
Synthesis and Biological Evaluation of 5′-O-Fatty Acyl Ester Derivatives of 3′-Fluoro-2′,3′-dideoxythymidine as Potential Anti-HIV Microbicides

 , ,
, , 
Abstract
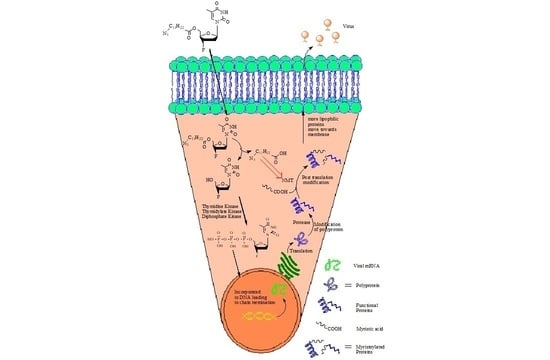
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Physicochemical Properties of 5
2.3. Biological Activities
2.3.1. Anti-HIV Activities against Cell-Free and Cell-Associated Virus
2.3.2. Anti-HIV Activities against MDR Isolates
2.3.3. Cellular Uptake Study
2.3.4. Cell Viability Study
2.3.5. Effect of Conjugates on Sperm Viability and Mobility
2.3.6. Effect of Conjugates on Vaginal Cell Viability
3. Materials and Methods
3.1. Materials
3.2. Chemistry
3.3. Physicochemical Properties (pKa, Log D., Solubility)
3.3.1. pKa
3.3.2. Log P and Log D
3.3.3. Solubility
3.4. Anti-HIV Assays
3.5. Cell Viability Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Center for Disease Control and Prevention (CDC). Morbidity and Morality Weekly Report. 1 June 2001; Volume 50, No. 21. Available online: https://www.cdc.gov/mmwr/pdf/wk/mm5021.pdf (accessed on 16 February 2022).
- Okoli, C.; Van de Velde, N.; Richman, B.; Allan, B.; Castellanos, E.; Young, B.; Brough, G.; Eremin, A.; Corbelli, G.M.; Mc Britton, M.; et al. Undetectable equals untransmittable (U = U): Awareness and associations with health outcomes among people living with HIV in 25 countries. Sex. Transm. Infect. 2021, 97, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Buttenheim, A.; Schmucker, L.; Bekker, L.G.; Thirumurthy, H.; Davey, D.L.J. Undetectable = Untransmittable (U = U) messaging increases uptake of HIV testing among men: Results from a pilot cluster randomized trial. AIDS Behav. 2021, 25, 3128–3136. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Allergy and Infectious Diseases (NIAID). HIV Prevention. 25 June 2020. Available online: https://www.niaid.nih.gov/diseases-conditions/hiv-prevention (accessed on 16 February 2022).
- Deng, W.; Sun, Y.; Yao, X.; Subramanian, K.; Ling, C.; Wang, H.; Chopra, S.S.; Xu, B.B.; Wang, J.X.; Chen, J.F.; et al. Masks for COVID-19. Adv. Sci. 2022, 9, 2102189. [Google Scholar] [CrossRef]
- Howarda, J.; Huangc, A.; Lid, Z.; Tufekcie, Z.; Zdimalf, V.; van der Westhuizen, H.M.; Delfth, A.V.; Pricej, A.; Fridmank, L.; Tangl, L.H.; et al. An evidence review of face masks against COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2014564118. [Google Scholar] [CrossRef]
- United Nations Program on HIV/AIDS (UNAIDS). Sustainable Goals Development, Goal 3: Ensure Healthy Lives and Promote Well-Being for All at All Ages. Available online: https://www.un.org/sustainabledevelopment/health/ (accessed on 16 February 2022).
- Assefa, Y.; Gilks, C.F. Ending the epidemic of HIV/AIDS by 2030: Will there be an endgame to HIV, or an endemic HIV requiring an integrated health systems response in many countries? Int. J. Infect. Dis. 2020, 100, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Parang, K.; Knaus, E.E.; Wiebe, L.I. Synthesis, in vitro anti-HIV activity, and biological stability of 5′-O-myristoyl analogue derivatives of 3′-fluoro-2′,3′-dideoxythymidine (FLT) as potential prodrugs of FLT. Nucleosides Nucleotides 1998, 17, 987–1008. [Google Scholar] [CrossRef]
- Parang, K.; Wiebe, L.I.; Knaus, E.E. Synthesis, in vitro anti-human immunodeficiency virus structure–activity relationships and biological stability of 5′-O-myristoyl analogue derivatives of 3′-azido-2′,3′-dideoxythymidine (AZT) as potential prodrugs. Antivir. Chem. Chemother. 1998, 9, 123–333. [Google Scholar] [CrossRef]
- Furuishi, K.; Matsuoko, H.; Takama, M.; Takahashi, I.; Misumi, S.; Shoji, S. Blockage of N-myristoylation of HIV-1 Gag induces the production of impotent progeny virus. Biochem. Biophys. Res. Commun. 1997, 237, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Langner, C.A.; Travis, J.K.; Caldwell, S.J.; Tianbao, J.E.; Li, Q.; Bryant, M.L.; Devadas, B.; Gokel, G.W.; Kobayashi, G.S.; Gordon, J.I. 4-Oxatetradecanoic acid is fungicidal for Cryptococcus neoformans and inhibits replication of human immunodeficiency virus I. J. Biol. Chem. 1992, 267, 17159–17169. [Google Scholar] [CrossRef]
- Parang, K.; Wiebe, L.I.; Knaus, E.E.; Huang, J.-S.; Tyrrell, D.L.; Csizmadia, F. In vitro antiviral activities of myristic acid analogs against human immunodeficiency and hepatitis B viruses. Antivir. Res. 1997, 34, 75–90. [Google Scholar] [CrossRef]
- Agarwal, H.K.; Loethan, K.; Mandal, D.; Gustavo, D.F.; Parang, K. Synthesis and biological evaluation of fatty acyl ester derivatives of 2′, 3′-didehydro-2′,3′-dideoxythymidine. Bioorg. Med. Chem. Lett. 2011, 7, 1917–1921. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, H.K.; Chhikara, B.S.; Hanley, M.; Ye, G.; Doncel, G.F.; Parang, K. Synthesis and biological evaluation of fatty acyl ester derivatives of (−)-2′,3′-dideoxy-3′-thiacytidine. J. Med. Chem. 2012, 55, 4861–4871. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, H.K.; Chhikara, B.S.; Quiterio, M.; Doncel, G.F.; Parang, K. Synthesis and anti-HIV activities of glutamate and peptide conjugates of nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2012, 55, 2672–2687. [Google Scholar] [CrossRef]
- Agarwal, H.K.; Chhikara, B.S.; Bhavaraju, S.; Mandal, D.; Doncel, G.F.; Parang, K. Emtricitabine prodrugs with improved anti-HIV activity and cellular uptake. Mol. Pharm. 2013, 10, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.B.; Zhu, Q.Y.; Vidal, P.M.; Watanabe, K.A.; Polsky, B.; Armstrong, D.; Ostrander, M.; Lang, S.A.; Muchmore, E.; Chou, T.C. Comparisons of anti-human immunodeficiency virus activities, cellular transport, and plasma and intracellular pharmacokinetics of 3′-fluoro-3′-deoxythymidine and 3′-azido-3′-deoxythymidine. Antimicrob. Agents Chemother. 1992, 36, 808–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinazi, R.F.; Boudinot, F.D.; Doshi, K.J.; Mcclure, H.M. Pharmacokinetics of 3′-fluoro-3′-deoxythymidine and 3′-deoxy-2′,3′-didehydrothymidine in rhesus monkeys. Antimicrob. Agents Chemother. 1990, 34, 1214–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudinot, F.D.; Smith, S.G.; Funderburg, E.D.; Schinazi, R.F. Pharmacokinetics of 3′-fluoro-3′-deoxythymidine and 3′-deoxy-2′,3′-didehydrothymidine in rats. Antimicrob. Agents Chemother. 1991, 35, 747–749. [Google Scholar] [CrossRef] [Green Version]
- Rusconi, S.; Moonis, M.; Merrill, D.P.; Pallai, P.V.; Neidhardt, E.A.; Singh, S.K.; Willis, K.J.; Osburne, M.S.; Profy, A.T.; Jenson, J.C.; et al. Naphthalene sulfonate polymers with CD4-blocking and anti-human immunodeficiency virus type 1 activities. Antimicrob. Agents Chemother. 1996, 40, 234–236. [Google Scholar] [CrossRef] [Green Version]
- Sundseth, R.; Joyner, S.S.; Moore, J.T.; Dornsife, R.E.; Dev, I.K. The anti-human immunodeficiency virus agent 3′-fluorothymidine induces DNA damage and apoptosis in human lymphoblastoid cells. Antimicrob. Agents Chemother. 1996, 40, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Ghosn, J.; Quinson, A.-M.; Sabo, N.D.; Cotte, L.; Piketty, C.; Dorléacq, N.; Bravo, M.-L.; Mayers, D.; Harmenberg, J.; Mårdh, G.; et al. Antiviral activity of low-dose alovudine in antiretroviral experienced patients: Results from a 4-week randomized, double-blind, placebo-controlled dose-ranging trial. HIV Med. 2007, 8, 142–147. [Google Scholar] [CrossRef]
- Velanguparackel, W.; Hamon, N.; Balzarini, J.; McGuigan, C.; Westwell, A.D. Synthesis, anti-HIV and cytostatic evaluation of 3′-deoxy-3′-fluorothymidine (FLT) pro-nucleotides. Bioorg. Med. Chem. Lett. 2014, 24, 2240–2243. [Google Scholar] [CrossRef] [PubMed]
- Kazmierska, J.; Cholewinski, W.; Piotrowski, T.; Sowinska, A.; Bak, B.; Cegła, P.; Malicki, J. Assessment of tumour hypoxia, proliferation and glucose metabolism in head and neck cancer before and during treatment. Br. J. Radiol. 2020, 93, 20180781. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.; Zanotti-Fregonara, P.; Eimer, S.; Gimbert, E.; Monteil, P.; Penchet, G.; Lamare, F.; Perez, P.; Vimont, D.; Ledure, S.; et al. Combining 3′-deoxy-3′-[18F] fluorothymidine and MRI increases the sensitivity of glioma volume detection. Nucl. Med. Commun. 2019, 40, 1066–1071. [Google Scholar] [CrossRef]
- Scarpelli, M.; Zahm, C.; Perlman, S.; McNeel, D.G.; Jeraj, R.; Liu, G. FLT PET/CT imaging of metastatic prostate cancer patients treated with pTVG-HP DNA vaccine and pembrolizumab. J. Immunother. Cancer 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Chhikara, B.S.; Tiwari, R.; Parang, K. N-Myristoylglutamic acid derivative of 3′-fluoro-3′-deoxythymidine as an organogel. Tetrahedron Lett. 2012, 53, 5335–5337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, M.L.; McWherter, C.A.; Kishore, N.S.; Gokel, G.W.; Gordon, J.I. MyristoylCoA: Protein N-myristoyltransferase as a therapeutic target for inhibiting replication of human immunodeficiency virus-1. Perspect. Drug Dis. Des. 1993, 1, 193–209. [Google Scholar] [CrossRef]
- Takamune, N.; Hamada, H.; Misumi, S.; Shoji, S. Novel strategy for anti-HIV-1 action: Selective cytotoxic effect of N-myristoyltransferase inhibitor on HIV-1 infected cells. FEBS Lett. 2002, 527, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Farazi, T.A.; Waksman, G.; Gordon, J.I. The biology and enzymology of protein N-myristoylation. J. Biol. Chem. 2001, 276, 39501–39504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Alexandratos, J.; Ericksen, B.; Lubkowshi, J.; Gallo, R.C.; Lu, W. Total chemical synthesis of N-myristoylated HIV-1 matrix protein p17: Structural and mechanistic implications of p17 myristoylation. Proc. Natl. Acad. Sci. USA 2004, 101, 11587–11592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herdewijn, P.; Balzarini, J.; De Clercq, E.; Pauwels, R.; Baba, M.; Broder, S.; Vanderhaeghe, H. 3′-Substituted 2′,3′-dideoxynucleoide analogues as potential anti-HIV (HTLV-III/LAV) agents. J. Med. Chem. 1987, 30, 1270–1278. [Google Scholar] [CrossRef]
- Tam, K.Y.; Takács-Novák, K. Multi-wavelength spectrophotometric determination of acid dissociation constants: A validation study. Anal. Chim. Acta 2001, 434, 157–167. [Google Scholar] [CrossRef]
- Krebs, F.C.; Miller, S.R.; Malamud, D.; Howett, M.K.; Wigdahl, B. Inactivation of human immunodeficiency virus type 1 by nonoxynol-9, C21G, or an alkyl sulfate, sodium dodecyl sulfate. Antivir. Res. 1999, 43, 147–163. [Google Scholar] [CrossRef]
- Buckheit, R.W., Jr.; Swanstrom, R. Characterization of an HIV-1 isolate displaying an apparent absence of virion-associated reverse transcriptase activity. AIDS Res. Hum. Retrovir. 1991, 7, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Doncel, G.F.; Chandra, N.; Fichorova, R.N. Preclinical assessment of the proinflammatory potential of microbicide candidates. J. Acquir. Immune Defic. Syndr. 2004, 37 (Suppl. S3), S174–S180. [Google Scholar] [CrossRef]
- Shah, V.; Doncel, G.F.; Seyoum, T.; Eaton, K.M.; Zalenskaya, I.; Hagver, R.; Azim, A.; Gross, R. Sophorolipids, microbial glycolipids with anti-human immunodeficiency virus and sperm-immobilizing activities. Antimicrob. Agents Chemother. 2005, 49, 4093–4100. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
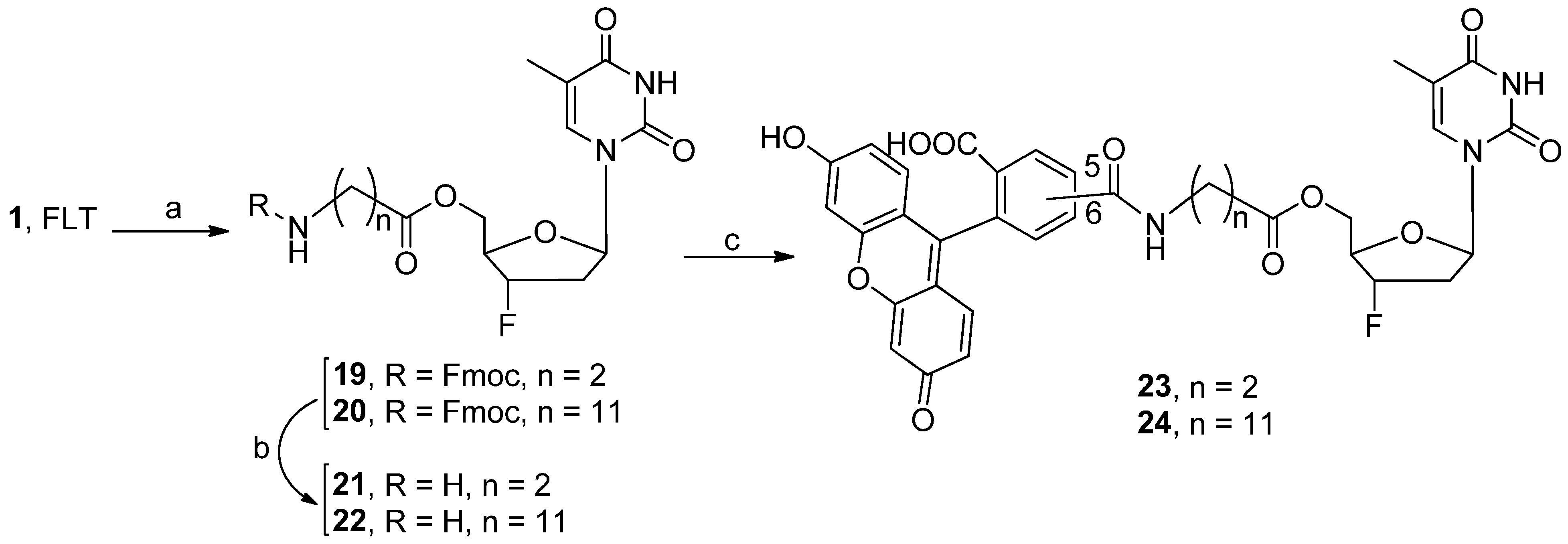{kind=link}

| Compd. | R1 | R2 | Compd. | R1 | R2 |
|---|---|---|---|---|---|
| FLT (1) | H | F | 12 | Br(CH2)11CO | N3 |
| AZT (2) | H | N3 | 13 | CH3(CH2)11CH(OMe)CO | F |
| 3 | CH3(CH2)13O | F | 14 | CH3(CH2)11CH(OMe)CO | N3 |
| 4 | CH3(CH2)13O | N3 | 15 | CH3(CH2)4S(CH2)7CO | F |
| 5 | N3(CH2)11CO | F | 16 | CH3(CH2)4S(CH2)7CO | N3 |
| 6 | CH3(CH2)12CO | F | 17 | CH3(CH2)9O(CH2)2CO | N3 |
| 7 | CH3(CH2)12CO | N3 | 18 | CH3(CH2)4O(CH2)7CO | N3 |
| 8 | CH3CH2S(CH2)11CO | F | 21 | NH2(CH2)2CO | F |
| 9 | CH3CH2S(CH2)11CO | N3 | 22 | NH2(CH2)11CO | F |
| 10 | CH3(CH2)13CO | N3 | 23 | FAM-NH2(CH2)2CO | F |
| 11 | Br(CH2)11CO | F | 24 | FAM-NH2(CH2)11CO | F |
| Compd. | Cytotoxicity a EC50 b (μM) | (X4) c EC50 (μM) | (R5) d EC50 (μM) | (CA) e EC50 (μM) |
|---|---|---|---|---|
| FLT, 1 | >410 | 0.8 | 0.4 | >410 |
| AZT, 2 | >375 | 10.9 | 14.2 | >375 |
| 3 | 179.0 | 180 | 176 | >227 |
| 4 | 205.0 | 125 | 27.6 | >216 |
| 5 | 1598.0 | 0.9 | 0.4 | 12.6 |
| 6 | 606.0 | 0.7 | 1.1 | 6.4 |
| 7 | >629 | 3.1 | 5.0 | >629 |
| 8 | >2000 | 1.0 | <0.2 | 2.3 |
| 9 | >101 | 7.7 | 5.2 | 90.7 |
| 10 | >611 | 17.9 | 4.5 | >611 |
| 11 | >198 | 1.8 | <0.2 | >198 |
| 12 | >190 | 14.8 | 4.6 | >190 |
| 13 | >206 | 1.0 | 0.2 | >206 |
| 14 | >197 | 9.7 | 6.7 | >197 |
| 15 | >64 | 11.4 | 4.4 | >64 |
| 16 | >202 | 9.3 | 12.9 | >202 |
| 17 | >209 | 6.7 | 2.1 | >209 |
| 18 | >209 | 4.8 | 6.1 | >209 |
| 22 | >226 | 3.4 | 1.6 | >227 |
| 24 | >125 | 6.6 | 3.6 | >123 |
| C-2f | >1000 | 1.6 | 85.9 | 5.1 |
| DMSO g | >1000 | >1000 | >1000 | >1000 |
| Chemical Name | Cytotoxicity a EC50 b (μM) | (X4) c EC50 (μM) | (R5) d EC50 (μM) | (CA) e EC50 (μM) |
|---|---|---|---|---|
| FLT (1) | >410 | 0.8 | 0.4 | >410 |
| AZT (2) | >374 | 10.9 | 14.2 | >374 |
| C13H27-COO-FLT (6) | 606.0 | 0.7 | 1.1 | 6.4 |
| C13H27-COOH + FLT (50:50) (25) | >106 | <0.2 | 0.8 | 33.0 |
| C13H27-COO-AZT (7) | >629 | 3.1 | 5.0 | >629 |
| C13H27-COOH + AZT (50:50) (26) | >202 | 1.4 | 46.2 | >202 |
| Br-C11H22-COO-AZT (12) | >190 | 14.8 | 4.6 | >190 |
| Br-C11H22-COOH + AZT (50:50) (27) | >183 | 34.8 | 8.8 | >183 |
| Compd. | Virus | Clade/Resistance | IC50 b (nM) | IC90 c (nM) |
|---|---|---|---|---|
| FLT | 94US3393IN | B-Wild Type | 2.0 | 20.4 |
| 98USMSC5016 | C-Wild Type | 2.0 | 12.3 | |
| A-17 MDR | B-NNRTI | 26.0 | 87.2 | |
| 4755-5 MDR | B-MDR | 107.3 | 1065.0 | |
| 5 | 94US3393IN | B-Wild Type | 1.1 | 6.4 |
| 98USMSC5016 | C-Wild Type | 2.1 | 8.6 | |
| A-17 MDR | B-NNRTI | 4.4 | 14.1 | |
| 4755-5 MDR | B-MDR | 39.6 | 507.2 | |
| 8 | 94US3393IN | B-Wild Type | 0.6 | 10.3 |
| 98USMSC5016 | C-Wild Type | 1.0 | 8.2 | |
| A-17 MDR | B-NNRTI | 13.6 | 45.7 | |
| 4755-5 MDR | B-MDR | 111.5 | 662.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarwal, H.K.; Chhikara, B.S.; Ye, G.; Bhavaraju, S.; Dixit, A.; Kumar, A.; Doncel, G.F.; Parang, K. Synthesis and Biological Evaluation of 5′-O-Fatty Acyl Ester Derivatives of 3′-Fluoro-2′,3′-dideoxythymidine as Potential Anti-HIV Microbicides. Molecules 2022, 27, 3352. https://doi.org/10.3390/molecules27103352
Agarwal HK, Chhikara BS, Ye G, Bhavaraju S, Dixit A, Kumar A, Doncel GF, Parang K. Synthesis and Biological Evaluation of 5′-O-Fatty Acyl Ester Derivatives of 3′-Fluoro-2′,3′-dideoxythymidine as Potential Anti-HIV Microbicides. Molecules. 2022; 27(10):3352. https://doi.org/10.3390/molecules27103352
Chicago/Turabian StyleAgarwal, Hitesh K., Bhupender S. Chhikara, Guofeng Ye, Sitaram Bhavaraju, Ajay Dixit, Anil Kumar, Gustavo F. Doncel, and Keykavous Parang. 2022. "Synthesis and Biological Evaluation of 5′-O-Fatty Acyl Ester Derivatives of 3′-Fluoro-2′,3′-dideoxythymidine as Potential Anti-HIV Microbicides" Molecules 27, no. 10: 3352. https://doi.org/10.3390/molecules27103352
APA StyleAgarwal, H. K., Chhikara, B. S., Ye, G., Bhavaraju, S., Dixit, A., Kumar, A., Doncel, G. F., & Parang, K. (2022). Synthesis and Biological Evaluation of 5′-O-Fatty Acyl Ester Derivatives of 3′-Fluoro-2′,3′-dideoxythymidine as Potential Anti-HIV Microbicides. Molecules, 27(10), 3352. https://doi.org/10.3390/molecules27103352