
High-Energy Long-Lived Emitting Mixed Excitons in Homopolymeric Adenine-Thymine DNA Duplexes


Abstract
:1. Introduction
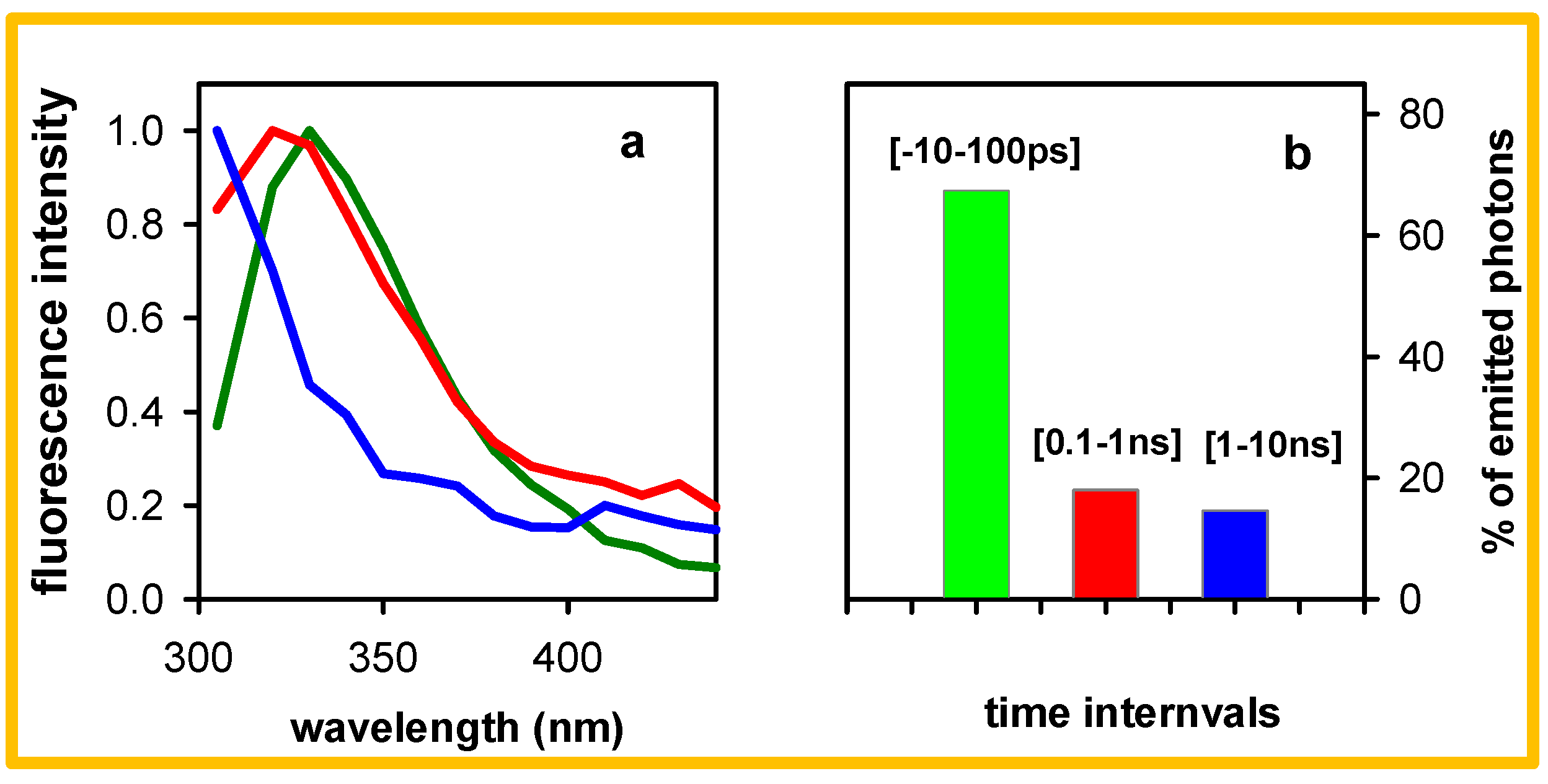
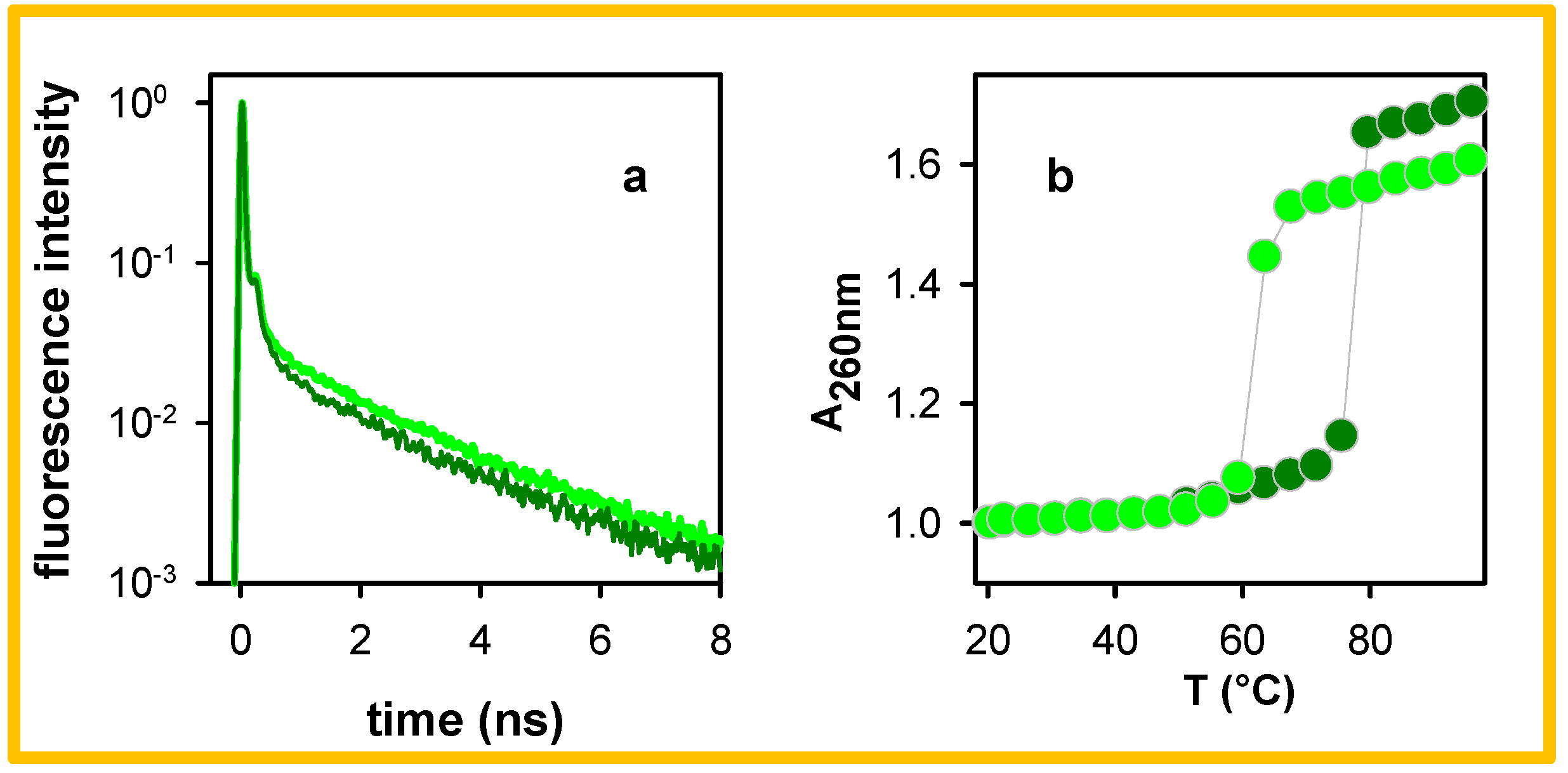
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials and DNA Handling
4.2. Time-Resolved Measurements
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Markovitsi, D.; Sharonov, A.; Onidas, D.; Gustavsson, T. Effect of molecular organisation in DNA oligomers studied by femtosecond fluorescence spectroscopy. ChemPhysChem 2003, 3, 303–305. [Google Scholar] [CrossRef]
- Schwalb, N.K.; Temps, F. Base sequence and higher-order structure induce the complex excited-state dynamics in DNA. Science 2008, 322, 243–245. [Google Scholar] [CrossRef]
- Markovitsi, D.; Gustavsson, T.; Vayá, I. Fluorescence of DNA Duplexes: From Model Helices to Natural DNA. J. Phys. Chem. Lett. 2010, 1, 3271–3276. [Google Scholar] [CrossRef] [Green Version]
- Kwok, W.M.; Ma, C.S.; Phillips, D.L. “Bright” and “Dark” excited states of an alternating AT oligomer characterized by femtosecond broadband spectroscopy. J. Phys. Chem. B 2009, 113, 11527–11534. [Google Scholar] [CrossRef]
- Gustavsson, T.; Markovitsi, D. Fundamentals of the Intrinsic DNA Fluorescence. Acc. Chem. Res. 2021, 54, 1226–1235. [Google Scholar] [CrossRef]
- Crespo-Hernández, C.E.; Cohen, B.; Kohler, B. Base stacking controls excited state dynamics in A-T DNA. Nature 2005, 436, 1141–1144. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.; Kohler, B. Excited states in DNA strands investigated by ultrafast laser spectroscopy. Top. Curr. Chem. 2015, 356, 39–87. [Google Scholar]
- Schreier, W.J.; Gilch, P.; Zinth, W. Early Events of DNA Photodamage. Annu. Rev. Phys. Chem. 2015, 66, 497–519. [Google Scholar] [CrossRef]
- Hanes, A.; Zhang, Y.; Kohler, B. Tracking Excited States in DNA from Formation to Deactivation. In DNA Photodamage: From Light Absorption to Cellular Responses and Skin Cancer; Importa, R., Douk, T., Eds.; RSC: Cambridge, UK, 2021; pp. 37–54. [Google Scholar]
- Cadet, J.; Vigny, P. The Photochemistry of Nucleic Acids. In Bioorganic Photochemistry; Morrison, H., Ed.; John Wiley & Sons: New York, NY, USA, 1990; 272p. [Google Scholar]
- Ge, G.; Georghiou, S. Room-temperature fluorescence properties of the polynucleotide poly(dA).poly(dT). Photochem. Photobiol. 1991, 54, 477–480. [Google Scholar] [CrossRef]
- Ge, G.; Georghiou, S. Excited-state properties of the alternating polynucleotide poly(dA-dT)poly (dA-dT). Photochem. Photobiol. 1991, 54, 301–305. [Google Scholar] [CrossRef]
- Ballini, J.P.; Daniels, M.; Vigny, P. Time-resolved fluorescence emission and excitation spectroscopy of d(TA) and d(AT) using synchrotron radiation. Biophys. Chem. 1991, 39, 253–265. [Google Scholar] [CrossRef]
- Huang, C.-R.; Georghiou, S. Room-temperature steady-state fluorescence properties of poly(dG-dC).poly(dG-dC). Photochem. Photobiol. 1992, 56, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Callis, P.R. Electronic states and luminescence of nucleic acid systems. Annu. Rev. Phys. Chem. 1983, 34, 329–357. [Google Scholar] [CrossRef]
- Ibele, L.; Sánchez-Murcia, P.; Mai, S.; Nogueira, J.; González, L. Excimer Intermediates en Route to Long-Lived Charge-Transfer States in Single-Stranded Adenine DNA as Revealed by Nonadiabatic Dynamics. J. Phys. Chem. Lett. 2020, 11, 7483–7488. [Google Scholar] [CrossRef]
- Zhang, Y.; de La Harpe, K.; Beckstead, A.A.; Improta, R.; Kohler, B. UV-Induced Proton Transfer between DNA Strands. J. Am. Chem. Soc. 2015, 137, 7059–7062. [Google Scholar] [CrossRef] [Green Version]
- Improta, R.; Barone, V. Interplay between “neutral” and “charge-transfer” excimers rules the excited state decay in adenine-rich polynucleotides. Angew. Chem. Int. Ed. 2011, 50, 12016–12019. [Google Scholar] [CrossRef]
- Spata, V.A.; Matsika, S. Role of Excitonic Coupling and Charge-Transfer States in the Absorption and CD Spectra of Adenine-Based Oligonucleotides Investigated through QM/MM Simulations. J. Phys. Chem. A 2014, 118, 12021–12030. [Google Scholar] [CrossRef]
- Improta, R.; Santoro, F.; Blancafort, L. Quantum Mechanical Studies on the Photophysics and the Photochemistry of Nucleic Acids and Nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef]
- Martinez-Fernandez, L.; Changenet, P.; Banyasz, A.; Gustavsson, T.; Markovitsi, D.; Improta, R. A Comprehensive Study of Guanine Excited State Relaxation and Photoreactivity in G-Quadruplexes. J. Phys. Chem. Lett. 2019, 10, 6873–6877. [Google Scholar] [CrossRef]
- Vayá, I.; Miannay, F.A.; Gustavsson, T.; Markovitsi, D. High energy long-lived excited states in DNA double strands. ChemPhysChem 2010, 11, 987–989. [Google Scholar] [CrossRef] [Green Version]
- Brazard, J.; Thazhathveetil, A.; Vayá, I.; Lewis, F.; Gustavsson, T.; Markovitsi, D. Electronic excited states of guanine-cytosine hairpins and duplexes studied by fluorescence spectroscopy. Photochem. Photobiol. Sci. 2013, 12, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Huix-Rotllant, M.; Brazard, J.; Improta, R.; Burghardt, I.; Markovitsi, D. Stabilization of mixed Frenkel-charge transfer excitons extended across both strands of guanine-cytosine DNA duplexes. J. Phys. Chem. Lett. 2015, 6, 2247–2251. [Google Scholar] [CrossRef] [PubMed]
- Vayá, I.; Brazard, J.; Huix-Rotllant, M.; Thazhathveetil, A.; Lewis, F.; Gustavsson, T.; Burghardt, I.; Improta, R.; Markovitsi, D. High energy long-lived mixed Frenkel—Charge transfer excitons: From double-stranded (AT)n to natural DNA. Chem. Eur. J. 2016, 22, 4904–4914. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.C.-T.; Ma, C.; Wong, A.K.-W.; Chan, C.T.-L.; Chow, J.C.-L.; Kwok, W.-M. Dual Time-Scale Proton Transfer and High-Energy, Long-Lived Excitons Unveiled by Broadband Ultrafast Time-Resolved Fluorescence in Adenine-Uracil RNA Duplexes. J. Phys. Chem. Lett. 2022, 13, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Markovitsi, D.; Onidas, D.; Gustavsson, T.; Talbot, F.; Lazzarotto, E. Collective behavior of Franck-Condon excited states and energy transfer in DNA double helices. J. Am. Chem. Soc. 2005, 127, 17130–17131. [Google Scholar] [CrossRef]
- Markovitsi, D.; Talbot, F.; Gustavsson, T.; Onidas, D.; Lazzarotto, E.; Marguet, S. Complexity of excited state dynamics in DNA. Nature 2006, 441, E7. [Google Scholar] [CrossRef] [Green Version]
- Markovitsi, D.; Gustavsson, T.; Talbot, F. Excited states and energy transfer among DNA bases in double helices. Photochem. Photobiol. Sci. 2007, 6, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Onidas, D.; Gustavsson, T.; Lazzarotto, E.; Markovitsi, D. Fluorescence of the DNA double helix (dA)20.(dT)20 studied by femtosecond spectroscopy—Effect of the duplex size on the properties of the excited states. J. Phys. Chem. B 2007, 111, 9644–9650. [Google Scholar] [CrossRef] [Green Version]
- Nelson, H.C.M.; Finch, J.T.; Luisi, B.F.; Klug, A. The structure of an oligo(dA).oligo(dT) tract and its biological implications. Nature 1987, 330, 221–226. [Google Scholar] [CrossRef]
- Alexeev, D.G.; Lipanov, A.A.; Skuratovskii, I.Y. poly(dA).poly(dT) is a B-type double helix with a distinctively norrow minor grrove. Nature 1987, 325, 821–823. [Google Scholar] [CrossRef]
- Kypr, J.; Kejnovska, I.; Renciuk, D.; Vorlickova, M. Circular dichroism and conformational polymorphism of DNA. Nucl. Acids Res. 2009, 37, 1713–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onidas, D. Effets Coopératifs dans les Etats Excités Singulets des Doubles Hélices Modèles d’ADN. Ph.D. Thesis, Université Paris 11, en Chimie—Chimie Physique, Orsay, France, 2004. [Google Scholar]
- Bouvier, B.; Gustavsson, T.; Markovitsi, D.; Millié, P. Dipolar coupling between electronic transitions of the DNA bases and its relevance to exciton states in double helices. Chem. Phys. 2002, 275, 75–92. [Google Scholar] [CrossRef]
- Bouvier, B.; Dognon, J.P.; Lavery, R.; Markovitsi, D.; Millié, P.; Onidas, D.; Zakrzewska, K. Influence of conformational dynamics on the exciton states of DNA oligomers. J. Phys. Chem. B 2003, 107, 13512–13522. [Google Scholar] [CrossRef]
- Lange, A.W.; Herbert, J.M. Both intra- and interstrand charge-transfer excited states in aqueous B-DNA are present at energies comparable to, or just above, the 1ππ* excitonic bright states. J. Am. Chem. Soc. 2009, 131, 3913–3922. [Google Scholar] [CrossRef] [PubMed]
- Voityuk, A.A. Conformational dependence of the electronic coupling for singlet excitation energy transfer in DNA. An INDO/S study. Phys. Chem. Chem. Phys. 2010, 12, 7403–7408. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.-J.; Cui, G.-L.; Fang, Q.; Fang, W.-H.; Luo, Y. Exploring Concerted Effects of Base Pairing and Stacking on the Excited-State Nature of DNA Oligonucleotides by DFT and TD-DFT Studies. Int. J. Quantum Chem. 2011, 111, 2366–2377. [Google Scholar] [CrossRef]
- Plasser, F.; Aquino, A.; Lischka, H.; Nachtigallová, D. Electronic Excitation Processes in Single-Strand and Double-Strand DNA: A Computational Approach. Top. Curr. Chem. 2015, 356, 1–38. [Google Scholar]
- Lu, Y.; Lan, Z.; Thiel, W. Computational modeling of photoexcitation in DNA single and double strands. Top. Curr. Chem. 2015, 356, 89–122. [Google Scholar]
- Markovitsi, D. Interaction of UV radiation with DNA helices. Pure Appl. Chem. 2009, 81, 1635–1644. [Google Scholar] [CrossRef]
- Starikov, E.B. Importance of charge transfer excitations in DNA electron spectrum: A ZINDO semiempirical quantum-chemical study. Mod. Phys. Lett. B 2004, 18, 825–831. [Google Scholar] [CrossRef]
- Varsano, D.; Di Felice, R.; Marques, M.A.L.; Rubio, A. A TDDFT study of the excited states of DNA bases and their assemblies. J. Phys. Chem. B 2006, 110, 7129–7138. [Google Scholar] [CrossRef] [PubMed]
- Santoro, F.; Barone, V.; Improta, R. Influence of base stacking on excited-state behavior of polyadenine in water, base on time-dependent density functional calculations. Proc. Natl. Acad. Sci. USA 2007, 104, 9931–9936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, C.R.; Kistler, K.A.; Lu, Z.; Matsika, S. Excited-State Energies and Electronic Couplings of DNA Base Dimers. J. Phys. Chem. B 2010, 114, 1674–1683. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, J.J.; Plasser, F.; Gonzalez, L. Electronic delocalization, charge transfer and hypochromism in the UV absorption spectrum of polyadenine unravelled by multiscale computations and quantitative wavefunction analysis. Chem. Sci. 2017, 8, 5682–5691. [Google Scholar] [CrossRef] [Green Version]
- Bittner, E.R. Frenkel exciton model of ultrafast excited state dynamics in AT DNA double helices. J. Photochem. Photobiol. A Chem. 2007, 190, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, A.G.; Tanimura, Y. Correlated fluctuations in the exciton dynamics and spectroscopy of DNA. New J. Phys. 2010, 12, 055005. [Google Scholar] [CrossRef]
- Chen, J.; Thazhathveetil, A.K.; Lewis, F.D.; Kohler, B. Ultrafast Excited-State Dynamics in Hexaethyleneglycol-Linked DNA Homoduplexes Made of A.T Base Pairs. J. Am. Chem. Soc. 2014, 135, 10290–10293. [Google Scholar] [CrossRef]
- Onidas, D.; Markovitsi, D.; Marguet, S.; Sharonov, A.; Gustavsson, T. Fluorescence properties of DNA nucleosides and nucleotides: A refined steady-state and femtosecond investigation. J. Phys. Chem. B 2002, 106, 11367–11374. [Google Scholar] [CrossRef]
- Zuo, X.; Cui, G.; Merz, K.M., Jr.; Zhang, L.; Lewis, F.D.; Tiede, D.M. X-ray diffraction “fingerprinting” of DNA structure in solution for quantitative evaluation of molecular dynamics simulation. Proc. Natl. Acad. Sci. USA 2006, 103, 3534–3539. [Google Scholar] [CrossRef] [Green Version]
- Hithell, G.; Donaldson, P.M.; Greetham, G.M.; Towrie, M.; Parker, A.W.; Burley, G.A.; Hunt, N.T. Effect of oligomer length on vibrational coupling and energy relaxation in double-stranded DNA. Chem. Phys. 2018, 512, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, A.C. Polarizations and assignments of transitions—Method of photoselection. J. Mol. Spectrosc. 1961, 6, 84–108. [Google Scholar] [CrossRef]
- McCullagh, M.; Lewis, F.; Markovitsi, D.; Douki, T.; Schatz, G.C. Conformational control of TT dimerization in DNA conjugates. A molecular dynamics study. J. Phys. Chem. B 2010, 114, 5215–5221. [Google Scholar] [CrossRef]
- Banyasz, A.; Ketola, T.; Muñoz-Losa, A.; Rishi, S.; Adhikary, A.; Sevilla, M.D.; Martinez-Fernandez, L.; Improrta, R.; Markovitsi, D. UV-induced Adenine Radicals Induced in DNA A-tracts: Spectral and Dynamical Characterization. J. Phys. Chem. Lett. 2016, 7, 3949–3953. [Google Scholar] [CrossRef] [Green Version]
- Markovitsi, D.; Onidas, D.; Talbot, F.; Marguet, S.; Gustavsson, T.; Lazzarotto, E. UVB/UVC induced processes in model DNA helices studied by time-resolved spectroscopy: Pitfalls and tricks. J. Photochem. Photobiol. A Chem. 2006, 183, 1–8. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Emission Wavelength | pA●pT (Standard Buffer) | A20●T20 (Standard Buffer) | pA●pT (Sixfold Diluted Buffer) |
|---|---|---|---|
| 305 nm | 27.5% | 50.1% | 31.7% |
| 330 nm | 12.5% | 30.6% | 15.7% |
| Excitation | pA●pT | A20●T20 |
|---|---|---|
| 267 nm | 5.6 (2.6 ns) | - |
| 285 nm | 3.6 (2.2 ns) | 3.7 (2.7 ns) 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vayá, I.; Gustavsson, T.; Markovitsi, D. High-Energy Long-Lived Emitting Mixed Excitons in Homopolymeric Adenine-Thymine DNA Duplexes. Molecules 2022, 27, 3558. https://doi.org/10.3390/molecules27113558
Vayá I, Gustavsson T, Markovitsi D. High-Energy Long-Lived Emitting Mixed Excitons in Homopolymeric Adenine-Thymine DNA Duplexes. Molecules. 2022; 27(11):3558. https://doi.org/10.3390/molecules27113558
Chicago/Turabian StyleVayá, Ignacio, Thomas Gustavsson, and Dimitra Markovitsi. 2022. "High-Energy Long-Lived Emitting Mixed Excitons in Homopolymeric Adenine-Thymine DNA Duplexes" Molecules 27, no. 11: 3558. https://doi.org/10.3390/molecules27113558
APA StyleVayá, I., Gustavsson, T., & Markovitsi, D. (2022). High-Energy Long-Lived Emitting Mixed Excitons in Homopolymeric Adenine-Thymine DNA Duplexes. Molecules, 27(11), 3558. https://doi.org/10.3390/molecules27113558