
Beyond Moco Biosynthesis―Moonlighting Roles of MoaE and MOCS2


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Discovery of MoaE in a Histone Acetyltransferase Complex in Drosophila
3. Evolution of MoaE and MBIP
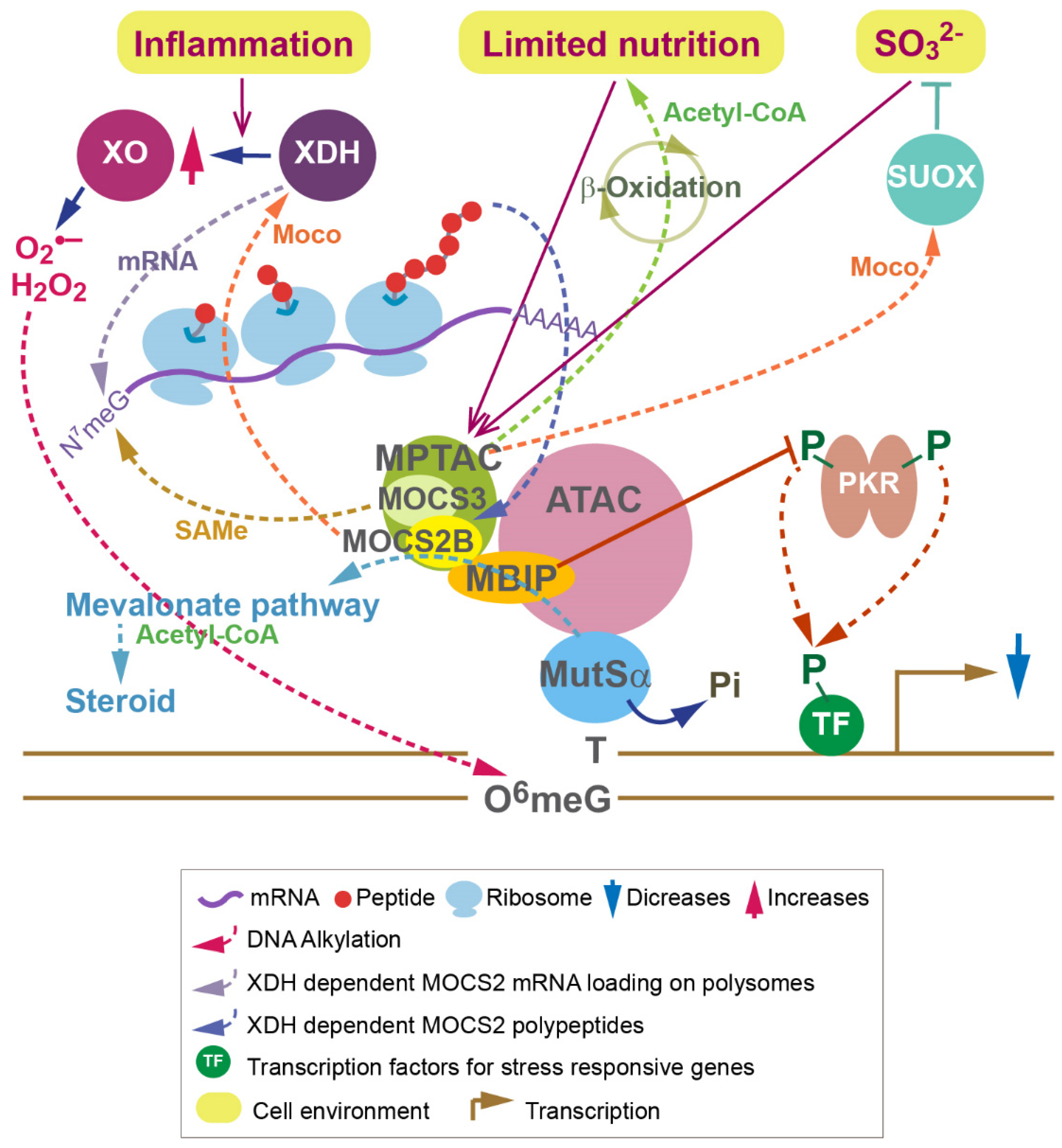
4. Roles of MoaE and MOCS2 in Signaling in Drosophila and Humans
5. MPT Synthase and ATAC Coordinate Transcription and Translation in Humans
6. Functions of MPTAC in Metabolism Are Disrupted in Dementia
7. The Association of ATAC with MPTAC Is Lost in Neurodegenerative Diseases
8. Prospects
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suganuma, T.; Workman, J.L. Chromatin and metabolism. Annu. Rev. Biochem. 2018, 87, 27–49. [Google Scholar] [CrossRef] [PubMed]
- McGhee, J.D.; Felsenfeld, G. Nucleosome structure. Annu. Rev. Biochem. 1980, 49, 1115–1156. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D.; Lorch, Y. Chromatin structure and transcription. Annu. Rev. Cell Biol. 1992, 8, 563–587. [Google Scholar] [CrossRef]
- Bennett, R.L.; Licht, J.D. Targeting epigenetics in cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 187–207. [Google Scholar] [CrossRef]
- Suganuma, T.; Workman, J.L. Nucleotide metabolism behind epigenetics. Front. Endocrinol. 2021, 12, 731648. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, T.; Workman, J.L. Signals and combinatorial functions of histone modifications. Annu. Rev. Biochem. 2011, 80, 473–499. [Google Scholar] [CrossRef]
- Suganuma, T.; Workman, J.L. Crosstalk among histone modifications. Cell 2008, 135, 604–607. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Swanson, S.K.; Gogol, M.; Florens, L.; Washburn, M.P.; Workman, J.L.; Suganuma, T. Serine and SAM responsive complex SESAME regulates histone modification crosstalk by Sensing cellular metabolism. Mol. Cell 2015, 60, 408–421. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, T.; Gutierrez, J.L.; Li, B.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Abmayr, S.M.; Workman, J.L. ATAC is a double histone acetyltransferase complex that stimulates nucleosome sliding. Nat. Struct. Mol. Biol. 2008, 16515, 364–372. [Google Scholar] [CrossRef]
- Leimkuhler, S. Shared function and moonlighting proteins in molybdenum cofactor biosynthesis. Biol. Chem. 2017, 398, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Reiss, J.; Johnson, J.L. Mutations in the molybdenum cofactor biosynthetic genes MOCS1, MOCS2, and GEPH. Hum. Mutat. 2003, 21, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Yamamoto, K.; Staub, A.; Tora, L. Identification of TATA-binding protein-free TAFII-containing complex subunits suggests a role in nucleosome acetylation and signal transduction. J. Biol. Chem. 1999, 274, 18285–18289. [Google Scholar] [CrossRef] [Green Version]
- Grant, P.A.; Duggan, L.; Cote, J.; Roberts, S.M.; Brownell, J.E.; Candau, R.; Ohba, R.; Owen-Hughes, T.; Allis, C.D.; Winston, F.; et al. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: Characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. 1997, 11, 1640–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pray-Grant, M.G.; Schieltz, D.; McMahon, S.J.; Wood, J.M.; Kennedy, E.L.; Cook, R.G.; Workman, J.L.; Yates, J.R., 3rd; Grant, P.A. The novel SLIK histone acetyltransferase complex functions in the yeast retrograde response pathway. Mol. Cell. Biol. 2002, 22, 8774–8786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, E.; Kundu, T.K.; Fu, J.; Roeder, R.G. A human SPT3-TAFII31-GCN5-L acetylase complex distinct from transcription factor IID. J. Biol. Chem. 1998, 273, 23781–23785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.L.; Faiola, F.; Xu, M.; Pan, S.; Martinez, E. Human ATAC Is a GCN5/PCAF-containing acetylase complex with a novel NC2-like histone fold module that interacts with the TATA-binding protein. J. Biol. Chem. 2008, 283, 33808–33815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guelman, S.; Kozuka, K.; Mao, Y.; Pham, V.; Solloway, M.J.; Wang, J.; Wu, J.; Lill, J.R.; Zha, J. The double-histone-acetyltransferase complex ATAC is essential for mammalian development. Mol. Cell. Biol. 2009, 29, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, T.; Mushegian, A.; Swanson, S.K.; Abmayr, S.M.; Florens, L.; Washburn, M.P.; Workman, J.L. The ATAC acetyltransferase complex coordinates MAP kinases to regulate JNK target genes. Cell 2010, 142, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, M.J.; Wuebbens, M.M.; Rajagopalan, K.V.; Schindelin, H. Crystal structure of molybdopterin synthase and its evolutionary relationship to ubiquitin activation. Nat. Struct. Biol. 2001, 8, 42–46. [Google Scholar] [CrossRef]
- Suganuma, T.; Mushegian, A.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Workman, J.L. A metazoan ATAC acetyltransferase subunit that regulates mitogen-activated protein kinase signaling is related to an ancient molybdopterin synthase component. Mol. Cell. Proteom. 2012, 11, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nadal, E.; Alepuz, P.M.; Posas, F. Dealing with osmostress through MAP kinase activation. EMBO Rep. 2002, 3, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Edmunds, J.W.; Mahadevan, L.C. MAP kinases as structural adaptors and enzymatic activators in transcription complexes. J. Cell Sci. 2004, 117, 3715–3723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayali, A.G.; Austin, D.A.; Webster, N.J. Stimulation of MAPK cascades by insulin and osmotic shock: Lack of an involvement of p38 mitogen-activated protein kinase in glucose transport in 3T3-L1 adipocytes. Diabetes 2000, 49, 1783–1793. [Google Scholar] [CrossRef] [Green Version]
- Thomson, S.; Mahadevan, L.C.; Clayton, A.L. MAP kinase-mediated signalling to nucleosomes and immediate-early gene induction. Semin. Cell Dev. Biol. 1999, 10, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.C.; Shao, C.; McGlynn, K.; Naziruddin, B.; Levy, M.F.; Cobb, M.H. Multiple chromatin-bound protein kinases assemble factors that regulate insulin gene transcription. Proc. Natl. Acad. Sci. USA 2009, 106, 22181–22186. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Yoshida, M.; Yamashita, A.; Deyama, T.; Baba, M.; Suzuki, A.; Mohri, H.; Ikezawa, Z.; Nakajima, H.; Hirai, S.; et al. MAPK upstream kinase (MUK)-binding inhibitory protein, a negative regulator of MUK/dual leucine zipper-bearing kinase/leucine zipper protein kinase. J. Biol. Chem. 2000, 275, 21247–21254. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Jiang, F.; Sluss, H.K.; Zhang, C.; Shokat, K.M.; Flavell, R.A.; Davis, R.J. Suppression of p53-dependent senescence by the JNK signal transduction pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 15759–15764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexaki, V.I.; Javelaud, D.; Mauviel, A. JNK supports survival in melanoma cells by controlling cell cycle arrest and apoptosis. Pigment. Cell Melanoma Res. 2008, 21, 429–438. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Schuierer, G.; Kurlemann, G.; Bick, U.; Stephani, U. Molybdenum-cofactor deficiency: CT and MR findings. Neuropediatrics 1995, 26, 51–54. [Google Scholar] [CrossRef]
- Nguyen, L.; Thomas, K.L.; Lucke-Wold, B.P.; Cavendish, J.Z.; Crowe, M.S.; Matsumoto, R.R. Dextromethorphan: An update on its utility for neurological and neuropsychiatric disorders. Pharmacol. Ther. 2016, 159, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Netzer, R.; Pflimlin, P.; Trube, G. Dextromethorphan blocks N-methyl-D-aspartate-induced currents and voltage-operated inward currents in cultured cortical neurons. Eur. J. Pharmacol. 1993, 238, 209–216. [Google Scholar] [CrossRef]
- Schenkel, L.B.; Molina, J.R.; Swinger, K.K.; Abo, R.; Blackwell, D.J.; Lu, A.Z.; Cheung, A.E.; Church, W.D.; Kunii, K.; Kuplast-Barr, K.G.; et al. A potent and selective PARP14 inhibitor decreases protumor macrophage gene expression and elicits inflammatory responses in tumor explants. Cell Chem. Biol. 2021, 28, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Church, M.C.; Workman, J.L.; Suganuma, T. Macrophages, Metabolites, and nucleosomes: Chromatin at the intersection between aging and inflammation. Int. J. Mol. Sci. 2021, 22, 10274. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Nakamura, T.; Furuhashi, M.; Li, P.; Cao, H.; Tuncman, G.; Sonenberg, N.; Gorgun, C.Z.; Hotamisligil, G.S. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010, 140, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Samuel, C.E. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 1993, 268, 7603–7606. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011, 75, 434–467. [Google Scholar] [CrossRef] [Green Version]
- Aitken, C.E.; Lorsch, J.R. A mechanistic overview of translation initiation in eukaryotes. Nat. Struct. Mol. Biol. 2012, 19, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, T.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Workman, J.L. Moco biosynthesis and the ATAC acetyltransferase engage translation initiation by inhibiting latent PKR activity. J. Mol. Cell Biol. 2016, 8, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganuma, T.; Swanson, S.K.; Gogol, M.; Garrett, T.J.; Florens, L.; Workman, J.L. MOCS2 links nucleotide metabolism to nucleoli function. J. Mol. Cell Biol. 2021, 13, 838–840. [Google Scholar] [CrossRef] [PubMed]
- Noma, A.; Sakaguchi, Y.; Suzuki, T. Mechanistic characterization of the sulfur-relay system for eukaryotic 2-thiouridine biogenesis at tRNA wobble positions. Nucleic Acids Res. 2009, 37, 1335–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, Y.; Nakai, M.; Lill, R.; Suzuki, T.; Hayashi, H. Thio modification of yeast cytosolic tRNA is an iron-sulfur protein-dependent pathway. Mol. Cell. Biol. 2007, 27, 2841–2847. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.; Chowdhury, M.M.; Hanzelmann, P.; Nimtz, M.; Lee, E.Y.; Schindelin, H.; Leimkuhler, S. The sulfurtransferase activity of Uba4 presents a link between ubiquitin-like protein conjugation and activation of sulfur carrier proteins. Biochemistry 2008, 47, 6479–6489. [Google Scholar] [CrossRef]
- Schlieker, C.D.; Van der Veen, A.G.; Damon, J.R.; Spooner, E.; Ploegh, H.L. A functional proteomics approach links the ubiquitin-related modifier Urm1 to a tRNA modification pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 18255–18260. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, M.M.; Dosche, C.; Lohmannsroben, H.G.; Leimkuhler, S. Dual role of the molybdenum cofactor biosynthesis protein MOCS3 in tRNA thiolation and molybdenum cofactor biosynthesis in humans. J. Biol. Chem. 2012, 287, 17297–17307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Wuebbens, M.M.; Rajagopalan, K.V.; Fitzgerald, M.C. Thermodynamic analysis of subunit interactions in Escherichia coli molybdopterin synthase. Biochemistry 2005, 44, 2595–2601. [Google Scholar] [CrossRef]
- Rudolph, M.J.; Wuebbens, M.M.; Turque, O.; Rajagopalan, K.V.; Schindelin, H. Structural studies of molybdopterin synthase provide insights into its catalytic mechanism. J. Biol. Chem. 2003, 278, 14514–14522. [Google Scholar] [CrossRef] [Green Version]
- Laxman, S.; Sutter, B.M.; Wu, X.; Kumar, S.; Guo, X.; Trudgian, D.C.; Mirzaei, H.; Tu, B.P. Sulfur amino acids regulate translational capacity and metabolic homeostasis through modulation of tRNA thiolation. Cell 2013, 154, 416–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, K.; Mizushima, N.; Noda, T.; Ohsumi, Y. A protein conjugation system in yeast with homology to biosynthetic enzyme reaction of prokaryotes. J. Biol. Chem. 2000, 275, 7462–7465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganuma, T.; Swanson, S.K.; Gogol, M.; Garrett, T.J.; Conkright-Fincham, J.; Florens, L.; Washburn, M.P.; Workman, J.L. MPTAC Determines APP fragmentation via sensing sulfur amino acid catabolism. Cell Rep. 2018, 24, 1585–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chari, A.; Golas, M.M.; Klingenhager, M.; Neuenkirchen, N.; Sander, B.; Englbrecht, C.; Sickmann, A.; Stark, H.; Fischer, U. An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell 2008, 135, 497–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, K.; Hynan, L.S.; Baskin, F.; Rosenberg, R.N. Platelet amyloid precursor protein processing: A bio-marker for Alzheimer’s disease. J. Neurol. Sci. 2006, 240, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Li, Y.; Wang, R. Drebrin and cognitive impairment. Clin. Chim. Acta 2015, 451, 121–124. [Google Scholar] [CrossRef]
- Gao, M.; Fritz, D.T.; Ford, L.P.; Wilusz, J. Interaction between a poly(A)-specific ribonuclease and the 5′ cap influences mRNA deadenylation rates in vitro. Mol. Cell. 2000, 5, 479–488. [Google Scholar] [CrossRef]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine oxidoreductase-catalyzed reactive species generation: A process in critical need of reevaluation. Redox Biol. 2013, 1, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Soll, J.M.; Sobol, R.W.; Mosammaparast, N. Regulation of DNA alkylation damage repair: Lessons and therapeutic opportunities. Trends Biochem. Sci. 2017, 42, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, T.; Workman, J.L. MPTAC links alkylation damage signaling to sterol biosynthesis. Redox Biol. 2022, 51, 102270. [Google Scholar] [CrossRef]
- Kaliyaperumal, S.; Patrick, S.M.; Williams, K.J. Phosphorylated hMSH6: DNA mismatch versus DNA damage recognition. Mutat. Res. 2011, 706, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Hagerman, P.J.; Hagerman, R.J. Fragile X-associated tremor/ataxia syndrome. Ann. N. Y. Acad. Sci. 2015, 1338, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.C.; Acuna, J.M.; Sherman, S.L. FMR1 and the fragile X syndrome: Human genome epidemiology review. Genet. Med. Off. J. Am. Coll. Med. Genet. 2001, 3, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Levin, R.; Shah, H.; Mathur, S.; Darnell, J.C.; Ouyang, B. Cholesterol levels in fragile X syndrome. Am. J. Med. Genet. Part. A 2015, 167, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalnapurkar, I.; Rafika, N.; Tassone, F.; Hagerman, R. Immune mediated disorders in women with a fragile X expansion and FXTAS. Am. J. Med. Genet. Part. A 2015, 167, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Westmark, C.J.; Westmark, P.R.; O’Riordan, K.J.; Ray, B.C.; Hervey, C.M.; Salamat, M.S.; Abozeid, S.H.; Stein, K.M.; Stodola, L.A.; Tranfaglia, M.; et al. Reversal of fragile X phenotypes by manipulation of AbetaPP/Abeta levels in Fmr1KO mice. PLoS ONE 2011, 6, e26549. [Google Scholar] [CrossRef] [Green Version]
- Feron, F.; Gepner, B.; Lacassagne, E.; Stephan, D.; Mesnage, B.; Blanchard, M.P.; Boulanger, N.; Tardif, C.; Deveze, A.; Rousseau, S.; et al. Olfactory stem cells reveal MOCOS as a new player in autism spectrum disorders. Mol. Psych. 2016, 21, 1215–1224. [Google Scholar] [CrossRef] [Green Version]
- Ichida, K.; Matsumura, T.; Sakuma, R.; Hosoya, T.; Nishino, T. Mutation of human molybdenum cofactor sulfurase gene is responsible for classical xanthinuria type II. Biochem. Biophys. Res. Commun. 2001, 282, 1194–1200. [Google Scholar] [CrossRef]
- Tejada-Jimenez, M.; Chamizo-Ampudia, A.; Calatrava, V.; Galvan, A.; Fernandez, E.; Llamas, A. From the eukaryotic molybdenum cofactor biosynthesis to the moonlighting enzyme mARC. Molecules 2018, 23, 3287. [Google Scholar] [CrossRef] [Green Version]
- Rontani, P.; Perche, O.; Greetham, L.; Jullien, N.; Gepner, B.; Feron, F.; Nivet, E.; Erard-Garcia, M. Impaired expression of the COSMOC/MOCOS gene unit in ASD patient stem cells. Mol. Psych. 2021, 26, 1606–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, S.J.; Sass, J.O.; Vry, J.; Kirschner, J.; Mader, I.; Hovener, J.B.; Reiss, J.; Santamaria-Araujo, J.A.; Schwarz, G.; Grunert, S.C. A mild case of molybdenum cofactor deficiency defines an alternative route of MOCS1 protein maturation. J. Inherit. Metab. Dis. 2018, 41, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Ngu, L.H.; Afroze, B.; Chen, B.C.; Affandi, O.; Zabedah, M.Y. Molybdenum cofactor deficiency in a Malaysian child. Singap. Med. J. 2009, 50, e365–e367. [Google Scholar]
- Toczek, M.; Zielonka, D.; Zukowska, P.; Marcinkowski, J.T.; Slominska, E.; Isalan, M.; Smolenski, R.T.; Mielcarek, M. An impaired metabolism of nucleotides underpins a novel mechanism of cardiac remodeling leading to Huntington’s disease related cardiomyopathy. Biochim. Biophys. Acta 2016, 1862, 2147–2157. [Google Scholar] [CrossRef]
- Everett, C.M.; Wood, N.W. Trinucleotide repeats and neurodegenerative disease. Brain A J. Neurol. 2004, 127, 2385–2405. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Aihara, Y.; Endo, W.; Hasegawa, H.; Ichida, K.; Uematsu, M.; Kure, S. The effect of dietary protein restriction in a case of molybdenum cofactor deficiency with MOCS1 mutation. Mol. Genet. Metab. Rep. 2021, 26, 100716. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suganuma, T. Beyond Moco Biosynthesis―Moonlighting Roles of MoaE and MOCS2. Molecules 2022, 27, 3733. https://doi.org/10.3390/molecules27123733
Suganuma T. Beyond Moco Biosynthesis―Moonlighting Roles of MoaE and MOCS2. Molecules. 2022; 27(12):3733. https://doi.org/10.3390/molecules27123733
Chicago/Turabian StyleSuganuma, Tamaki. 2022. "Beyond Moco Biosynthesis―Moonlighting Roles of MoaE and MOCS2" Molecules 27, no. 12: 3733. https://doi.org/10.3390/molecules27123733
APA StyleSuganuma, T. (2022). Beyond Moco Biosynthesis―Moonlighting Roles of MoaE and MOCS2. Molecules, 27(12), 3733. https://doi.org/10.3390/molecules27123733