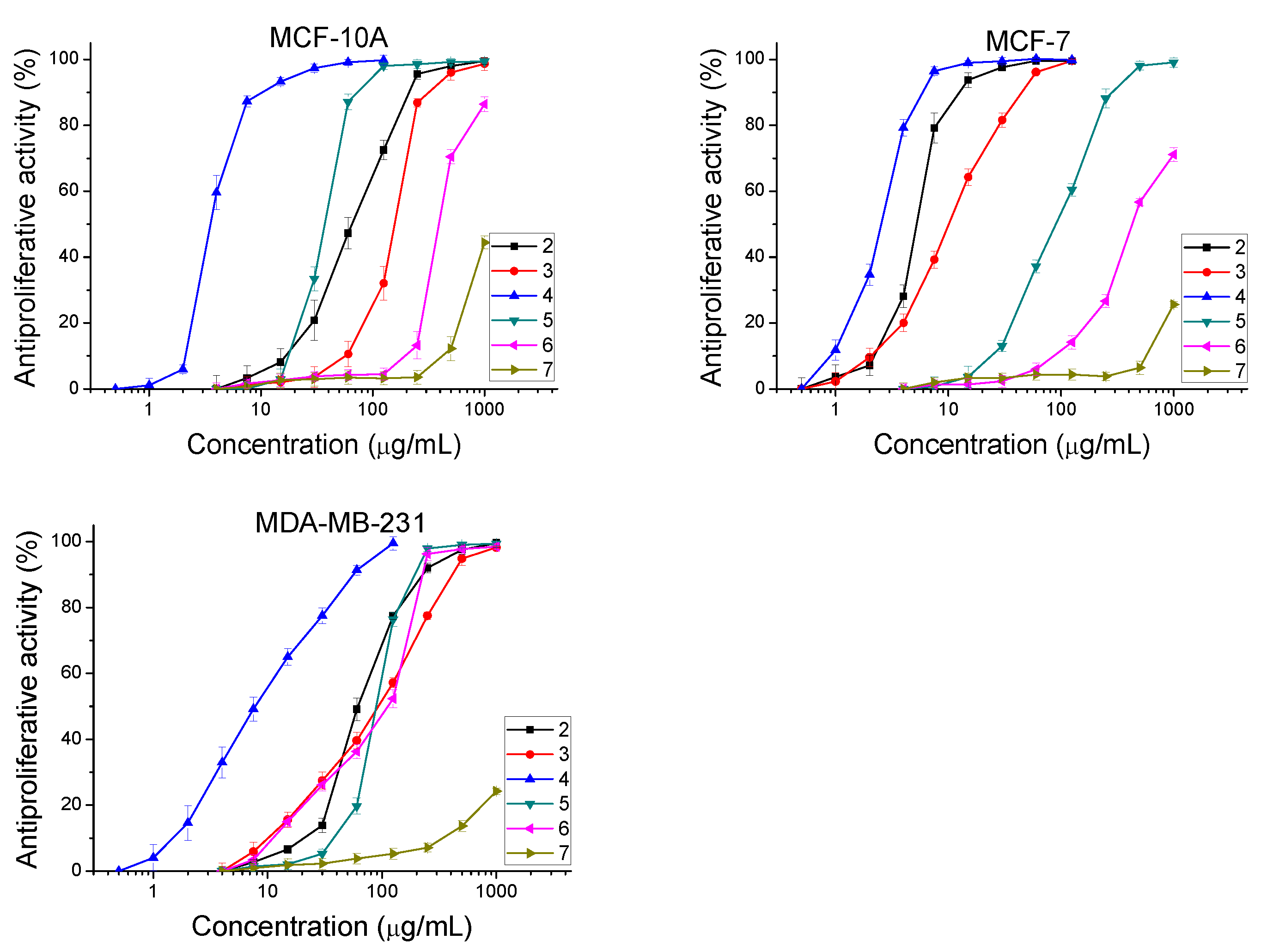
2.3.2. Antiproliferative Activity
2.3.3. Ligand Efficiency
3. Materials and Methods
3.1. Synthesis
3.1.1. General Procedure for the Synthesis of Ethyl 2-Amino-3-cyano-4-methylthiophene-5-carboxylate1
3.1.2. General Procedure for the Synthesis of 2-Substituted 4-Amino Thieno [2,3-d]Pyrimidines: 2–7
3.1.3. Ethyl 4-Amino-2-benzyl-5-methyl-thieno[2,3-d]pyrimidine-6-carboxylate (2)
3.1.4. Ethyl 4-Amino-5-methyl-2-(4-nitrobenzyl) Thieno [2,3-d] Pyrimidine-6-carboxylate (3)
3.1.5. Ethyl 4-Amino-5-methyl-2-(3,4,5-trimethoxyphenyl)thieno [2,3-d]Pyrimidine-6-carboxylate (4)
3.1.6. Ethyl 4-Amino-5-methyl-2-(3-(trifluoromethyl) phenyl) Thieno [2,3-d] Pyrimidine-6-carboxylate (5)
3.1.7. Ethyl 4-Amino-5-methyl-2-(pyridin-4-yl)thieno[2,3-d]pyrimidine-6-carboxylate hydrochloride (6)
3.1.8. Ethyl 4-Amino-5-methyl-2-(pyridin—2-yl) Thieno[2,3-d]pyrimidine-6-carboxylate (7)
3.1.9. Ethyl 4-Amino-5-methyl-2-(pyridin-3-yl)thieno [2,3-d]Pyrimidine-6-carboxylate (8)
3.2. Light Source
3.3. Chemicals
3.4. Cell Lines
3.5. Cytotoxicity and Phototoxicity Testing
3.6. In Vitro Antiproliferative Activity
3.7. Molecular Calculations
Quantum Chemical Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Abdel-Rahman, E.-G.; Hoda, H.; Hend, H. Synthesis and biological evaluation of thieno[2,3-d]pyrimidine derivatives for anti-inflammatory, analgesic and ulcerogenic activity. Acta Pharm. 2007, 57, 395–411. [Google Scholar]
- Kanawade, S.B.; Toche, R.B.; Rajani, D.P. Synthetic tactics of new class of 4-aminothieno[2,3-d]pyrimidine-6-carbonitrile derivatives acting as antimicrobial agents. Eur. J. Med. Chem. 2013, 64, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, T.J.; Zarzycka, B.; Lim, H.D.; Kellam, B.; Mistry, S.N.; Katrich, V.; Scammells, P.J.; Lane, J.R.; Capuano, B. A thieno[2,3-d]pyrimidine Scaffold Is a Novel Negative Allosteric Modulator of the Dopamine D2 Receptor. J. Med. Chem. 2019, 62, 174–206. [Google Scholar] [CrossRef] [PubMed]
- Harrison, G.A.; Mayer Bridwell, A.E.; Singh, M.; Jayaraman, K.; Weiss, L.A.; Kinsella, R.L.; Aneke, J.S.; Flentie, K.; Schene, M.E.; Gaggioli, M.; et al. Identification of 4-aminothieno[2,3-d]pyrimidines as QcrB inhibitors in Mycobacterium tuberculosis. mSpher 2019, 4, e00606-19. [Google Scholar] [CrossRef] [Green Version]
- Carreras, C.W.; Santi, D.V. The Catalytic Mechanism and Structure of Thymidylate Synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A.; Qiu, Y.; Kisliuk, R.L. Synthesis of classical and nonclassical 2-amino-4-oxo-6-benzylthieno-[2,3-d]pyrimidines as potential thymidylate synthase inhibitors. J. Heterocycl. Chem. 1995, 41, 941–946. [Google Scholar] [CrossRef]
- Arooj, M.; Lee, K.W. Binding mode analysis of dual inhibitors of human thymidylate synthase and dihydrofolate reductase as antitumour agents via molecular docking and DFT studies. Mol. Simul. 2014, 41, 311–314. [Google Scholar] [CrossRef]
- Mercer, K.E.; Pritchard, C.A. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim. Biophys. Acta 2003, 1653, 25–40. [Google Scholar] [CrossRef]
- Packard, G.K.; Papa, P.; Riggs, J.R.; Erdman, P.; Tehrani, L.; Robinson, D.; Harris, R.; Shevlin, G.; Perrin-Ninkovic, S.; Hilgraf, R.; et al. Discovery and optimization of thieno[2,3-d] pyrimidines as B-Raf inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 747–752. [Google Scholar] [CrossRef]
- Pédeboscq, S.; Gravier, D.; Casadebaig, F.; Hou, G.; Gissot, A.; Rey, C.; Ichas, F.; De Giorgi, F.; Lartigue, L.; Pometan, J.-P. Synthesis and evaluation of apoptosis induction of thienopyrimidine compounds on KRAS and BRAF mutated colorectal cancer cell lines. Bioorg. Med. Chem. 2012, 20, 6724–6731. [Google Scholar] [CrossRef]
- Yu, Q. The dynamic roles of angiopoietins in tumor angiogenesis. Future Oncol. 2005, 1, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Jo, G.; Bae, J.; Hong, J.H.; Han, A.; Kim, D.; Hong, S.P.; Kim, J.A.; Lee, S.; Koh, G.Y.; Kim, H.M. Structural insights into the clustering and activation of Tie2 receptor mediated by Tie2 agonistic antibody. Nat. Commun. 2021, 12, 6287. [Google Scholar] [CrossRef] [PubMed]
- Luke, R.W.A.; Ballard, P.; Buttar, D.; Campbell, L.; Curwen, J.; Emery, S.C.; Griffen, A.M.; Hassall, L.; Hayter, B.R.; Jones, C.D.; et al. Novel thienopyrimidine and thiazolopyrimidine kinase inhibitors with activity against Tie-2 in vitro and in vivo. Bioorg. Med. Chem. Lett. 2009, 19, 6670–6674. [Google Scholar] [CrossRef]
- McClellan, W.J.; Dai, Y.; Abad-Zapatero, C.; Albert, D.H.; Bouska, J.J.; Glaser, K.B.; Magoc, T.J.; Marcotte, P.A.; Osterling, D.J.; Stewart, K.D.; et al. Discovery of potent and selective thienopyrimidine inhibitors of Aurora kinases. Bioorg. Med. Chem. Lett. 2011, 21, 5620–5624. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Coumar, M.S.; Chu, C.Y.; Lin, W.H.; Chen, Y.R.; Chen, C.T.; Shiao, H.Y.; Rafi, S.; Wang, S.Y.; Hsu, H.; et al. Design and synthesis of tetrahydropyridothieno[2,3-d]pyrimidine scaffold based epidermal growth factor receptor (EGFR) kinase inhibitors: The role of side chain chirality and Michael acceptor group for maximal potency. J. Med. Chem. 2010, 53, 7316–7326. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Hung, M.-C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Elmetwally, S.A.; Saied, K.F.; Eissa, I.H.; Elkaeed, E.B. Design, synthesis and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers. Bioorg. Chem. 2019, 88, 102944. [Google Scholar] [CrossRef] [PubMed]
- Adly, M.E.; Gedawy, E.M.; El-Malah, A.A.; El-Telbany, F.A. Synthesis of novel thieno[2,3-d]pyrimidine derivatives and evaluation of their cytotoxicity and EGFR inhibitory activity. Anticancer Agents Med. Chem. 2018, 18, 747–756. [Google Scholar] [CrossRef]
- El-Ansary, A.K.; Kamal, A.M.; Al-Ghorafi, M.A. Design, Synthesis and Biological Evaluation of Some 5-Arylthieno[2,3-d]pyrimidines as potential anti-cancer agents. Chem. Pharm. Bull. 2016, 64, 1172–1180. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, Y.; Liu, J.; Chen, Q.; Pang, D.; Jiang, Y. Effects of FGFR1 Gene Polymorphisms on the Risk of Breast Cancer and FGFR1 Protein Expression. Cell. Physiol. Biochem. 2018, 47, 2569–2578. [Google Scholar] [CrossRef]
- Gryshchenko, A.A.; Bdzhola, V.G.; Balanda, A.O.; Briukhovetska, N.V.; Kotey, I.M.; Golub, G.A.; Rubana, T.P.; Lukash, L.; Yarmoluk, S.M. Design, synthesis and biological evaluation of N-phenylthieno[2,3-d]pyrimidin-4-amines as inhibitors of FGFR1. Bioorg. Med. Chem. 2015, 23, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Ghith, A.; Youssef, K.M.; Ismail, N.S.M.; Abouzid, K.A.M. Design, synthesis and molecular modeling study of certain VEGFR-inhibitors based on thienopyrimidne scaffold as cancer targeting agents. Bioorg. Chem. 2019, 83, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Elrazaz, E.Z.; Serya, R.A.T.; Ismail, N.S.M.; Albohy, A.; Abou El Ella, D.A.; Abouzid, K.A.M. Discovery of potent thieno[2,3-d]pyrimidine VEGFR-2 inhibitors: Design, synthesis and enzyme inhibitory evaluation supported by molecular dynamics simulations. Bioorg. Chem. 2021, 113, 105019. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.J.; Kulkarni, V.M. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med. Drug Disc. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Dai, Y.; Guo, Y.; Frey, R.R.; Ji, Z.; Curtin, M.L.; Ahmed, A.A.; Albert, D.H.; Arnold, L.; Arries, S.S.; Barlozzari, T.; et al. Thienopyrimidine ureas as novel and potent multitargeted receptor tyrosine kinase inhibitors. J. Med. Chem. 2005, 48, 6066e6083. [Google Scholar] [CrossRef] [PubMed]
- Häcker, H.G.; de la Haye, A.; Sterz, K.; Schnakenburg, G.; Wiese, M.; Gütschow, M. Analogs of a 4-aminothieno[2,3-d]pyrimidine lead (QB13) as modulators of P-glycoprotein substrate specificity. Bioorg. Med. Chem. Lett. 2009, 19, 6102–6105. [Google Scholar] [CrossRef] [PubMed]
- Sharaky, M.; Kamel, M.; Aziz, M.A.; Omran, M.; Rageh, M.M.; Abouzid, K.A.M.; Shouman, S.A. Design, synthesis and biological evaluation of a new thieno[2,3-d]pyrimidine-based urea derivative with potential antitumor activity against tamoxifen sensitive and resistant breast cancer cell lines. J. Enzyme Inhib. Med. Chem. 2020, 35, 1641–1656. [Google Scholar] [CrossRef]
- Dimov, S.; Mavrova, A.T.; Yancheva, D.; Nikolova, B.; Tsoneva, I. Thieno[2,3-d]pyrimidin-4(3H)-one Derivatives of Benzimidazole as Potential Anti- Breast Cancer (MDA-MB-231, MCF-7) Agents. Anti-Cancer Agents Med. Chem. 2021, 21, 1441–1450. [Google Scholar] [CrossRef]
- Mavrova, A.T.; Dimov, S.; Yancheva, D.; Rangelov, M.; Wesselinova, D.; Naydenova, E. New C2- and N3-Modified Thieno[2,3-d]Pyrimidine Conjugates with Cytotoxicity in the Nanomolar Range. Anti-Cancer Agents Med. Chem. 2021, 22, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Gewald, K. Heterocyclen aus CH-aciden Nitrilen, VII. 2-Amino-thiophene aus α-Oxo-mercaptanen und methylenaktiven Nitrilen. Chem. Ber. 1965, 98, 3571–3577. [Google Scholar] [CrossRef]
- Gewald, K.; Schinke, E.; Heterocyclen aus CH-aciden Nitrilen, X. Notiz zur Reaktion von Aceton mit Cyanessigester und Schwefel. Chem. Ber. 1966, 99, 2712–2715. [Google Scholar] [CrossRef]
- Sabnis, R.W. The gewald synthesis. Sulfur. Rep. 1994, 16, 1–17. [Google Scholar] [CrossRef]
- Sabnis, R.; Rangnekar, D.; Sonawane, N. 2-Aminothiophenes by the Gewald reaction. J. Heterocycl. Chem. 1999, 36, 333–345. [Google Scholar] [CrossRef]
- Puterová, Z.; Krutošíková, A.; Végh, D. Gewald reaction: Synthesis, properties and applications of substituted 2-aminothiophenes. Arkivoc 2010, 1, 209–246. [Google Scholar] [CrossRef]
- Shishoo, C.; Devani, M.; Bhadti, V.; Jain, K.; Ananthan, S. Reaction of nitriles under acidic conditions. Part VI. Synthesis of condensed 4-chloro-and 4-aminopyrimidines from ortho-aminonitriles. J. Heterocycl. Chem. 1990, 27, 119–126. [Google Scholar] [CrossRef]
- Mavrova, A.; Dimov, S.; Vuchev, D.; Anichina, K.; Yancheva, D. Antihelminthic Activity of Some 2-substituted Thieno[2,3-d]pyrimidin-4-ones. Lett. Drug Des. Discov. 2018, 15, 887–894. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Hall, C.D.; El-Gendy, B.E.D.M.; Draghici, B. Tautomerism in drug discovery. J. Comput. Aided Mol. Des. 2010, 24, 475–484. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision A.1. B01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ghiviriga, I.; El-Gendy, E.D.B.; Steel, P.J.; Katritzky, A.R. Tautomerism of guanidines studied by 15N NMR: 2-hydrazono-3-phenylquinazolin-4(3H)-ones and related compounds. Org. Biomol. Chem. 2009, 7, 4110–4119. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Abad-Zapatero, C. Ligand efficiency indices for effective drug discovery. Exp. Opin. Drug Discov. 2007, 2, 469–488. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Tkocz, A.; Kucia, U. Beware of ligand efficiency (LE): Understanding LE data in modeling structure-activity and structure-economy relationships. J. Cheminform. 2017, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shultz, M.D. The thermodynamic basis for the use of lipophilic efficiency (LipE) in enthalpic optimizations. Bioorg. Med. Chem. Lett. 2013, 23, 5992–6000. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O.; Kogej, T. Compound properties and their influence on drug quality. In The Practice of Medicinal Chemistry, 4th ed.; Wermuth, C., Aldous, D., Raboisson, P., Rognan, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 379–393. [Google Scholar]
- Kenny, P.W.; Leitão, A.; Montanari, C.A. Ligand efficiency metrics considered harmful. J. Comput. Aided Mol. Des. 2014, 28, 699–710. [Google Scholar] [CrossRef]
- Kenny, P.W. The nature of ligand efficiency. J. Cheminform. 2019, 11, 8. [Google Scholar] [CrossRef]
- Halle, W. The Registry of Cytotoxicity: Toxicity testing in cell cultures to predict acute toxicity (LD50) and to reduce testing in animals. Altern. Lab. Anim. 2003, 31, 89–198. [Google Scholar] [CrossRef]
- ESAC. Statement on the scientific validity of the 3T3 NRU PT test (an in vitro test for phototoxic potential). In Proceedings of the 9th Meeting of ECVAM Scientific Advisory Committee, Hamburg, Germany, 1–2 October 1997. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Stephens, P.; Devlin, F.; Chabalowski, C.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Compounds | CC50 of Mean ± SD (µg/mL) | PIF * | |
|---|---|---|---|---|
| Cytotoxicity | Phototoxicity | |||
| BALB 3T3 | 2 | 1212 ± 103.3 | 632.3 ± 35.6 | 1.9 |
| 3 | 1155 ± 57.8 | 578 ± 58.7 | 2 | |
| 4 | 8.37 ± 0.6 | 11.85 ± 0.8 | 0.7 | |
| 5 | - | - | - | |
| 6 | - | - | - | |
| 7 | 2754 ± 64.3 | 2552 ± 75.1 | 1.08 | |
| Compounds | IC50 ± SD (μg/mL) Resp. in (µM) | ||
|---|---|---|---|
| Antiproliferative Activity | |||
| MCF-10A | MCF-7 | MDA-MB-231 | |
| 2 | 67.77 ± 9.1 (0.21 µM) | 4.3 ± 0.11 (0.013 µM) | 18.28 ± 1.58 (0.056 µM) |
| 3 | 165.4 ± 8.81 (0.44 µM) | 8.58 ± 0.55 (0.023 µM) | 98.25 ± 4.73 (0.26 µM) |
| 4 | 2.74 ± 0.13 (0.0068 µM) | 2.01 ± 0.07 (0.005 µM) | 6.43 ± 0.95 (0.016 µM) |
| 5 | 39.23 ± 1.8 (0.1 µM) | 95.94 ± 3.49 (0.25 µM) | 94.94 ± 1.48 0.25 µM) |
| 6 | 410.9 ± 5.1 (1.31 µM) | 444.5 ± 9.2 (1.41 µM) | 116 ± 9.63 (0.37 µM) |
| 7 | >1000 - | >1000 - | >1000 - |
| Compounds | Selective Index | |
|---|---|---|
| MCF-7 | MDA-MB-231 | |
| 2 | 15.8 | 3.7 |
| 3 | 19.3 | 1.7 |
| 4 | 1.4 | 0.4 |
| 5 | 0.41 | 0.41 |
| 6 | 0.92 | 3.54 |
| 7 | - | - |
| No | IC50 (µM)/ (pCI50) | LE kcal/mol | LEI | BEI | LLE | clogP | MW | TPSA | NHA | NHD |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 0.013 (7.88) | 0.476 | 0.347 | 0.024 | 3.984 | 3.986 | 327.41 | 78.11 | 5 | 2 |
| 3 | 0.023 (7.64) | 0.421 | 0.306 | 0.021 | 3.910 | 3.73 | 372.41 | 123.94 | 8 | 2 |
| 4 | 0.005 (8.30) | 0.425 | 0.321 | 0.022 | 4.83 | 3.469 | 403.46 | 105.81 | 8 | 2 |
| 5 | 0.25 (6.60) | 0.263 | 0.269 | 0.018 | 1.89 | 4.71 | 381.38 | 78.11 | 5 | 2 |
| 6 | 1.41 (5.85) | 0.372 | 0.27 | 0.019 | 3.247 | 2.603 | 314.37 | 91.00 | 6 | 2 |
| A | 0.074 (7.13) | 0.243 | 0.178 | 0.012 | 3.143 | 3.987 | 646.15 | 177.84 | 13 | 3 |
| No | IC50 (µM) | (pCI50) | LE kcal/mol | LEI | BEI | LLE | Vol |
|---|---|---|---|---|---|---|---|
| 2 | 0.056 | 7.25 | 0.476 | 0.346 | 0.024 | 4.014 | 287.82 |
| 3 | 0.26 | 6.585 | 0.369 | 0.269 | 0.019 | 3.27 | 311.16 |
| 4 | 0.016 | 7.796 | 0.391 | 0.286 | 0.002 | 4.531 | 347.66 |
| 5 | 0.25 | 6.60 | 0.384 | 0.280 | 0.018 | 4.71 | 302.32 |
| 6 | 0.37 | 6.431 | 0.436 | 0.318 | 0.022 | 4.37 | 266.87 |
| B * | 0.32 | 6.444 | 0.342 | 0.28 | 0.018 | 5.64 | 344.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mavrova, A.; Dimov, S.; Sulikovska, I.; Yancheva, D.; Iliev, I.; Tsoneva, I.; Staneva, G.; Nikolova, B. Design, Cytotoxicity and Antiproliferative Activity of 4-Amino-5-methyl-thieno[2,3-d]pyrimidine-6-carboxylates against MFC-7 and MDA-MB-231 Breast Cancer Cell Lines. Molecules 2022, 27, 3314. https://doi.org/10.3390/molecules27103314
Mavrova A, Dimov S, Sulikovska I, Yancheva D, Iliev I, Tsoneva I, Staneva G, Nikolova B. Design, Cytotoxicity and Antiproliferative Activity of 4-Amino-5-methyl-thieno[2,3-d]pyrimidine-6-carboxylates against MFC-7 and MDA-MB-231 Breast Cancer Cell Lines. Molecules. 2022; 27(10):3314. https://doi.org/10.3390/molecules27103314
Chicago/Turabian StyleMavrova, Anelia, Stephan Dimov, Inna Sulikovska, Denitsa Yancheva, Ivan Iliev, Iana Tsoneva, Galya Staneva, and Biliana Nikolova. 2022. "Design, Cytotoxicity and Antiproliferative Activity of 4-Amino-5-methyl-thieno[2,3-d]pyrimidine-6-carboxylates against MFC-7 and MDA-MB-231 Breast Cancer Cell Lines" Molecules 27, no. 10: 3314. https://doi.org/10.3390/molecules27103314
APA StyleMavrova, A., Dimov, S., Sulikovska, I., Yancheva, D., Iliev, I., Tsoneva, I., Staneva, G., & Nikolova, B. (2022). Design, Cytotoxicity and Antiproliferative Activity of 4-Amino-5-methyl-thieno[2,3-d]pyrimidine-6-carboxylates against MFC-7 and MDA-MB-231 Breast Cancer Cell Lines. Molecules, 27(10), 3314. https://doi.org/10.3390/molecules27103314