
Adamantane-Monoterpenoid Conjugates Linked via Heterocyclic Linkers Enhance the Cytotoxic Effect of Topotecan

 , , , ,
, , , ,  ,
,  ,
, 
Abstract
:1. Introduction
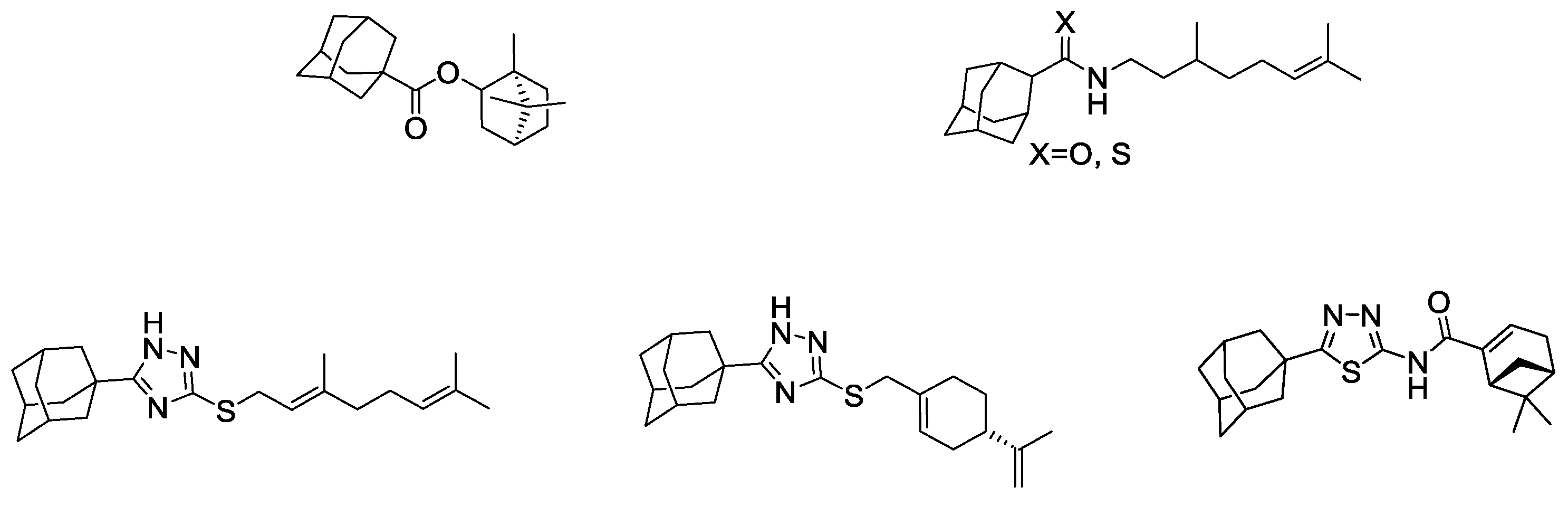
2. Results and Discussion
2.1. Chemistry
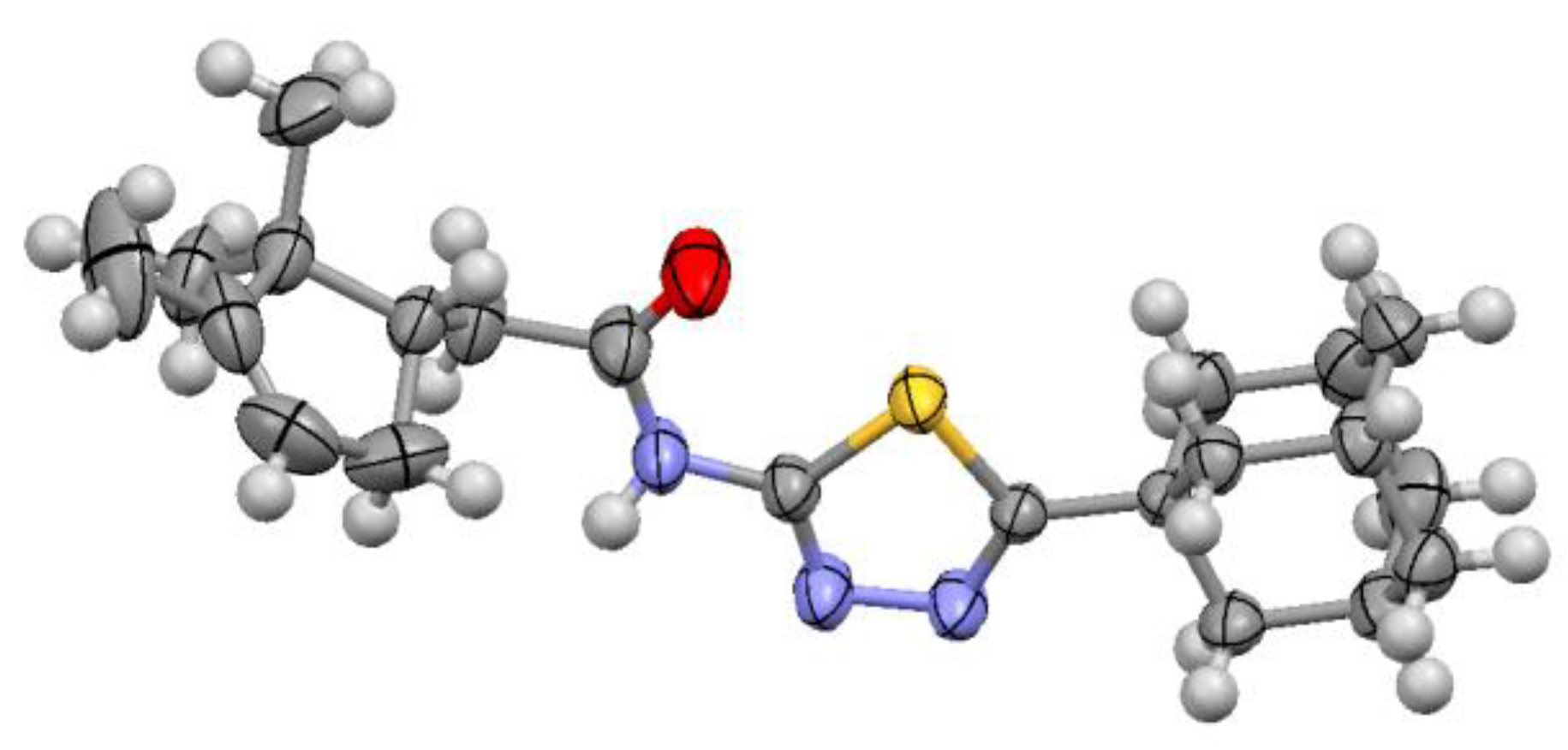
2.2. Biology
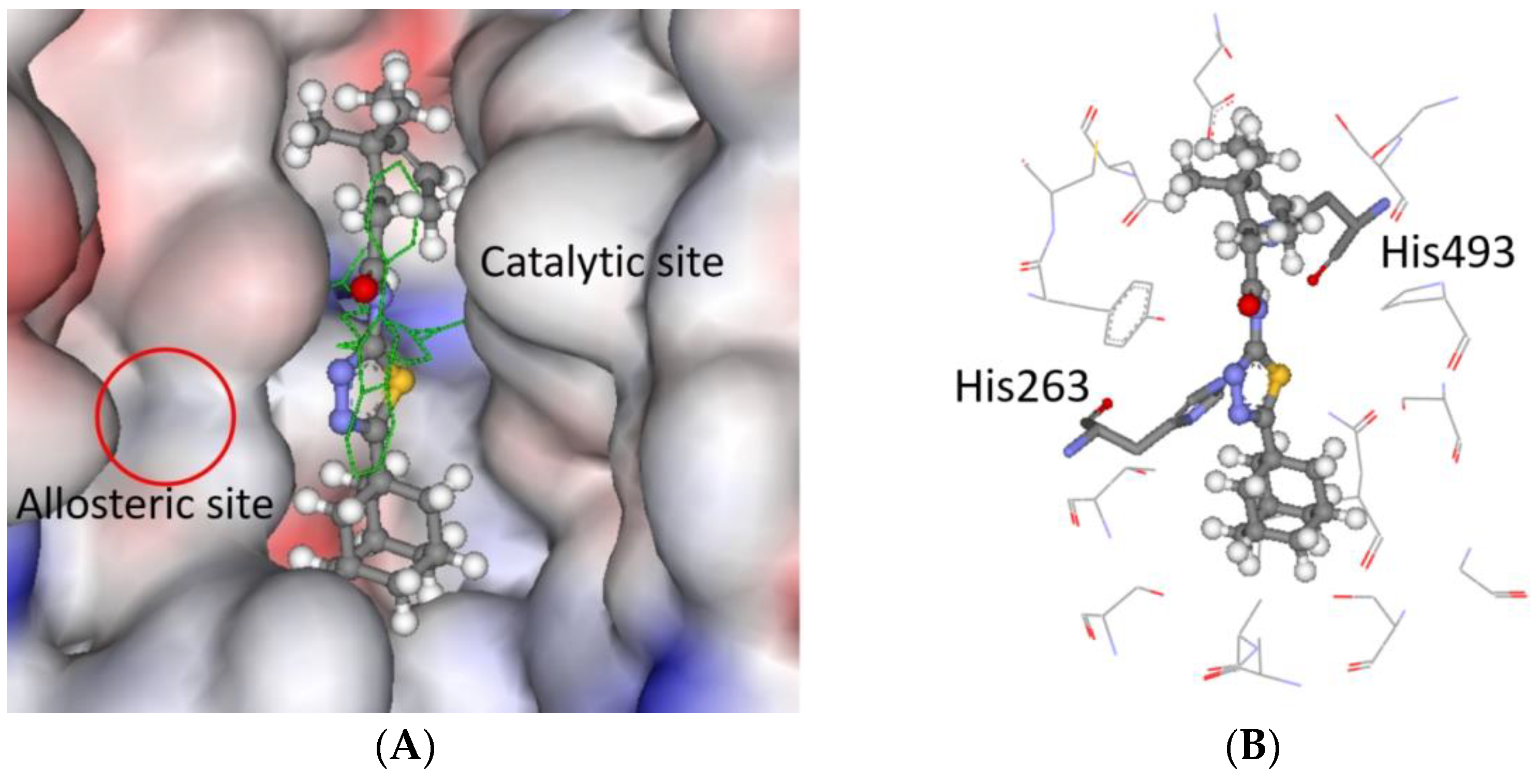
2.3. Molecular Modeling
2.4. Chemical Space
3. Materials and Methods
3.1. Chemistry
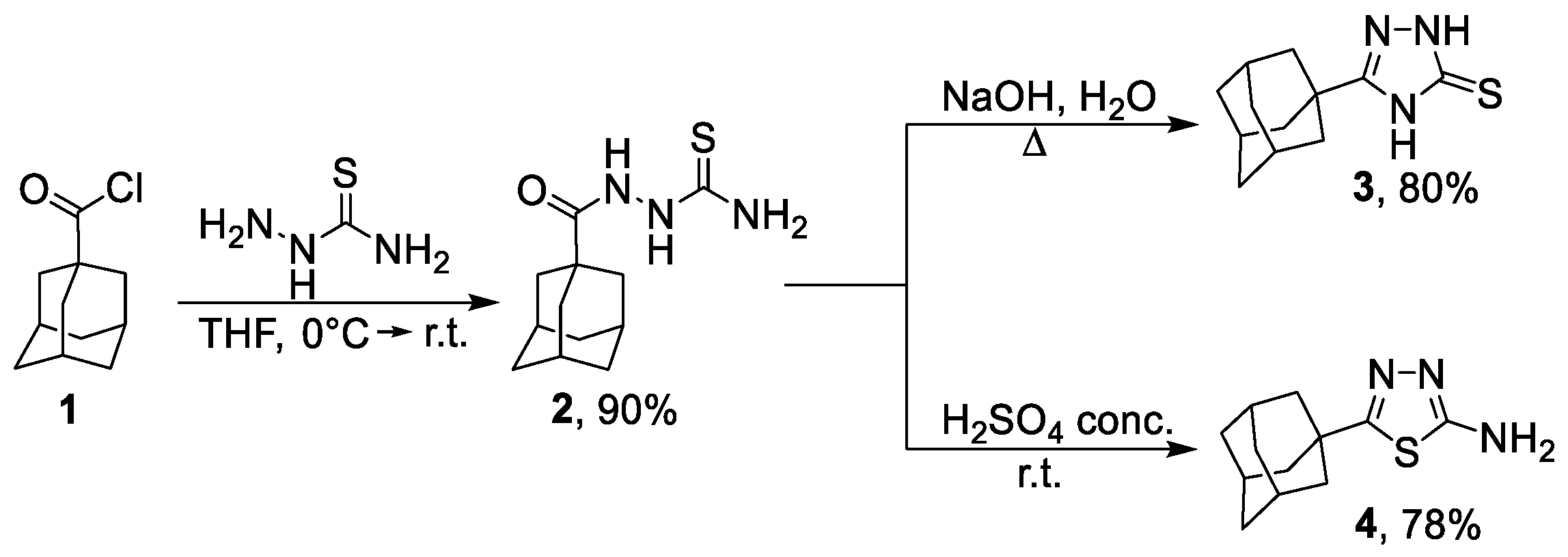
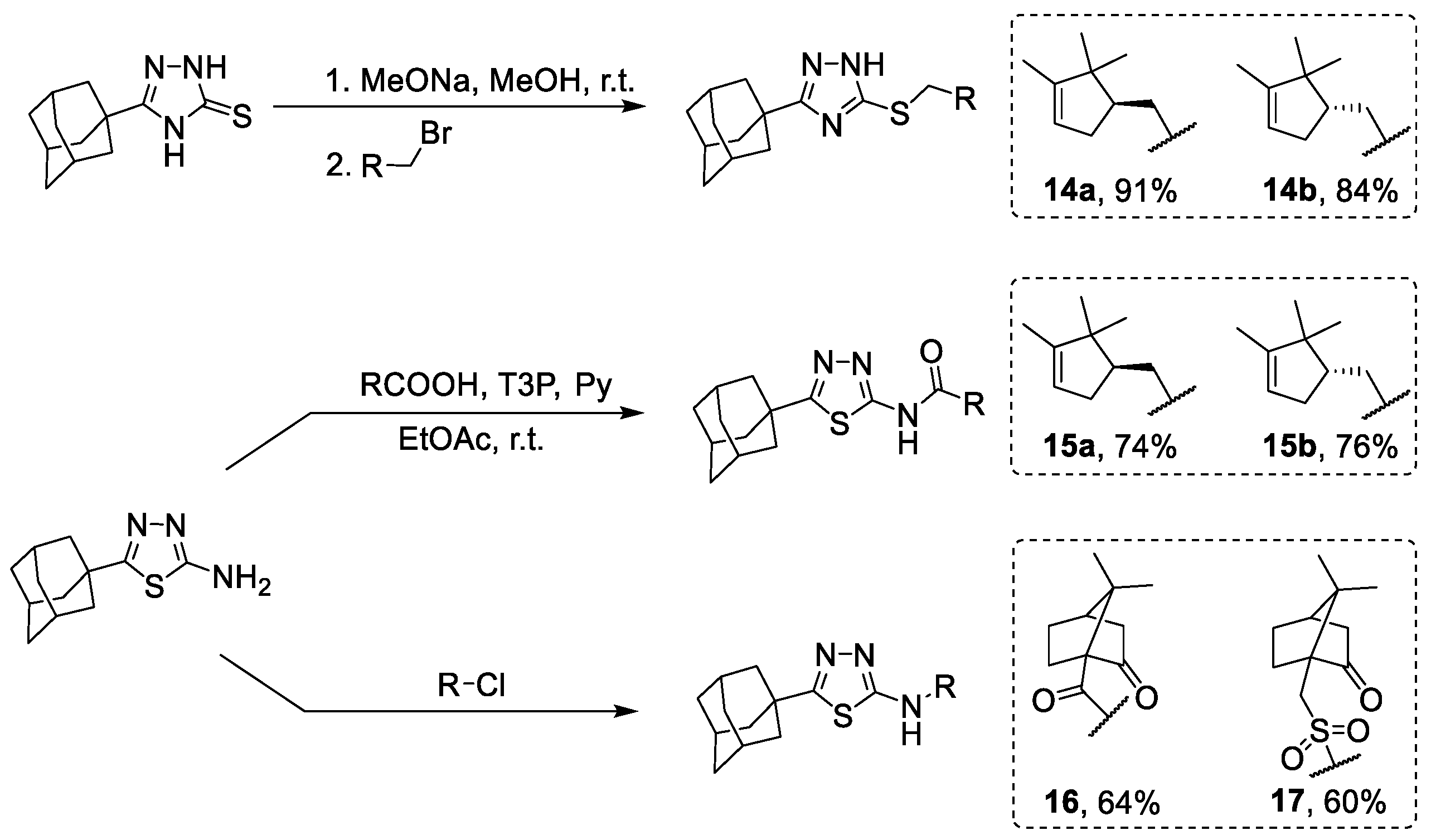
3.1.1. Synthesis of 2-(Adamantane-1-carbonyl)hydrazine-1-carbothioamide 2
3.1.2. Synthesis of 5-(Adamantan-1-yl)-2,4-dihydro-3H-1,2,4-triazole-3-thione 3
3.1.3. Synthesis of 5-(Adamantan-1-yl)-1,3,4-thiadiazol-2-amine 4
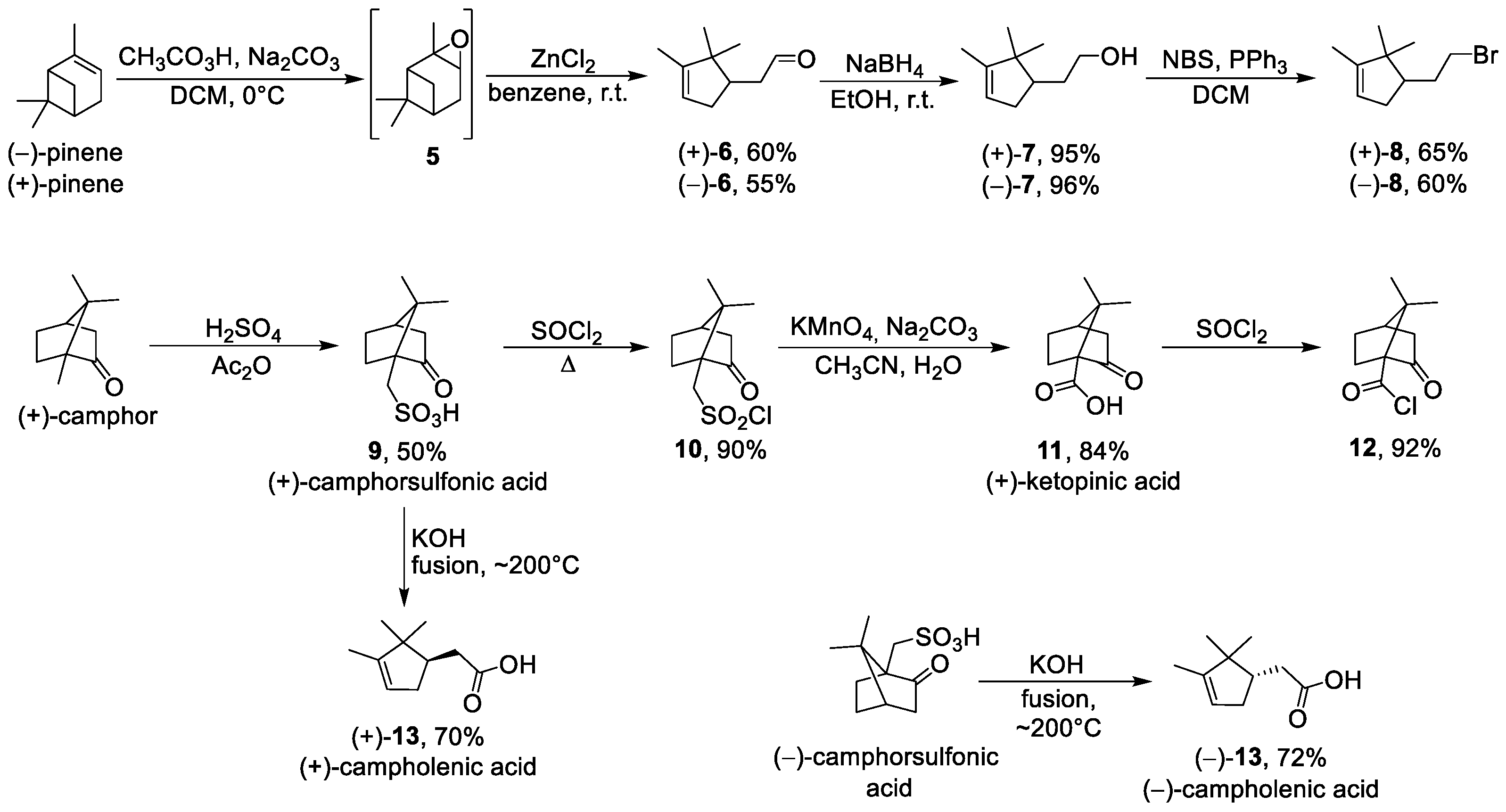
3.1.4. Synthesis of (+)-Campholenic Aldehyde 6
3.1.5. Synthesis of (+)-Campholenic Alcohol 7
3.1.6. General Procedure for Synthesis of Bromides 8
3.1.7. (R)-4-(2-Bromoethyl)-1,5,5-trimethylcyclopent-1-ene (+)-8
 1H-NMR (400 MHz, CDCl3) (ppm) δ: 0.75 (s, 3H, H-7), 0.97 (s, 3H, H-8), 1.58 (dt, 4J = 2.4 Hz, 3H, H-6), 1.75–2.03 (m, 4H, H-3, H-9), 2.23–2.33 (m, 1H, H-4), 3.33 (dt, J = 10 Hz, J = 8 Hz, 1H, H-10a), 3.48 (m, 1H, H-10b), 5.20 (m, 1H, H-2). 13C-NMR (100 MHz, CDCl3) (ppm) δ: 12.5 (q, C-6), 19.7 (q, C-7), 25.6 (q, C-8), 33.1 (t, C-10), 33.7 (t, C-9), 34.6 (t, C-3), 46.8 (s, C-5), 48.8 (d, C-4), 121.2 (d, C-2), 148.5 (s, C-1). HR MS: 216.0504 (M+, C10H16Br+; calc. 216.0508). = +24 (c 0.69 in CHCl3).
1H-NMR (400 MHz, CDCl3) (ppm) δ: 0.75 (s, 3H, H-7), 0.97 (s, 3H, H-8), 1.58 (dt, 4J = 2.4 Hz, 3H, H-6), 1.75–2.03 (m, 4H, H-3, H-9), 2.23–2.33 (m, 1H, H-4), 3.33 (dt, J = 10 Hz, J = 8 Hz, 1H, H-10a), 3.48 (m, 1H, H-10b), 5.20 (m, 1H, H-2). 13C-NMR (100 MHz, CDCl3) (ppm) δ: 12.5 (q, C-6), 19.7 (q, C-7), 25.6 (q, C-8), 33.1 (t, C-10), 33.7 (t, C-9), 34.6 (t, C-3), 46.8 (s, C-5), 48.8 (d, C-4), 121.2 (d, C-2), 148.5 (s, C-1). HR MS: 216.0504 (M+, C10H16Br+; calc. 216.0508). = +24 (c 0.69 in CHCl3).3.1.8. (S)-4-(2-Bromoethyl)-1,5,5-trimethylcyclopent-1-ene (−)-8
3.1.9. Synthesis of (+)-10-Camphorsulfonic Acid 9
3.1.10. Synthesis of (+)-10-Camphorsulfonyl Chloride 10
3.1.11. Synthesis of (+)-Ketopinic Acid 11
3.1.12. Synthesis of (+)-Ketopinic Acid Chloride 12
3.1.13. Synthesis of (+)-Campholenic Acid 13
3.1.14. General Procedure for Obtaining 1,2,4-Triazole Derivatives 14a-b
3.1.15. 3-(Adamantan-1-yl)-5-((2-((R)-2,2,3-trimethylcyclopent-3-en-1-yl)ethyl)thio)-1H-1,2,4-triazole 14a
 1H-NMR (600 MHz, CDCl3) (ppm) δ: 0.72 (s, 3H, H-24), 0.93 (s, 3H, H-25), 1.55–1.58 (m, 3H, H-23), 1.59–1.67 (m, 1H, H-17b), 1.70–1.78 (m, 6H, 2H-4, 2H-6, 2H-10), 1.78–1.88 (m, 3H, H-17a, H-18, H-19b), 1.98 (d, 3J = 2.9 Hz, 6H, 2H-2, 2H-8, 2H-9), 2.03–2.08 (m, 3H, H-3, H-5, H-7), 2.25–2.34 (m, 1H, H-19a), 3.01 (ddd, J = 12.6, 9.8, 6.4 Hz, 1H, H-16b), 3.16 (ddd, J = 12.6, 10.0, 4.9 Hz, 1H, H-16a), 5.17–5.20 (m, 1H, H-20). 13C-NMR (150 MHz, CDCl3) (ppm) δ: 34.2 (s, C-1), 40.7 (t, C-2, C-8, C-9), 27.9 (d, C-3, C-5, C-7), 36.3 (t, C-4, C-6, C-10), 166.6 (s, C-11), 159.2 (s, C-13), 31.7 (t, C-16), 30.3 (t, C-17), 49.5 (d, C-18), 35.1 (t, C-19), 121.3 (d, C-20), 148.4 (s, C-21), 46.8 (s, C-22), 12.5 (q, C-23), 19.6 (q, C-24), 25.6 (q, C-25). HR MS: 371.2389 (M+, C22H33N3S1+; calc. 371.2390). = +15 (c 0.43 in MeOH).
1H-NMR (600 MHz, CDCl3) (ppm) δ: 0.72 (s, 3H, H-24), 0.93 (s, 3H, H-25), 1.55–1.58 (m, 3H, H-23), 1.59–1.67 (m, 1H, H-17b), 1.70–1.78 (m, 6H, 2H-4, 2H-6, 2H-10), 1.78–1.88 (m, 3H, H-17a, H-18, H-19b), 1.98 (d, 3J = 2.9 Hz, 6H, 2H-2, 2H-8, 2H-9), 2.03–2.08 (m, 3H, H-3, H-5, H-7), 2.25–2.34 (m, 1H, H-19a), 3.01 (ddd, J = 12.6, 9.8, 6.4 Hz, 1H, H-16b), 3.16 (ddd, J = 12.6, 10.0, 4.9 Hz, 1H, H-16a), 5.17–5.20 (m, 1H, H-20). 13C-NMR (150 MHz, CDCl3) (ppm) δ: 34.2 (s, C-1), 40.7 (t, C-2, C-8, C-9), 27.9 (d, C-3, C-5, C-7), 36.3 (t, C-4, C-6, C-10), 166.6 (s, C-11), 159.2 (s, C-13), 31.7 (t, C-16), 30.3 (t, C-17), 49.5 (d, C-18), 35.1 (t, C-19), 121.3 (d, C-20), 148.4 (s, C-21), 46.8 (s, C-22), 12.5 (q, C-23), 19.6 (q, C-24), 25.6 (q, C-25). HR MS: 371.2389 (M+, C22H33N3S1+; calc. 371.2390). = +15 (c 0.43 in MeOH).3.1.16. 3-(Adamantan-1-yl)-5-((2-((S)-2,2,3-trimethylcyclopent-3-en-1-yl)ethyl)thio)-1H-1,2,4-triazole 14b
3.1.17. Synthesis of Amides 15a-b
3.1.18. Synthesis of Amides 16–17
3.1.19. N-(5-(Adamantan-1-yl)-1,3,4-thiadiazol-2-yl)-2-((R)-2,2,3-trimethylcyclopent-3-en-1-yl)acetamide 15a
 1H-NMR (600 MHz, CDCl3) (ppm) δ: 0.92 (s, 3H, H-24), 1.04 (s, 3H, H-25), 1.59–1.62 (m, 3H, H-23), 1.72–1.81 (m, 6H, 2H-4, 2H-6, 2H-10), 1.99–2.05 (m, 1H, H-19b), 2.04 (d, 3J = 2.8 Hz, 6H, 2H-2, 2H-8, 2H-9), 2.06–2.11 (m, 3H, H-3, H-5, H-7), 2.31–2.42 (m, 2H, H-18, H-19a), 2.77–2.87 (m, 2H, H-17), 5.19–5.22 (m, 1H, H-20), 13.53 (br.s, 1H, NH). 13C-NMR (150 MHz, CDCl3) (ppm) δ: 37.7 (s, C-1), 43.1 (t, C-2, C-8, C-9), 28.3 (d, C-3, C-5, C-7), 36.3 (t, C-4, C-6, C-10), 174.4 (s, C-11), 159.8 (s, C-13), 172.1 (s, C-16), 37.1 (t, C-17), 46.7 (d, C-18), 35.3 (t, C-19), 121.6 (d, C-20), 147.8 (s, C-21), 47.0 (s, C-22), 12.6 (q, C-23), 20.0 (q, C-24), 25.5 (q, C-25). HR MS: 385.2185 (M+, C22H31O1N3S1+; calc. 385.2182). = +49 (c 0.41 in CHCl3).
1H-NMR (600 MHz, CDCl3) (ppm) δ: 0.92 (s, 3H, H-24), 1.04 (s, 3H, H-25), 1.59–1.62 (m, 3H, H-23), 1.72–1.81 (m, 6H, 2H-4, 2H-6, 2H-10), 1.99–2.05 (m, 1H, H-19b), 2.04 (d, 3J = 2.8 Hz, 6H, 2H-2, 2H-8, 2H-9), 2.06–2.11 (m, 3H, H-3, H-5, H-7), 2.31–2.42 (m, 2H, H-18, H-19a), 2.77–2.87 (m, 2H, H-17), 5.19–5.22 (m, 1H, H-20), 13.53 (br.s, 1H, NH). 13C-NMR (150 MHz, CDCl3) (ppm) δ: 37.7 (s, C-1), 43.1 (t, C-2, C-8, C-9), 28.3 (d, C-3, C-5, C-7), 36.3 (t, C-4, C-6, C-10), 174.4 (s, C-11), 159.8 (s, C-13), 172.1 (s, C-16), 37.1 (t, C-17), 46.7 (d, C-18), 35.3 (t, C-19), 121.6 (d, C-20), 147.8 (s, C-21), 47.0 (s, C-22), 12.6 (q, C-23), 20.0 (q, C-24), 25.5 (q, C-25). HR MS: 385.2185 (M+, C22H31O1N3S1+; calc. 385.2182). = +49 (c 0.41 in CHCl3).3.1.20. N-(5-(Adamantan-1-yl)-1,3,4-thiadiazol-2-yl)-2-((S)-2,2,3-trimethylcyclopent-3-en-1-yl)acetamide 15b
3.1.21. (1S,4R)-N-(5-(Adamantan-1-yl)-1,3,4-thiadiazol-2-yl)-7,7-dimethyl-2-oxobicyclo[2.2.1]heptane-1-carboxamide 16
 1H-NMR (600 MHz, CDCl3) (ppm) δ: 1.00 (s, 3H, H-24), 1.28 (s, 3H, H-25), 1.49 (ddd, J = 12.8, 9.2, 4.0 Hz, 1H, H-21endo), 1.70–1.82 (m, 7H, 2H-4, 2H-6, 2H-10, H-22b), 2.02–2.11 (m, 10H, H-3, H-5, H-7, 2H-2, 2H-8, 2H-9, H-19endo), 2.12–2.16 (m, 1H, H-20), 2.15–2.23 (m, 1H, H-21exo), 2.46–2.53 (m, 1H, H-22endo), 2.59 (dm, 2J=18.8 Hz, 1H, H-19exo), 11.04 (br.s, 1H, NH). 13C-NMR (150 MHz, CDCl3) (ppm) δ: 37.6 (s, C-1), 43.1 (t, C-2, C-8, C-9), 28.3 (d, C-3, C-5, C-7), 36.3 (t, C-4, C-6, C-10), 175.4 (s, C-11), 156.6 (s, C-13), 167.3 (s, C-16), 64.1 (s, C-17), 215.5 (s, C-18), 43.4 (t, C-19), 43.3 (d, C-20), 27.7 (t, C-21), 29.1 (t, C-22), 50.7 (s, C-23), 20.2 (q, C-24), 20.6 (q, C-25). HR MS: 399.1973 (M+, C22H29O2N3S1+; calc. 399.1975). = +16 (c 0.47 in CHCl3).
1H-NMR (600 MHz, CDCl3) (ppm) δ: 1.00 (s, 3H, H-24), 1.28 (s, 3H, H-25), 1.49 (ddd, J = 12.8, 9.2, 4.0 Hz, 1H, H-21endo), 1.70–1.82 (m, 7H, 2H-4, 2H-6, 2H-10, H-22b), 2.02–2.11 (m, 10H, H-3, H-5, H-7, 2H-2, 2H-8, 2H-9, H-19endo), 2.12–2.16 (m, 1H, H-20), 2.15–2.23 (m, 1H, H-21exo), 2.46–2.53 (m, 1H, H-22endo), 2.59 (dm, 2J=18.8 Hz, 1H, H-19exo), 11.04 (br.s, 1H, NH). 13C-NMR (150 MHz, CDCl3) (ppm) δ: 37.6 (s, C-1), 43.1 (t, C-2, C-8, C-9), 28.3 (d, C-3, C-5, C-7), 36.3 (t, C-4, C-6, C-10), 175.4 (s, C-11), 156.6 (s, C-13), 167.3 (s, C-16), 64.1 (s, C-17), 215.5 (s, C-18), 43.4 (t, C-19), 43.3 (d, C-20), 27.7 (t, C-21), 29.1 (t, C-22), 50.7 (s, C-23), 20.2 (q, C-24), 20.6 (q, C-25). HR MS: 399.1973 (M+, C22H29O2N3S1+; calc. 399.1975). = +16 (c 0.47 in CHCl3).3.1.22. N-(5-(Adamantan-1-yl)-1,3,4-thiadiazol-2-yl)-1-((1S,4R)-7,7-dimethyl-2-oxobicyclo[2.2.1]heptan-1-yl)methanesulfonamide 17
 1H-NMR (600 MHz, CDCl3) (ppm) δ: 0.83 (s, 3H, H-24), 1.09 (s, 3H, H-25), 1.41 (ddd, J = 12.7, 9.4, 3.5 Hz, 1H, H-21endo), 1.66–1.79 (m, 7H, 2H-4, 2H-6, 2H-10, H-22b), 1.90–1.96 (m, 7H, 2H-2, 2H-8, 2H-9, H-19endo), 1.98–2.05 (m, 1H, H-21exo), 2.05–2.10 (m, 4H, H-3, H-5, H-7, H-20), 2.32 (ddd, J = 18.5, 4.8, 3.5 Hz, 1H, H-19exo), 2.55 (ddd, J = 14.5, 11.7, 4.0 Hz, 1H, H-22a), 3.00 (d, J = 14.8 Hz, 1H, H-16b), 3.56 (d, J = 14.8 Hz, 1H, H-16a). 13C-NMR (150 MHz, CDCl3) (ppm) δ: 38.2 (s, C-1), 41.9 (t, C-2, C-8, C-9), 28.0 (d, C-3, C-5, C-7), 36.1 (t, C-4, C-6, C-10), 168.1 (s, C-11), 167.9 (s, C-13), 49.8 (t, C-16), 58.2 (s, C-17), 215.7 (s, C-18), 42.5 (t, C-19), 42.5 (d, C-20), 26.8 (t, C-21), 24.4 (t, C-22), 48.1 (s, C-23), 19.6 (q, C-24), 19.8 (q, C-25). HR MS: 449.1803 (M+, C22H31O3N3S2+; calc. 449.1801). = +29 (c 0.47 in CHCl3).
1H-NMR (600 MHz, CDCl3) (ppm) δ: 0.83 (s, 3H, H-24), 1.09 (s, 3H, H-25), 1.41 (ddd, J = 12.7, 9.4, 3.5 Hz, 1H, H-21endo), 1.66–1.79 (m, 7H, 2H-4, 2H-6, 2H-10, H-22b), 1.90–1.96 (m, 7H, 2H-2, 2H-8, 2H-9, H-19endo), 1.98–2.05 (m, 1H, H-21exo), 2.05–2.10 (m, 4H, H-3, H-5, H-7, H-20), 2.32 (ddd, J = 18.5, 4.8, 3.5 Hz, 1H, H-19exo), 2.55 (ddd, J = 14.5, 11.7, 4.0 Hz, 1H, H-22a), 3.00 (d, J = 14.8 Hz, 1H, H-16b), 3.56 (d, J = 14.8 Hz, 1H, H-16a). 13C-NMR (150 MHz, CDCl3) (ppm) δ: 38.2 (s, C-1), 41.9 (t, C-2, C-8, C-9), 28.0 (d, C-3, C-5, C-7), 36.1 (t, C-4, C-6, C-10), 168.1 (s, C-11), 167.9 (s, C-13), 49.8 (t, C-16), 58.2 (s, C-17), 215.7 (s, C-18), 42.5 (t, C-19), 42.5 (d, C-20), 26.8 (t, C-21), 24.4 (t, C-22), 48.1 (s, C-23), 19.6 (q, C-24), 19.8 (q, C-25). HR MS: 449.1803 (M+, C22H31O3N3S2+; calc. 449.1801). = +29 (c 0.47 in CHCl3).3.2. Biological Assays
3.2.1. Detection of TDP1 Activity
3.2.2. Cytotoxicity Assays
3.3. Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Interthal, H.; Pouliot, J.J.; Champoux, J.J. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc. Natl. Acad. Sci. USA 2001, 98, 12009–12014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Pourquier, P.; Fan, Y.; Strumberg, D. Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochim. Biophys. Acta Gene Struct. Expr. 1998, 1400, 83–106. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X.; et al. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef]
- Comeaux, E.Q.; Van Waardenburg, R.C.A.M. Tyrosyl-DNA phosphodiesterase I resolves both naturally and chemically induced DNA adducts and its potential as a therapeutic target. Drug Metab. Rev. 2014, 46, 494–507. [Google Scholar] [CrossRef]
- Barthelmes, H.U.; Habermeyer, M.; Christensen, M.O.; Mielke, C.; Interthal, H.; Pouliot, J.J.; Boege, F.; Marko, D. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J. Biol. Chem. 2004, 279, 55618–55625. [Google Scholar] [CrossRef] [Green Version]
- Meisenberg, C.; Gilbert, D.C.; Chalmers, A.; Haley, V.; Gollins, S.; Ward, S.E.; El-Khamisy, S.F. Clinical and cellular roles for TDP1 and TOP1 in modulating colorectal cancer response to irinotecan. Mol. Cancer Ther. 2015, 14, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Katyal, S.; El-Khamisy, S.F.; Russell, H.R.; Li, Y.; Ju, L.; Caldecott, K.W.; McKinnon, P.J. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007, 26, 4720–4731. [Google Scholar] [CrossRef] [Green Version]
- Das, B.B.; Antony, S.; Gupta, S.; Dexheimer, T.S.; Redon, C.E.; Garfield, S.; Shiloh, Y.; Pommier, Y. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 2009, 28, 3667–3680. [Google Scholar] [CrossRef] [Green Version]
- Khomenko, T.; Zakharenko, A.; Odarchenko, T.; Arabshahi, H.J.; Sannikova, V.; Zakharova, O.; Korchagina, D.; Reynisson, J.; Volcho, K.; Salakhutdinov, N.; et al. New inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 7-hydroxycoumarin and monoterpenoid moieties. Bioorg. Med. Chem. 2016, 24, 5573–5581. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, K.Y.; Suslov, E.V.; Zakharenko, A.L.; Zakharova, O.D.; Rogachev, A.D.; Korchagina, D.V.; Zafar, A.; Reynisson, J.; Nefedov, A.A.; Volcho, K.P.; et al. Aminoadamantanes containing monoterpene-derived fragments as potent tyrosyl-DNA phosphodiesterase 1 inhibitors. Bioorg. Chem. 2018, 76, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, O.; Luzina, O.; Zakharenko, A.; Sokolov, D.; Filimonov, A.; Dyrkheeva, N.; Chepanova, A.; Ilina, E.; Ilyina, A.; Klabenkova, K.; et al. Synthesis and evaluation of aryliden- and hetarylidenfuranone derivatives of usnic acid as highly potent Tdp1 inhibitors. Bioorg. Med. Chem. 2018, 26, 4470–4480. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, A.S.; Chepanova, A.A.; Luzina, O.A.; Zakharenko, A.L.; Zakharova, O.D.; Ilina, E.S.; Dyrkheeva, N.S.; Kuprushkin, M.S.; Kolotaev, A.V.; Khachatryan, D.S.; et al. New Hydrazinothiazole Derivatives of Usnic Acid as Potent Tdp1 Inhibitors. Molecules 2019, 24, 3711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyrkheeva, N.S.; Filimonov, A.S.; Luzina, O.A.; Zakharenko, A.L.; Ilina, E.S.; Malakhova, A.A.; Medvedev, S.P.; Reynisson, J.; Volcho, K.P.; Zakian, S.M.; et al. New hybrid compounds combining fragments of usnic acid and monoterpenoids for effective tyrosyl-dna phosphodiesterase 1 inhibition. Biomolecules 2021, 11, 973. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Luzina, O.A.; Sokolov, D.N.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Patel, J.; Zakharova, O.D.; Chepanova, A.A.; Zafar, A.; et al. Novel tyrosyl-DNA phosphodiesterase 1 inhibitors enhance the therapeutic impact of topotecan on in vivo tumor models. Eur. J. Med. Chem. 2019, 161, 581–593. [Google Scholar] [CrossRef]
- Khomenko, T.M.; Zakharenko, A.L.; Chepanova, A.A.; Ilina, E.S.; Zakharova, O.D.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Korchagina, D.V.; Reynisson, J.; et al. Promising New Inhibitors of Tyrosyl-DNA Phosphodiesterase I (Tdp 1) Combining 4-Arylcoumarin and Monoterpenoid Moieties as Components of Complex Antitumor Therapy. Int. J. Mol. Sci. 2019, 21, 126. [Google Scholar] [CrossRef] [Green Version]
- Nikolin, V.P.; Popova, N.A.; Kaledin, V.I.; Luzina, O.A.; Zakharenko, A.L.; Salakhutdinov, N.F.; Lavrik, O.I. The influence of an enamine usnic acid derivative (a tyrosyl-DNA phosphodiesterase 1 inhibitor) on the therapeutic effect of topotecan against transplanted tumors in vivo. Clin. Exp. Metastasis 2021, 38, 431–440. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Filimonov, A.S.; Luzina, O.A.; Orlova, K.A.; Chernyshova, I.A.; Kornienko, T.E.; Malakhova, A.A.; Medvedev, S.P.; Zakharenko, A.L.; Ilina, E.S.; et al. New Hybrid Compounds Combining Fragments of Usnic Acid and Thioether Are Inhibitors of Human Enzymes TDP1, TDP2 and PARP1. Int. J. Mol. Sci. 2021, 22, 11336. [Google Scholar] [CrossRef]
- Luzina, O.; Filimonov, A.; Zakharenko, A.; Chepanova, A.; Zakharova, O.; Ilina, E.; Dyrkheeva, N.; Likhatskaya, G.; Salakhutdinov, N.; Lavrik, O. Usnic Acid Conjugates with Monoterpenoids as Potent Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. J. Nat. Prod. 2020, 83, 2320–2329. [Google Scholar] [CrossRef]
- Gladkova, E.D.; Chepanova, A.A.; Ilina, E.S.; Zakharenko, A.L.; Reynisson, J.; Luzina, O.A.; Volcho, K.P.; Lavrik, O.I.; Salakhutdinov, N.F. Discovery of novel sultone fused berberine derivatives as promising tdp1 inhibitors. Molecules 2021, 26, 1945. [Google Scholar] [CrossRef] [PubMed]
- Salomatina, O.V.; Popadyuk, I.I.; Zakharenko, A.L.; Zakharova, O.D.; Chepanova, A.A.; Dyrkheeva, N.S.; Komarova, N.I.; Reynisson, J.; Anarbaev, R.O.; Salakhutdinov, N.F.; et al. Deoxycholic acid as a molecular scaffold for tyrosyl-DNA phosphodiesterase 1 inhibition: A synthesis, structure–activity relationship and molecular modeling study. Steroids 2021, 165, 108771. [Google Scholar] [CrossRef] [PubMed]
- Sirivolu, V.R.; Vernekar, S.K.V.; Marchand, C.; Naumova, A.; Chergui, A.; Renaud, A.; Stephen, A.G.; Chen, F.; Sham, Y.Y.; Pommier, Y.; et al. 5-Arylidenethioxothiazolidinones as Inhibitors of Tyrosyl-DNA Phosphodiesterase i. J. Med. Chem. 2012, 55, 8671–8684. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, F.-T.; Wu, M.; Wang, W.; Agama, K.; Pommier, Y.; An, L.-K. Synthesis of 11-aminoalkoxy substituted benzophenanthridine derivatives as tyrosyl-DNA phosphodiesterase 1 inhibitors and their anticancer activity. Bioorg. Chem. 2022, 123, 105789. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wang, F.-T.; Si-Tu, M.-X.; Fan, H.; Hu, J.-S.; Yang, H.; Guan, S.-Y.; An, L.-K.; Zhang, C.-X. Pyranodipyran Derivatives with Tyrosyl DNA Phosphodiesterase 1 Inhibitory Activities and Fluorescent Properties from Aspergillus sp. EGF 15-0-3. Mar. Drugs 2022, 20, 211. [Google Scholar] [CrossRef]
- Leung, E.; Patel, J.; Hollywood, J.A.; Zafar, A.; Tomek, P.; Barker, D.; Pilkington, L.I.; van Rensburg, M.; Langley, R.J.; Helsby, N.A.; et al. Validating TDP1 as an Inhibition Target for the Development of Chemosensitizers for Camptothecin-Based Chemotherapy Drugs. Oncol. Ther. 2021, 9, 541–556. [Google Scholar] [CrossRef]
- Mozhaitsev, E.S.; Zakharenko, A.L.; Suslov, E.V.; Korchagina, D.V.; Zakharova, O.D.; Vasil’eva, I.A.; Chepanova, A.A.; Black, E.; Patel, J.; Chand, R.; et al. Novel Inhibitors of DNA Repair Enzyme TDP1 Combining Monoterpenoid and Adamantane Fragments. Anticancer Agents Med. Chem. 2018, 19, 463–472. [Google Scholar] [CrossRef]
- Chepanova, A.A.; Mozhaitsev, E.S.; Munkuev, A.A.; Suslov, E.V.; Korchagina, D.V.; Zakharova, O.D.; Zakharenko, A.L.; Patel, J.; Ayine-Tora, D.M.; Reynisson, J.; et al. The development of Tyrosyl-DNA phosphodiesterase 1 inhibitors. Combination of Monoterpene and Adamantine Moieties via Amide or Thioamide Bridges. Appl. Sci. 2019, 9, 2767. [Google Scholar] [CrossRef] [Green Version]
- Munkuev, A.A.; Mozhaitsev, E.S.; Chepanova, A.A.; Suslov, E.V.; Korchagina, D.V.; Zakharova, O.D.; Ilina, E.S.; Dyrkheeva, N.S.; Zakharenko, A.L.; Reynisson, J.; et al. Novel Tdp1 Inhibitors Based on Adamantane Connected with Monoterpene Moieties via Heterocyclic Fragments. Molecules 2021, 26, 3128. [Google Scholar] [CrossRef]
- Ponomarev, K.; Pavlova, A.; Suslov, E.; Ardashov, O.; Korchagina, D.; Nefedov, A.; Tolstikova, T.; Volcho, K.; Salakhutdinov, N. Synthesis and analgesic activity of new compounds combining azaadamantane and monoterpene moieties. Med. Chem. Res. 2015, 24, 4146–4156. [Google Scholar] [CrossRef]
- Huynh, U.; McDonald, S.L.; Lim, D.; Uddin, M.N.; Wengryniuk, S.E.; Dey, S.; Coltart, D.M. Formation, Alkylation, and Hydrolysis of Chiral Nonracemic N-Amino Cyclic Carbamate Hydrazones: An Approach to the Enantioselective α-Alkylation of Ketones. J. Org. Chem. 2018, 83, 12951–12964. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Faraldos, J.A.; Coates, R.M. Scope and mechanism of intramolecular aziridination of cyclopent-3-enyl-methylamines to 1-azatricyclo[2.2.1.02,6]heptanes with lead tetraacetate. J. Am. Chem. Soc. 2009, 131, 11998–12006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharenko, A.; Khomenko, T.; Zhukova, S.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V.; et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem. 2015, 23, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.W.; Falconi, M.; Kristoffersen, E.L.; Simonsen, A.T.; Cifuentes, J.B.; Marcussen, L.B.; Frøhlich, R.; Vagner, J.; Harmsen, C.; Juul, S.; et al. Real-time detection of TDP1 activity using a fluorophore-quencher coupled DNA-biosensor. Biosens. Bioelectron. 2013, 48, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Antony, S.; Marchand, C.; Stephen, A.G.; Thibaut, L.; Agama, K.K.; Fisher, R.J.; Pommier, Y. Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1. Nucleic Acids Res. 2007, 35, 4474–4484. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. The combination index (CI < 1) as the definition of synergism and of synergy claims. Synergy 2018, 7, 49–50. [Google Scholar] [CrossRef]
- Das, B.B.; Huang, S.N.; Murai, J.; Rehman, I.; Amé, J.C.; Sengupta, S.; Das, S.K.; Majumdar, P.; Zhang, H.; Biard, D.; et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014, 42, 4435–4449. [Google Scholar] [CrossRef]
- Lei, L.; Xie, X.; He, L.; Chen, K.; Lv, Z.; Zhou, B.; Li, Y.; Hu, W.; Zhou, Z. The bromodomain and extra-terminal domain inhibitor JQ1 synergistically sensitizes human colorectal cancer cells to topoisomerase I inhibitors through repression of Mre11-mediated DNA repair pathway. Investig. New Drugs 2021, 39, 362–376. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Kiselev, E.; Lountos, G.T.; Wang, W.; Tropea, J.E.; Needle, D.; Hilimire, T.A.; Schneekloth, J.S.; Waugh, D.S.; Pommier, Y.; et al. Small molecule microarray identifies inhibitors of tyrosyl-DNA phosphodiesterase 1 that simultaneously access the catalytic pocket and two substrate binding sites. Chem. Sci. 2021, 12, 3876–3884. [Google Scholar] [CrossRef]
- Zakharenko, A.; Luzina, O.; Koval, O.; Nilov, D.; Gushchina, I.; Dyrkheeva, N.; Švedas, V.; Salakhutdinov, N.; Lavrik, O. Tyrosyl-DNA Phosphodiesterase 1 Inhibitors: Usnic Acid Enamines Enhance the Cytotoxic Effect of Camptothecin. J. Nat. Prod. 2016, 79, 2961–2967. [Google Scholar] [CrossRef]
- Zhu, F.; Logan, G.; Reynisson, J. Wine compounds as a source for HTS screening collections. A feasibility study. Mol. Inform. 2012, 31, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Eurtivong, C.; Reynisson, J. The Development of a Weighted Index to Optimise Compound Libraries for High Throughput Screening. Mol. Inform. 2019, 38, 1800068. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, A.; Gowri, V.; Neetu, G. Enantioselective syntheses of diquinane and (cis, anti, cis)-linear triquinanes. Tetrahedron Asymmetry 2010, 21, 202–207. [Google Scholar] [CrossRef]
- Keller, F.; Weinmann, H.; Schurig, V. Chiral Polysiloxane-Fixed Metal 1,3-Diketonates (Chirasil-Metals) as Catalytic Lewis Acids for a Hetero Diels-Alder Reaction -Inversion of Enantioselectivity upon Catalyst-Polymer Binding. Chem. Ber. 1997, 130, 879–885. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Scigress Ultra, V.F. J 2.6. (EU 3.1.7); Fujitsu Limited: Tokyo, Japan, 2008–2016. [Google Scholar]
- Allinger, N.L.; Yuh, Y.H.; Lii, J.H. Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 1989, 111, 8551–8566. [Google Scholar] [CrossRef]
- Lii, J.H.; Allinger, N.L. Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 2. Vibrational Frequencies and Thermodynamics. J. Am. Chem. Soc. 1989, 111, 8566–8575. [Google Scholar] [CrossRef]
- Lii, J.H.; Allinger, N.L. Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 3. The van der Waals’ Potentials and Crystal Data for Aliphatic and Aromatic Hydrocarbons. J. Am. Chem. Soc. 1989, 111, 8576–8582. [Google Scholar] [CrossRef]
- Goto, H.; Osawa, E. An efficient algorithm for searching low-energy conformers of cyclic and acyclic molecules. J. Chem. Soc. Perkin Trans. 2 1993, 187–198. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking 1 1Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided. Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced Protein-Ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Mooij, W.T.M.; Verdonk, M.L. General and targeted statistical potentials for protein-ligand interactions. Proteins Struct. Funct. Genet. 2005, 61, 272–287. [Google Scholar] [CrossRef] [PubMed]
- QikProp; Version 6.2; Schrödinger: New York, NY, USA, 2009.
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the reliability of QikProp. correlation between experimental and predicted values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | TDP1 IC50, μM | HeLa CC50, μM | HEK293A CC50, μM | Tpc CC50 HeLa, µM | Tpc CC50 HEK293A, nM | |
|---|---|---|---|---|---|---|
| Tpc | 4.0 ± 1.3 | 66 ± 28 | ||||
| 14a |  | 5.5 ± 1.5 | 53 ± 8 | 47 ± 3 | 0.97 ± 0.23 | 240 ± 60 |
| 14b |  | 6.1 ± 0.1 | 54 ± 7 | 50 ± 3 | 1.2 ± 0.4 | 63 ± 23 |
| 15a |  | 6.1 ± 2.6 | >100 | >100 | 1.7 ± 0.6 | 79 ± 20 |
| 15b |  | 5.6 + 1.9 | >100 | >100 | 2.2 ± 0.8 | 140 ± 45 |
| 16 |  | >30 | 20 ± 8 | 23 ± 10 | 1.6 ± 0.4 | 120 ± 33 |
| 17 |  | >30 | 100 ± 20 | >100 | 1.9 ± 0.7 | 95 ± 25 |
| Furamidine | 1.2 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munkuev, A.A.; Dyrkheeva, N.S.; Kornienko, T.E.; Ilina, E.S.; Ivankin, D.I.; Suslov, E.V.; Korchagina, D.V.; Gatilov, Y.V.; Zakharenko, A.L.; Malakhova, A.A.; et al. Adamantane-Monoterpenoid Conjugates Linked via Heterocyclic Linkers Enhance the Cytotoxic Effect of Topotecan. Molecules 2022, 27, 3374. https://doi.org/10.3390/molecules27113374
Munkuev AA, Dyrkheeva NS, Kornienko TE, Ilina ES, Ivankin DI, Suslov EV, Korchagina DV, Gatilov YV, Zakharenko AL, Malakhova AA, et al. Adamantane-Monoterpenoid Conjugates Linked via Heterocyclic Linkers Enhance the Cytotoxic Effect of Topotecan. Molecules. 2022; 27(11):3374. https://doi.org/10.3390/molecules27113374
Chicago/Turabian StyleMunkuev, Aldar A., Nadezhda S. Dyrkheeva, Tatyana E. Kornienko, Ekaterina S. Ilina, Dmitry I. Ivankin, Evgeniy V. Suslov, Dina V. Korchagina, Yuriy V. Gatilov, Alexandra L. Zakharenko, Anastasia A. Malakhova, and et al. 2022. "Adamantane-Monoterpenoid Conjugates Linked via Heterocyclic Linkers Enhance the Cytotoxic Effect of Topotecan" Molecules 27, no. 11: 3374. https://doi.org/10.3390/molecules27113374
APA StyleMunkuev, A. A., Dyrkheeva, N. S., Kornienko, T. E., Ilina, E. S., Ivankin, D. I., Suslov, E. V., Korchagina, D. V., Gatilov, Y. V., Zakharenko, A. L., Malakhova, A. A., Reynisson, J., Volcho, K. P., Salakhutdinov, N. F., & Lavrik, O. I. (2022). Adamantane-Monoterpenoid Conjugates Linked via Heterocyclic Linkers Enhance the Cytotoxic Effect of Topotecan. Molecules, 27(11), 3374. https://doi.org/10.3390/molecules27113374