
The LAM of the Rings: Large Amplitude Motions in Aromatic Molecules Studied by Microwave Spectroscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
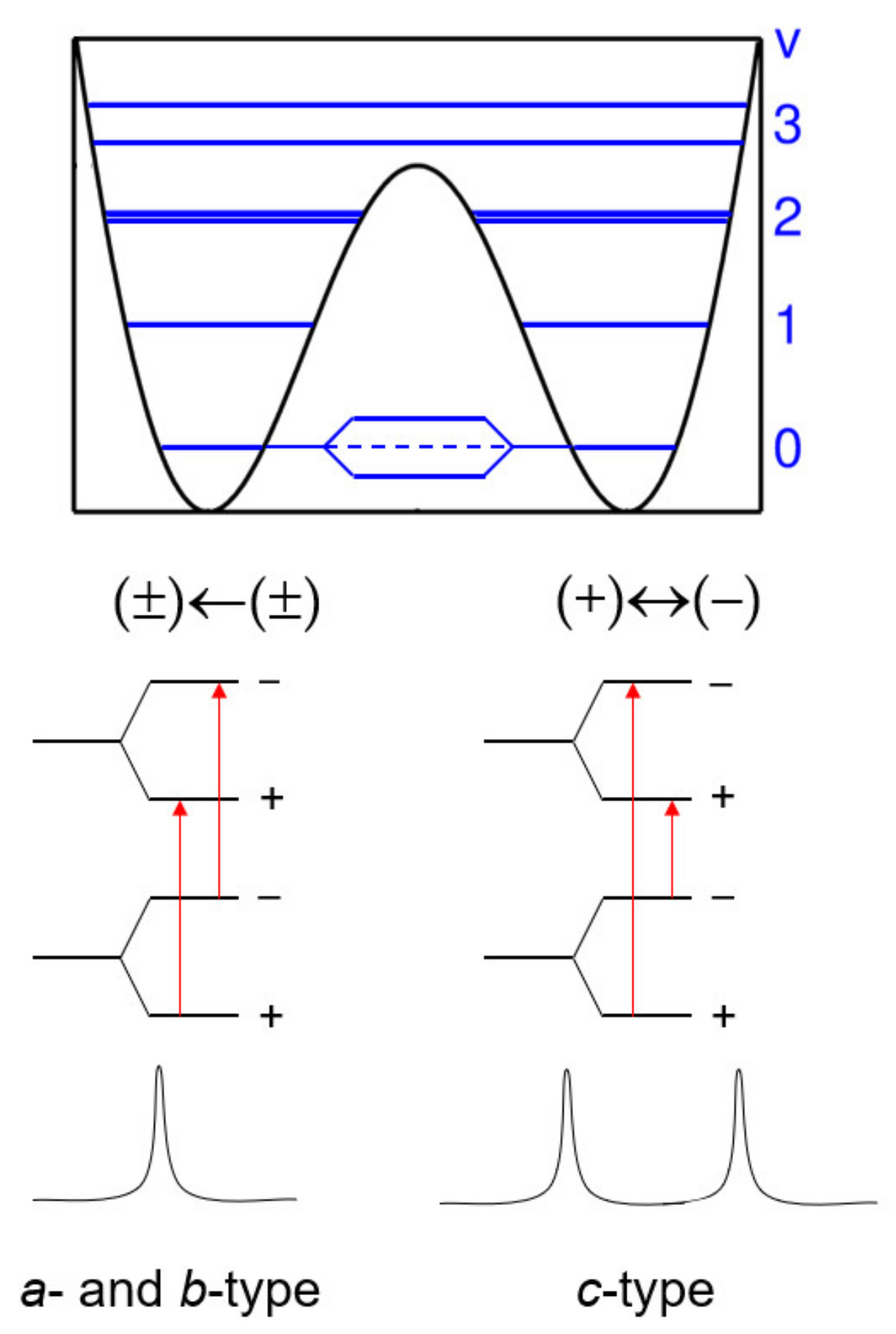
2. Internal Rotation
2.1. Monomethyl-Substituted (One-Top) Aromatic Rings
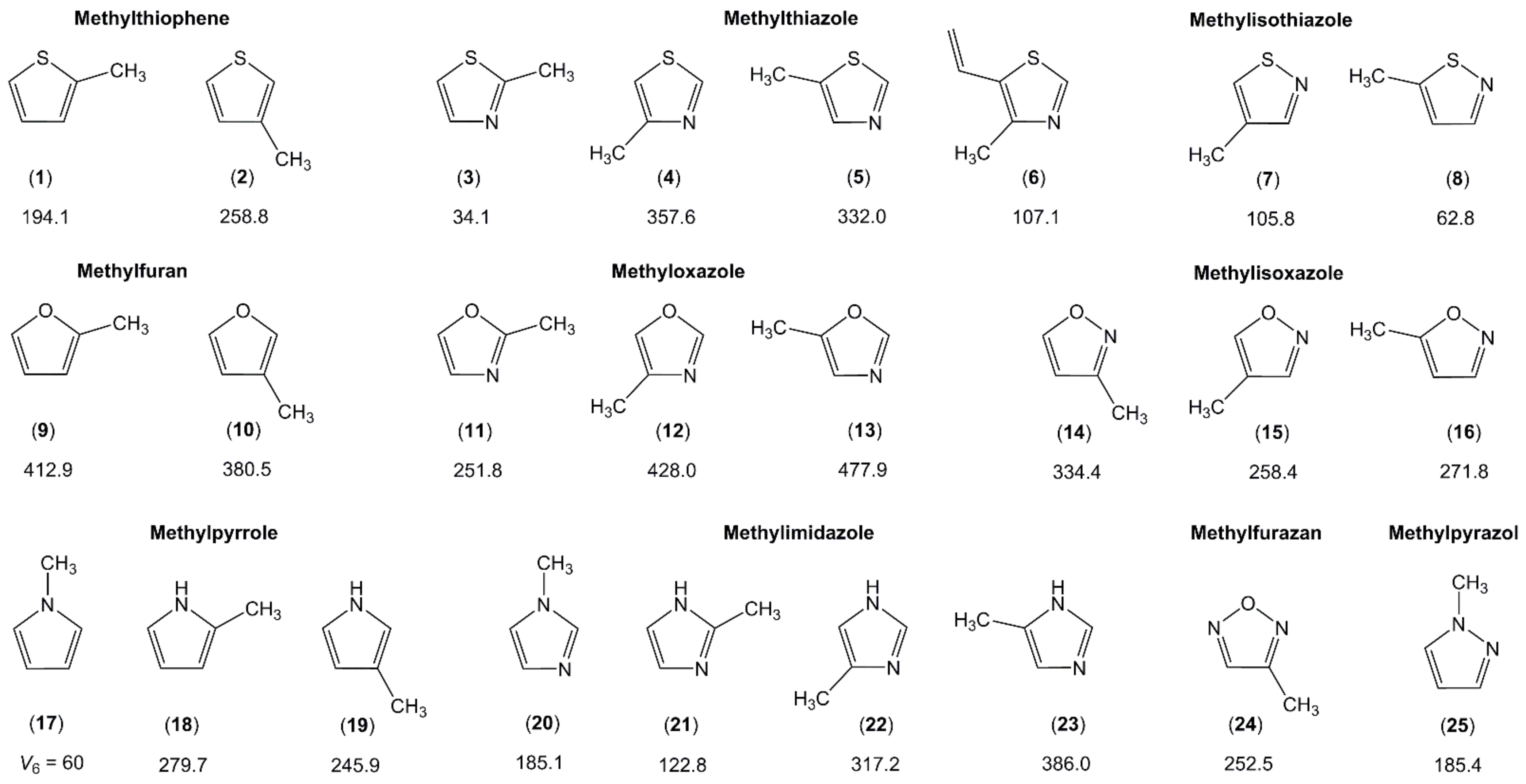
2.1.1. Five-Membered Rings with an (Extended) Conjugated Double-Bond System
2.1.1.1. Sole Methyl Substitution on the Ring
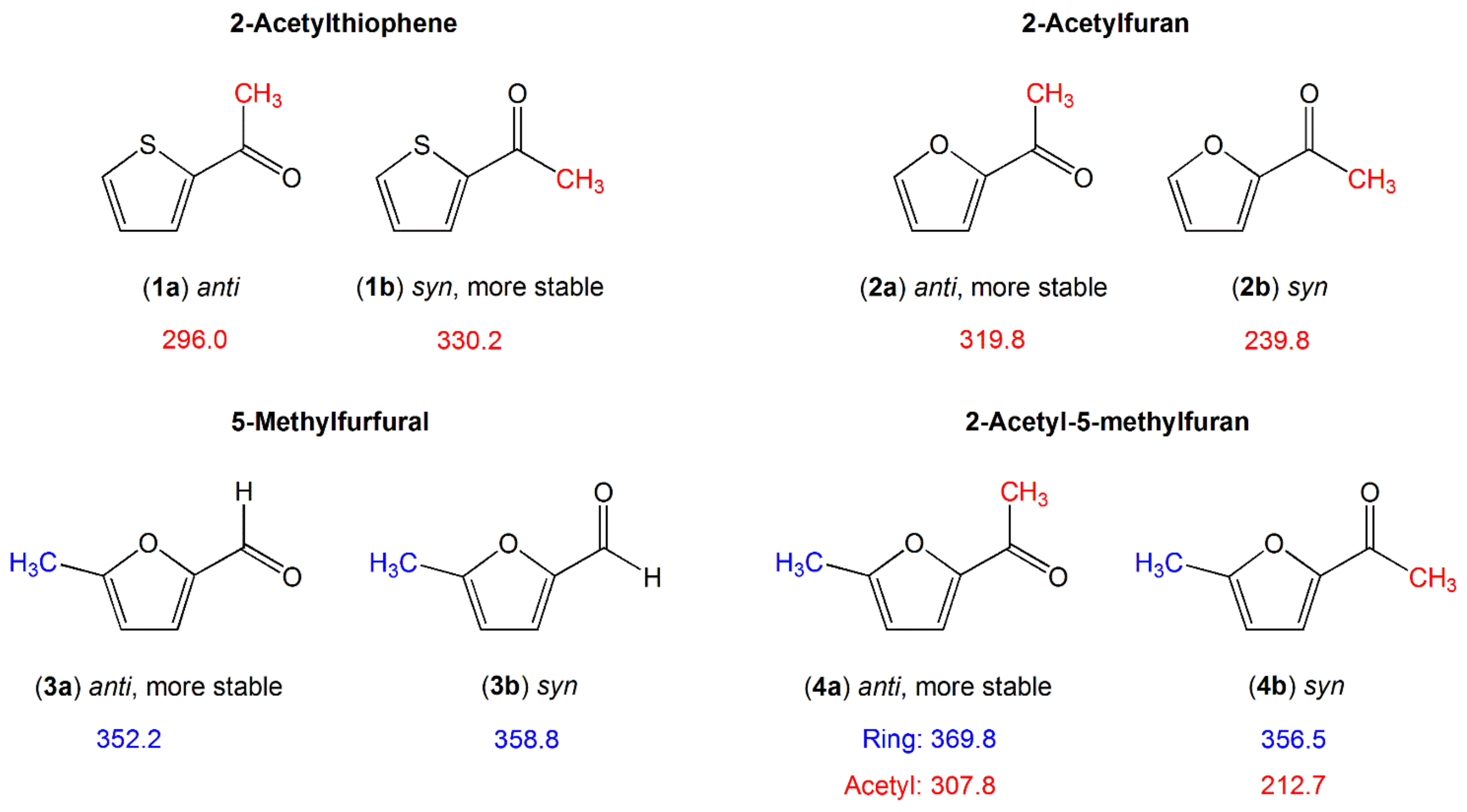
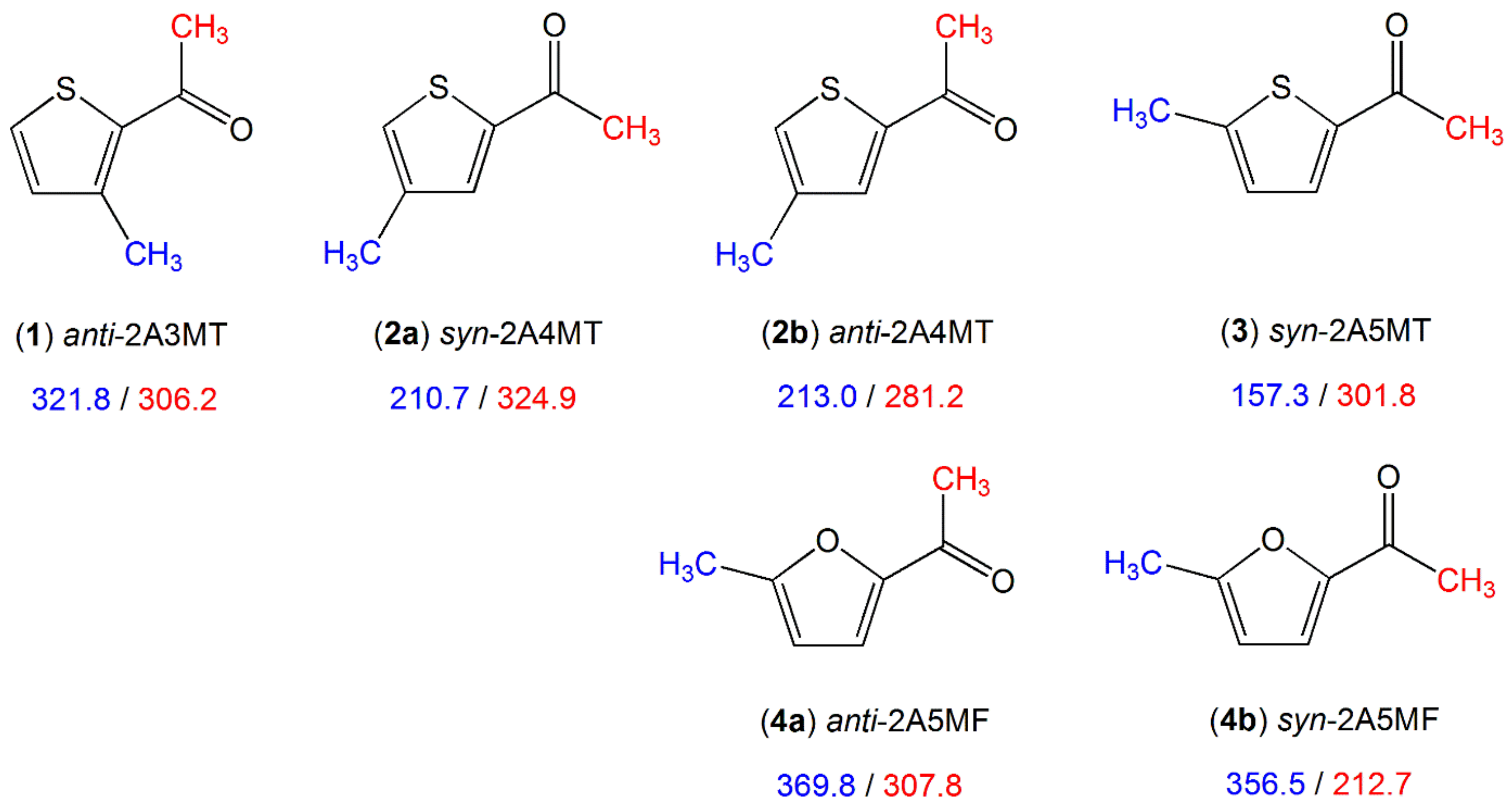
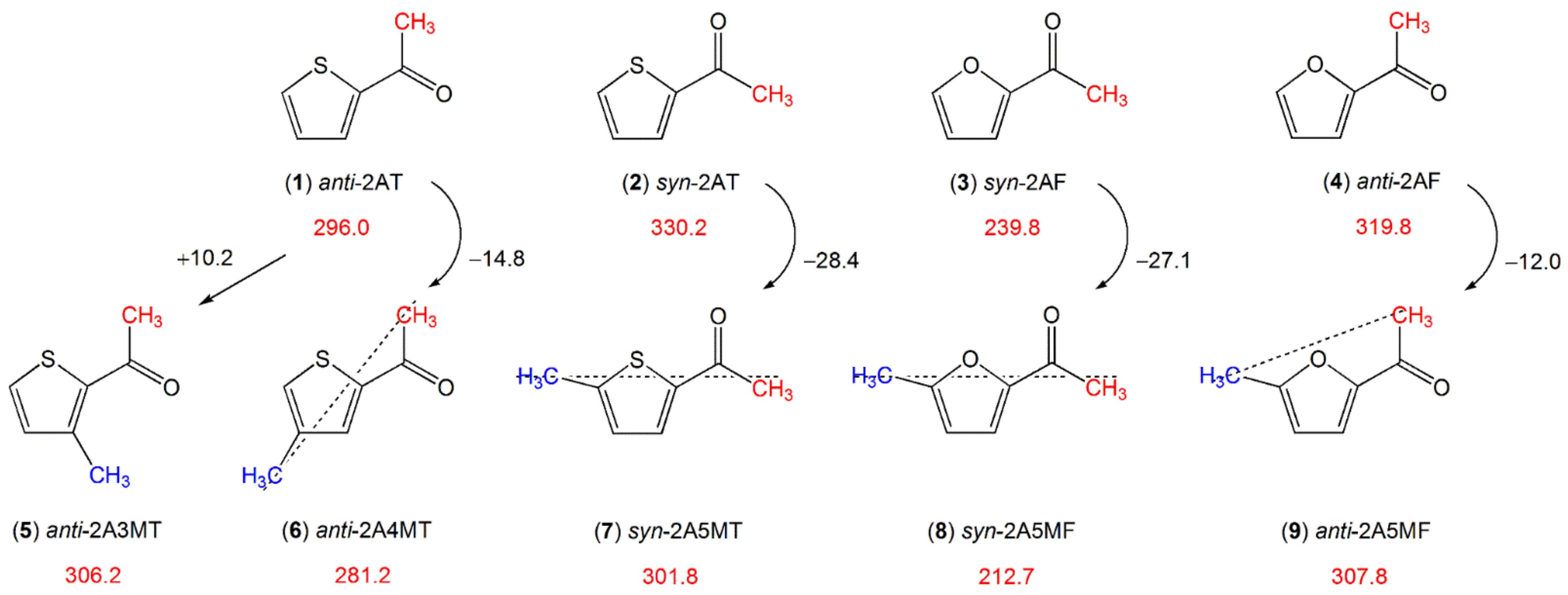
2.1.1.2. Carbonyl Substituent on the Ring
2.1.2. Six-Membered Rings with an (Extended) Conjugated Double-Bond System
2.1.2.1. Toluene
2.1.2.2. Cresol and Methylanisole Derivatives
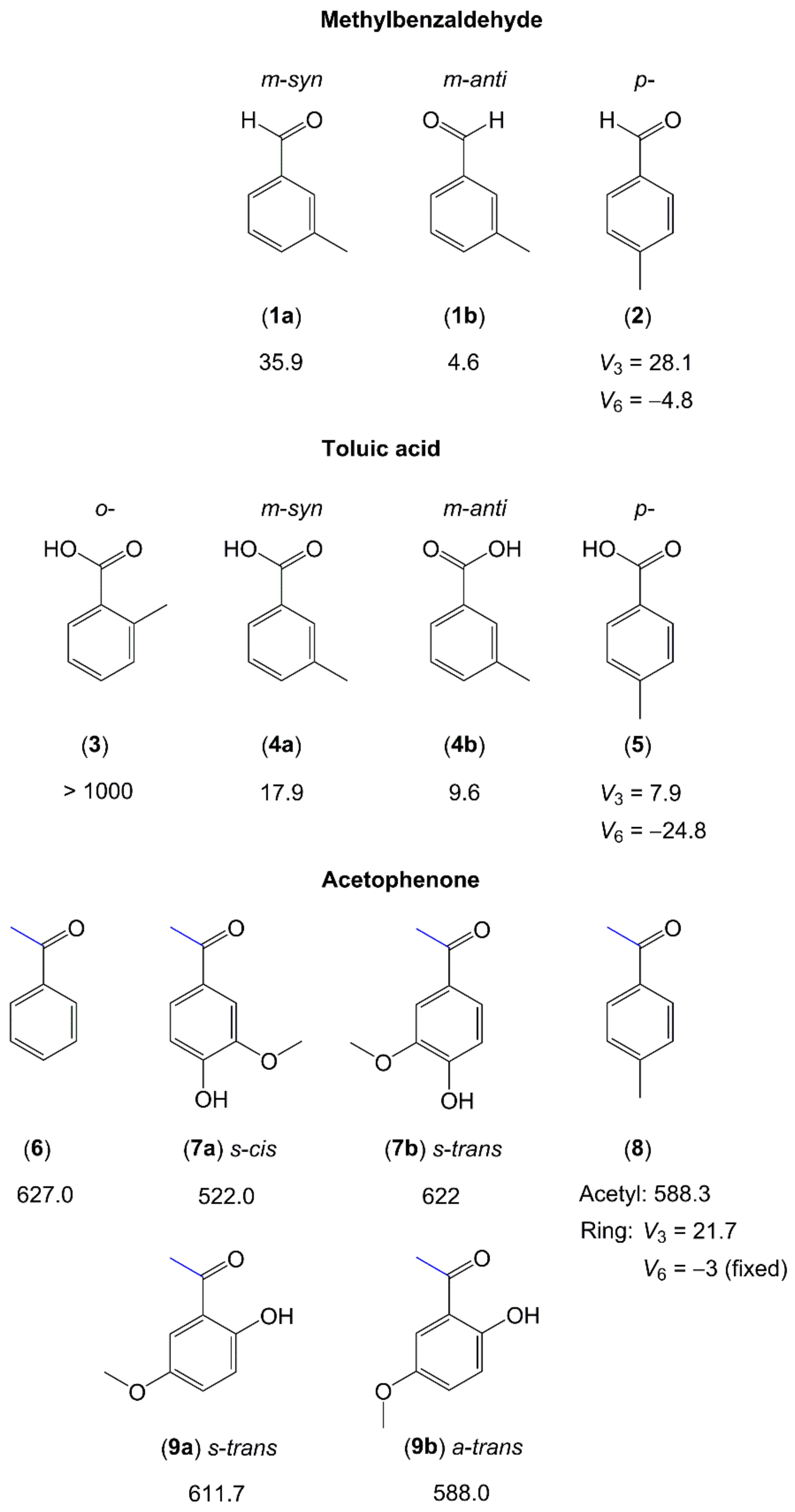
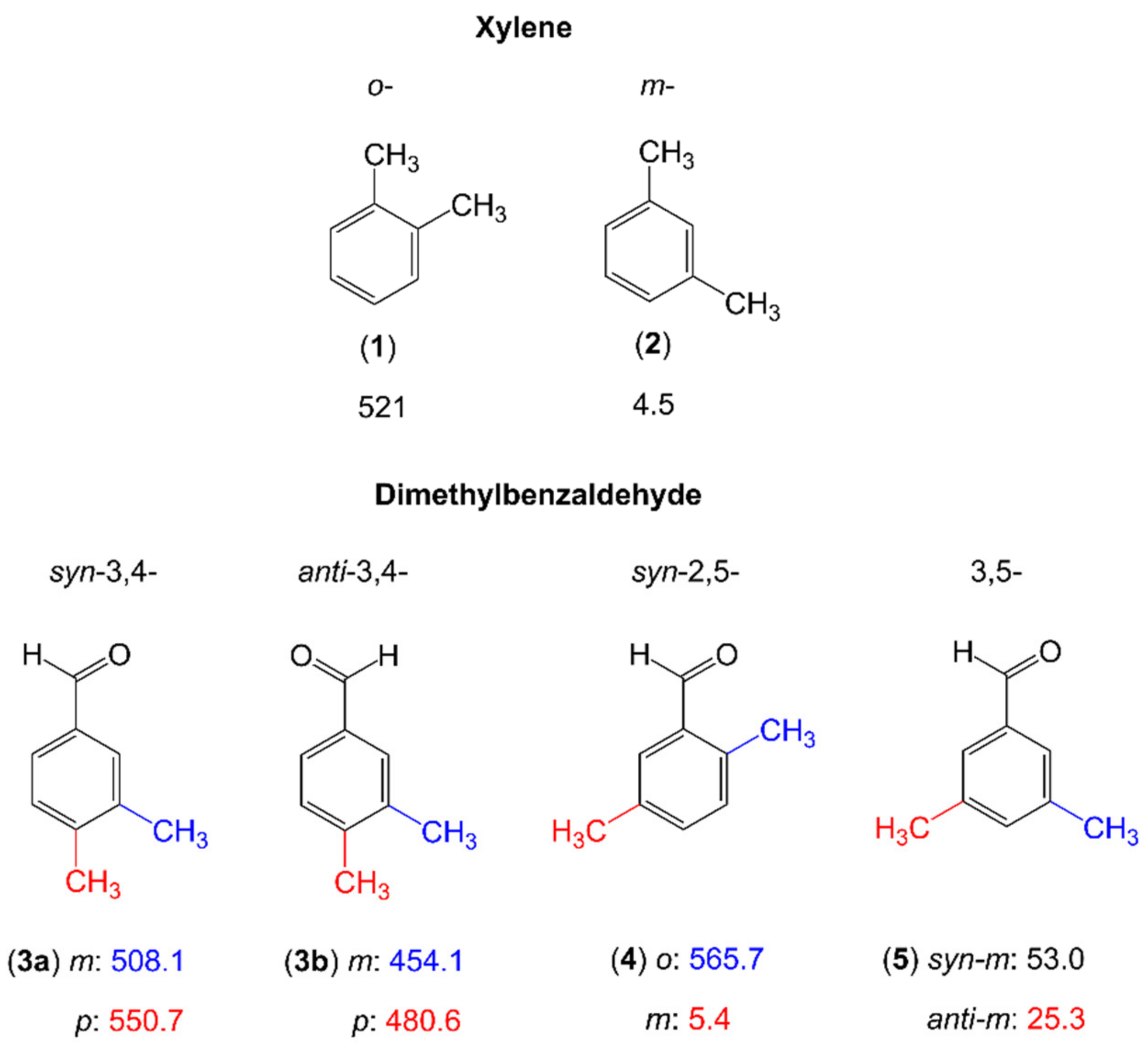
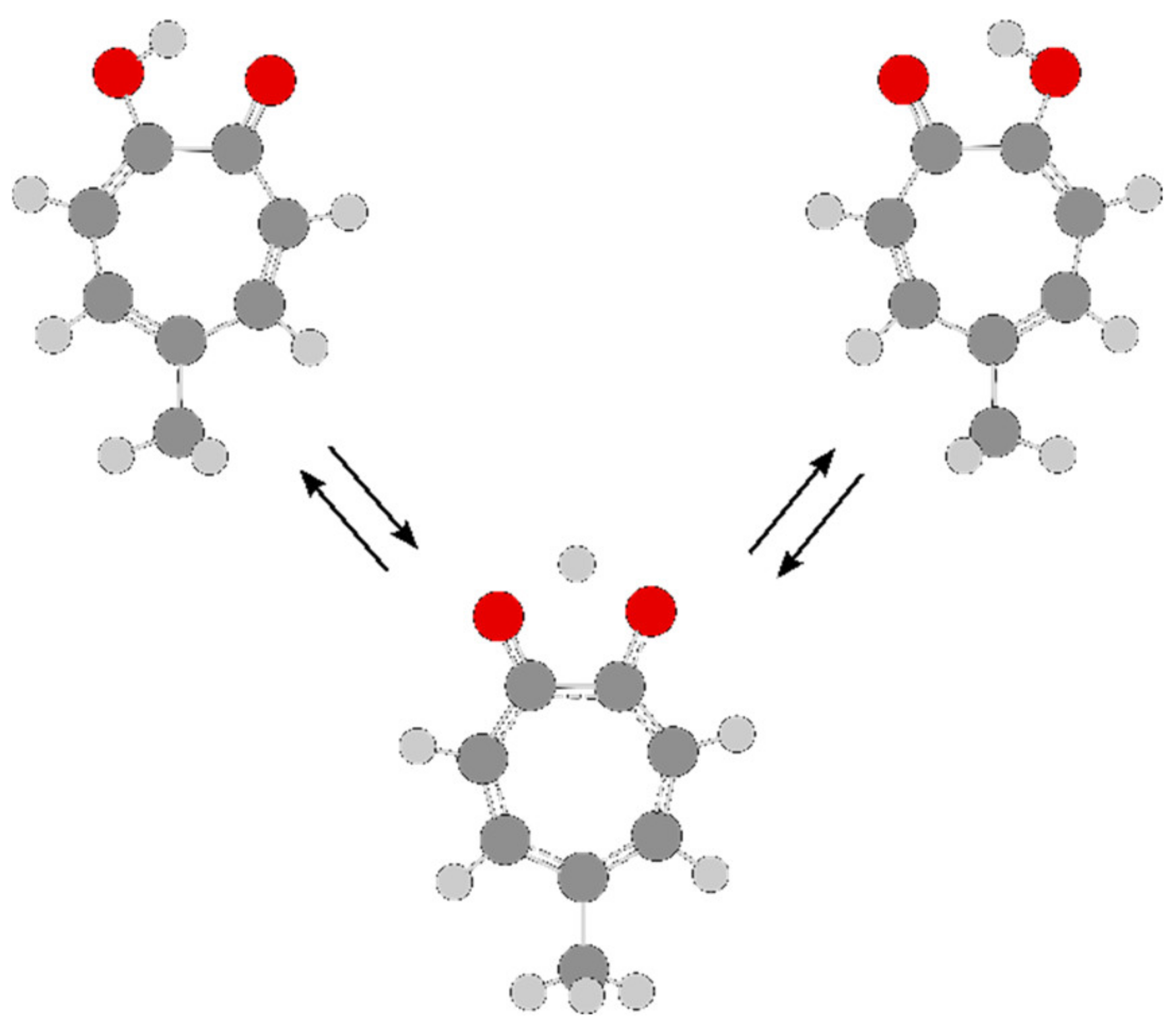
2.1.2.3. Carbonyl Substituent on the Ring
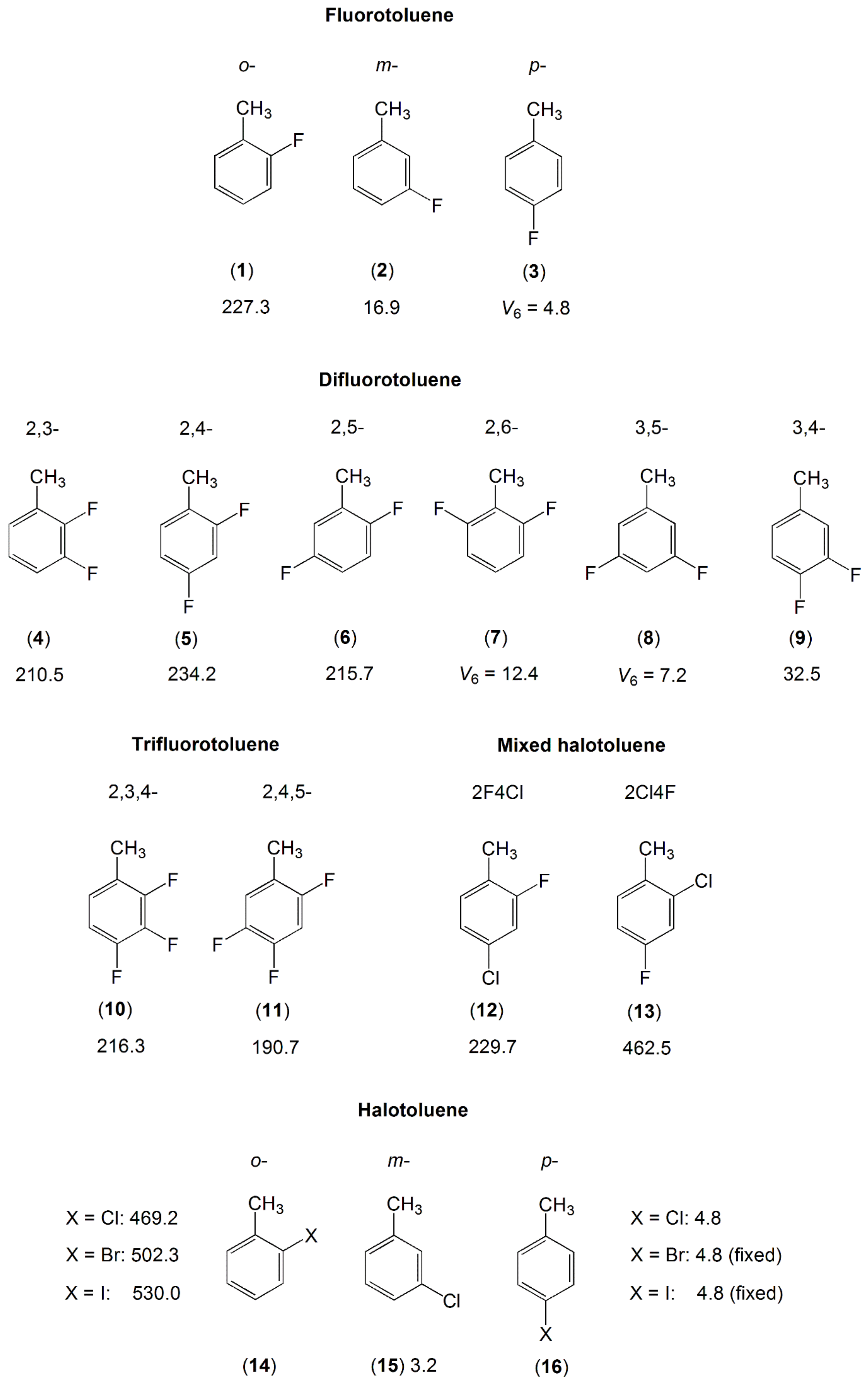
2.1.2.4. Halogen Substituent(s) on the Ring
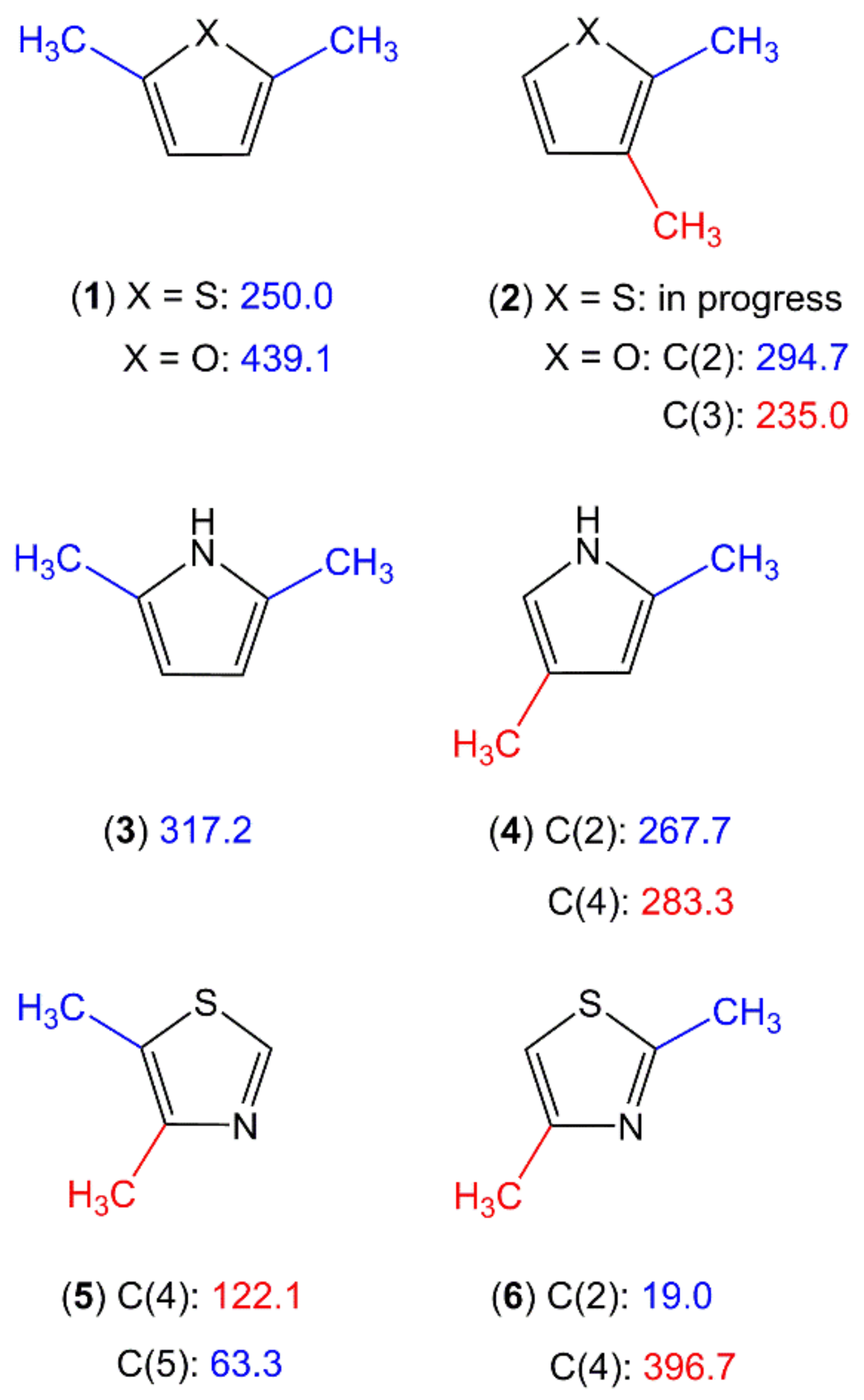
2.1.2.5. Nitrogen-Containing Aromatic Six-Membered Ring
2.1.3. Larger Methyl-Substituted Aromatic Rings
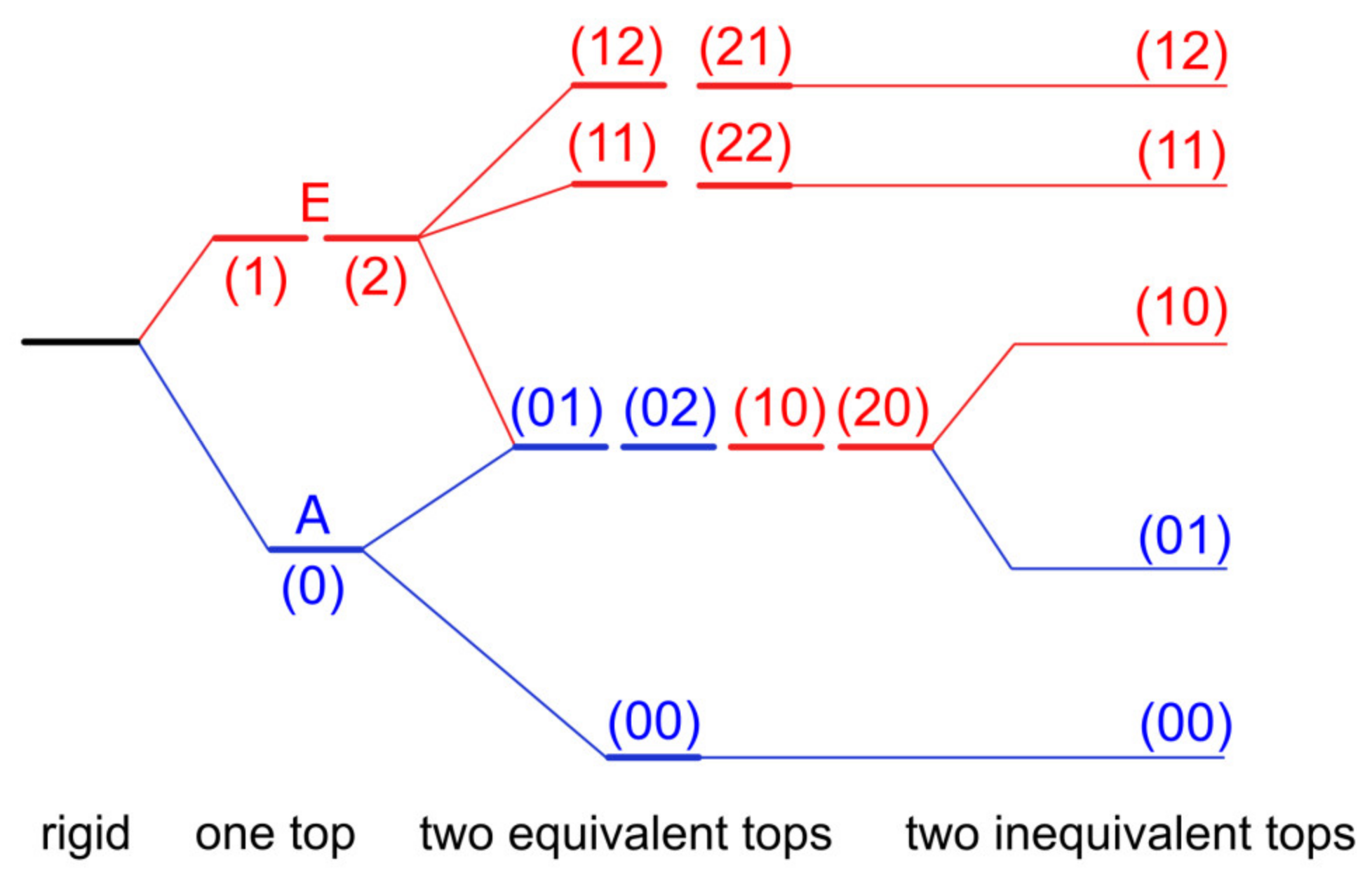
2.2. Dimethyl-Substituted Aromatic Rings
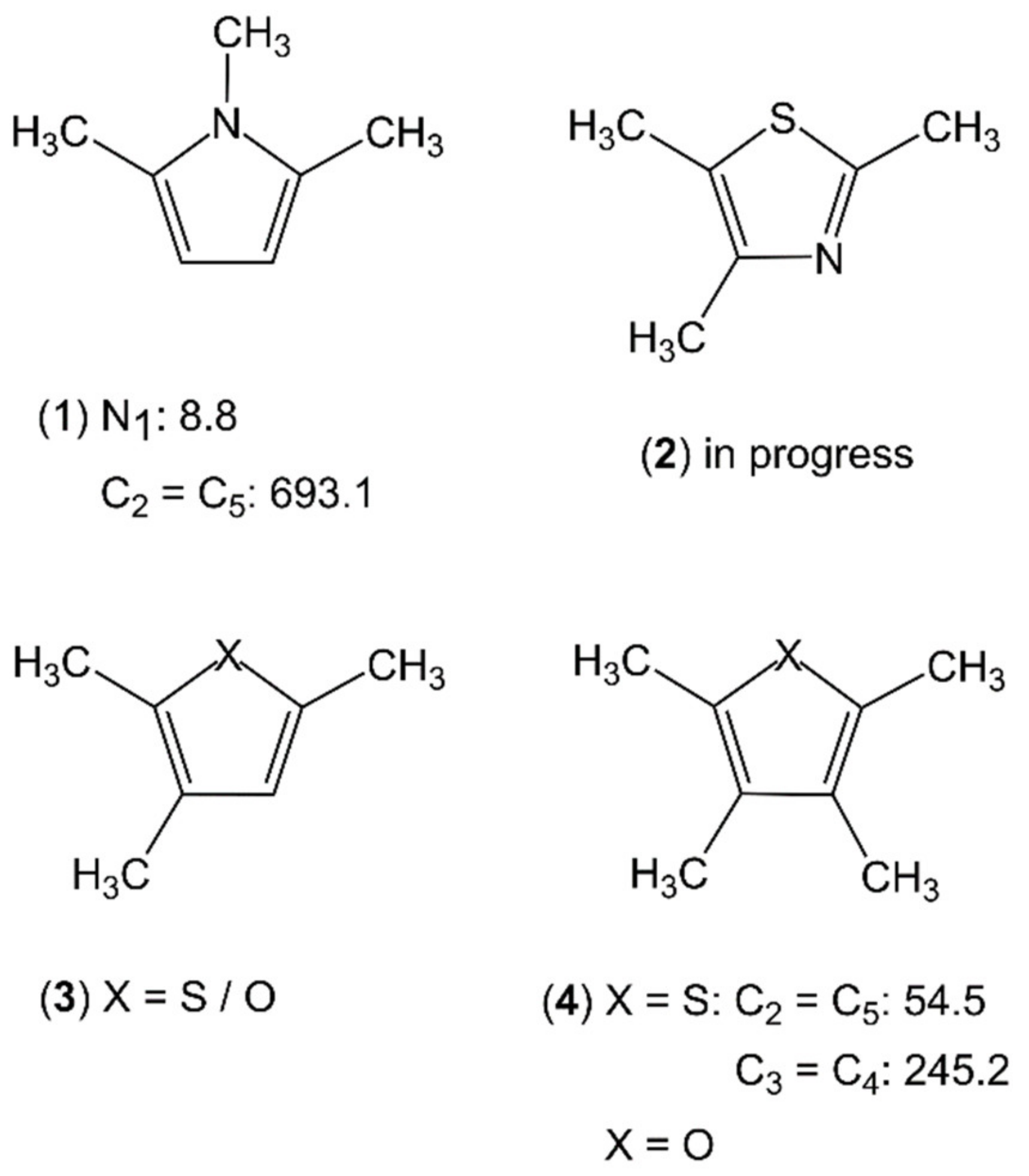
2.2.1. Dimethyl-Substituted Five-Membered Rings
Sole Dimethyl Substitutions on the Ring
Methyl Substitution on a Substituent
2.2.2. Dimethyl-Substituted Six-Membered Rings
2.2.2.1. Coupled Internal Rotations in Dimethylanisoles
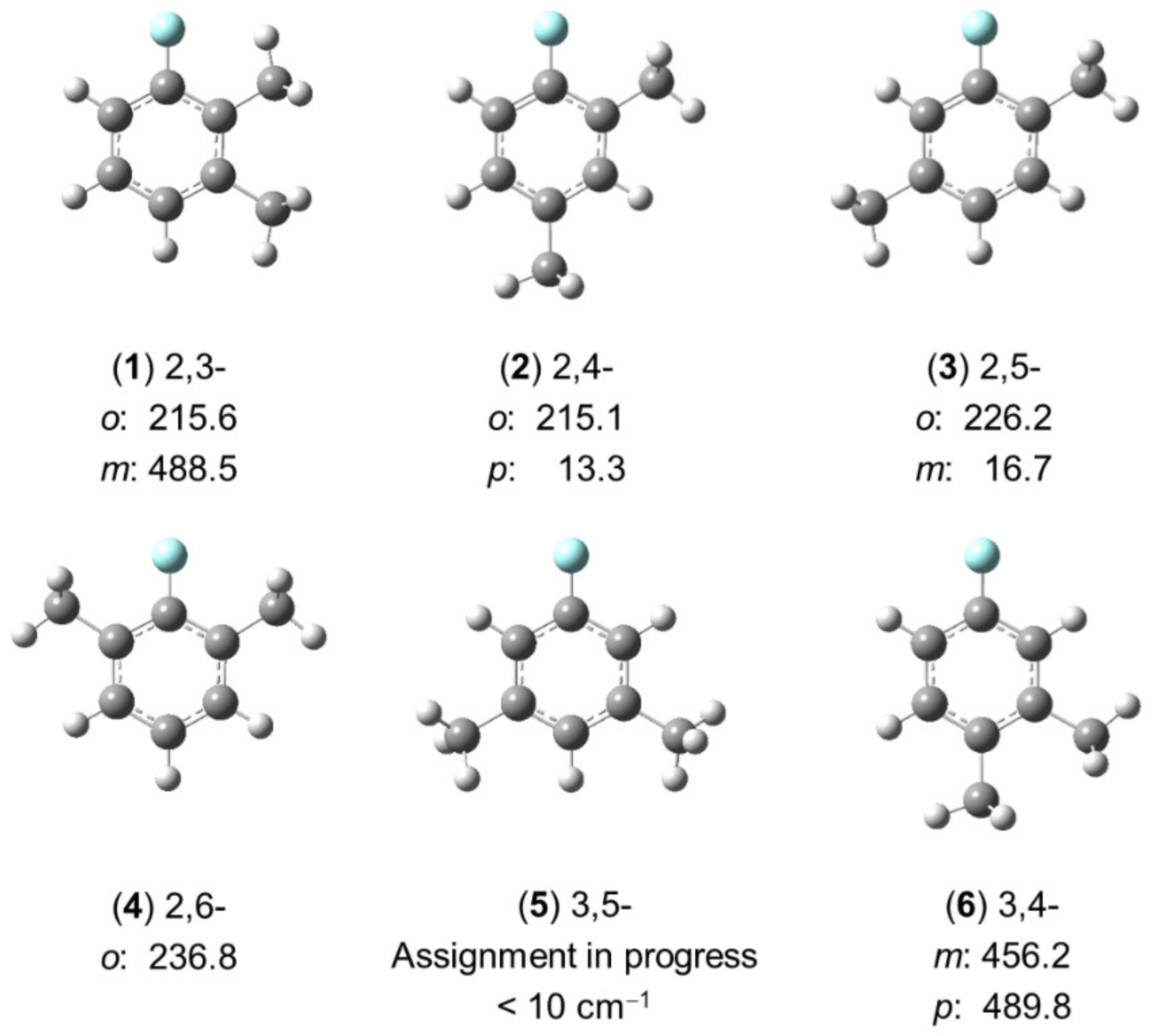
2.2.2.2. Six Isomers in the Dimethylfluorobenzene Family
2.3. Trimethyl- and Tetramethyl-Substituted Planar Five-Membered Rings
3. Ring Inversion Tunneling
4. Internal Rotation Coupled with Ring Inversion Tunneling
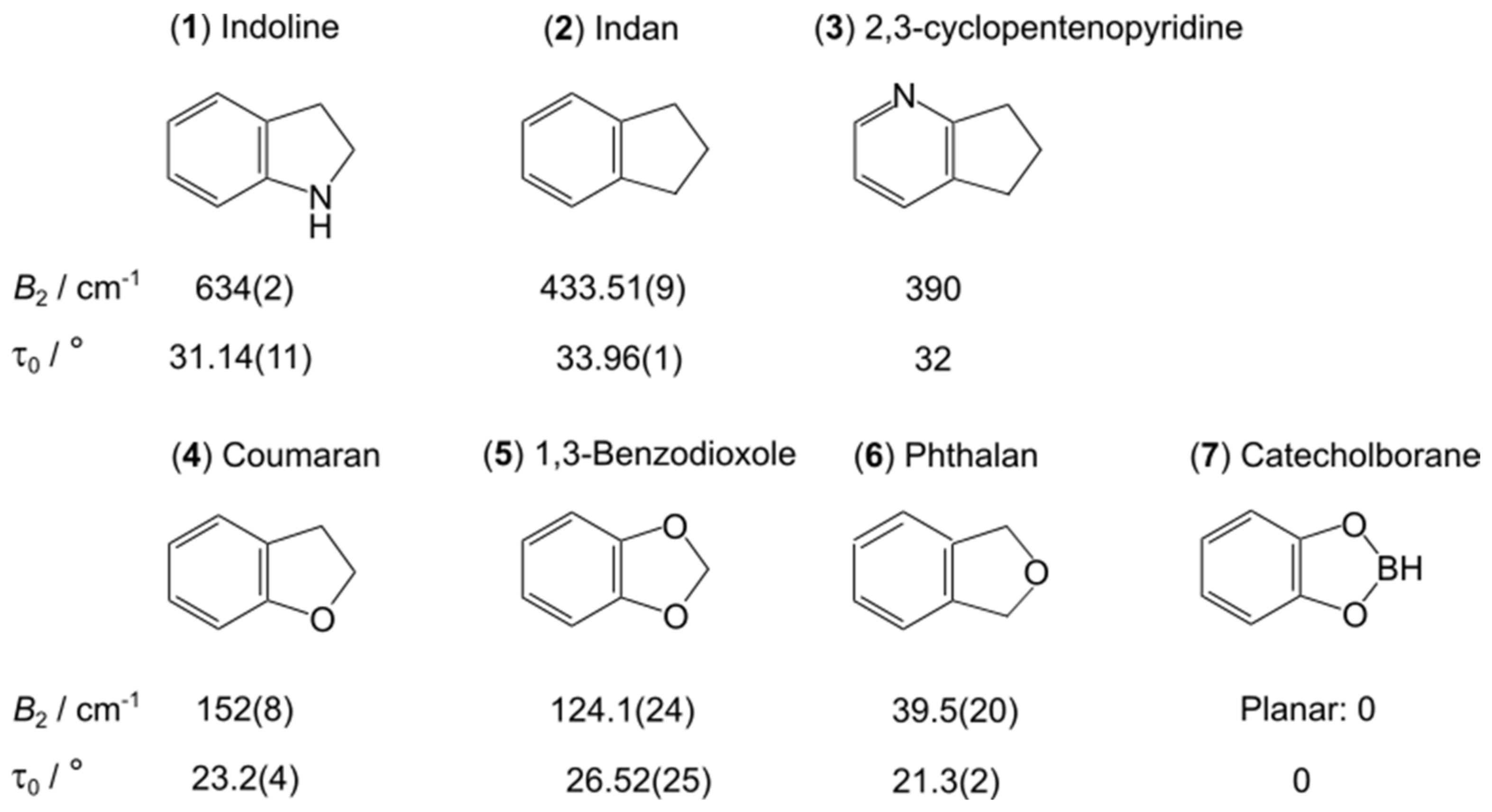
5. Ring Puckering
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lister, D.G.; Macdonald, J.N.; Owen, N.L. Internal Rotation and Inversion: An Introduction to Large Amplitude Motions in Molecules; Academic Press: New York, NY, USA, 1978. [Google Scholar]
- Legon, A.C. Equilibrium Conformations of Four- and Five-Membered Cyclic Molecules in the Gas Phase: Determination and Classification. Chem. Rev. 1980, 80, 231–262. [Google Scholar] [CrossRef]
- Nguyen, H.V.L.; Kleiner, I. Theoretical and Computational Chemistry; Gulaczyk, I., Tylkowski, B., Eds.; De Gruyter: Berlin, Germany; Boston, MA, USA, 2021; pp. 41–78. [Google Scholar]
- Nguyen, H.V.L.; Gulaczyk, I.; Kręglewski, M.; Kleiner, I. Large Amplitude Inversion Tunneling Motion in Ammonia, Methylamine, Hydrazine, and Secondary Amines: From Structure Determination to Coordination Chemistry. Coord. Chem. Rev. 2021, 436, 213797. [Google Scholar] [CrossRef]
- Lin, C.C.; Swalen, J.D. Internal Rotation and Microwave Spectroscopy. Rev. Mod. Phys. 1959, 31, 841. [Google Scholar] [CrossRef]
- Kleiner, I. Asymmetric-Top Molecules Containing One Methyl-Like Internal Rotor: Methods and Codes for Fitting and Predicting Spectra. J. Mol. Spectrosc. 2010, 260, 1–18. [Google Scholar] [CrossRef]
- Kleiner, I. Spectroscopy of Interstellar Internal Rotors: An Important Tool for Investigating Interstellar Chemistry. ACS Earth Space Chem. 2019, 3, 1812–1842. [Google Scholar] [CrossRef]
- Cleeton, C.E.; Williams, N.H. Electromagnetic Waves of 1.1 cm Wave-Length and the Absorption Spectrum of Ammonia. Phys. Rev. 1934, 45, 234. [Google Scholar] [CrossRef]
- Dailey, B.P. First-Order Stark Effect in the Microwave Spectrum of Methyl Alcohol. Phys. Rev. 1947, 72, 84. [Google Scholar] [CrossRef]
- Hershberger, W.D.; Turkevich, J. Absorption of Methyl Alcohol and Methylamine for 1.25-Cm Waves. Phys. Rev. 1947, 71, 554. [Google Scholar] [CrossRef]
- Engerholm, G.G.; Luntz, A.C.; Gwinn, W.D.; Harris, D.O. Ring Puckering in Five-Membered Rings. II. The Microwave Spectrum, Dipole Moment, and Barrier to Pseudorotation in Tetrahydrofuran. J. Chem. Phys. 1969, 50, 2446. [Google Scholar] [CrossRef]
- Gordy, W.; Cook, R.L. Microwave Molecular Spectra, 3rd ed.; Wiley: New York, NY, USA, 1984; Volume 18. [Google Scholar]
- Blanco, S.; Sanz, M.E.; Lesarri, A.; López, J.C.; Alonso, J.L. Free Internal Rotation in CH3–CC–CF3. Chem. Phys. Lett. 2004, 397, 379–381. [Google Scholar] [CrossRef]
- Herbers, S.; Kraus, P.; Grabow, J.-U. Accurate Equilibrium Structures of Methyl Methacrylate and Methacrylic Acid by Microwave Spectroscopy and Dispersion Corrected Calculations. J. Chem. Phys. 2019, 150, 144308. [Google Scholar] [CrossRef]
- Wilcox, D.S.; Shirar, A.J.; Williams, O.L.; Dian, B.C. Additional Conformer Observed in the Microwave Spectrum of Methyl Vinyl Ketone. Chem. Phys. Lett. 2011, 508, 10–16. [Google Scholar] [CrossRef]
- Ilyushin, V.; Rizzato, R.; Evangelisti, L.; Feng, G.; Maris, A.; Melandri, S.; Caminati, W. Almost Free Methyl Top Internal Rotation: Rotational Spectrum of 2-Butynoic Acid. J. Mol. Spectr. 2011, 267, 186–190. [Google Scholar] [CrossRef]
- Kawashima, Y.; Usami, T.; Suenram, R.D.; Golubiatnikov, G.Y.; Hirota, E. Dynamical Structure of Peptide Molecules: Fourier Transform Microwave Spectroscopy and Ab Initio Calculations of N-Methylformamide. J. Mol. Spectrosc. 2010, 263, 11–20. [Google Scholar] [CrossRef]
- Weslley, C.; Silva, W.G.D.P.; van Wijngaarden, J. Rotational Spectrum and Quantum Chemical Calculations of Methyl Cyanoacetate: A Compound of Potential Astrochemical Interest. J. Mol. Spectrosc. 2021, 377, 111444. [Google Scholar]
- Nguyen, H.V.L.; Andresen, M.; Stahl, W. Conformational Sampling and Large Amplitude Motion of Methyl Valerate. Phys. Chem. Chem. Phys. 2021, 23, 2930–2937. [Google Scholar] [CrossRef]
- Dang, N.-N.; Pham, H.-N.; Kleiner, I.; Schwell, M.; Grabow, J.-U.; Nguyen, H.V.L. Methyl Internal Rotation in Fruit Esters: Chain-Length Effect Observed in the Microwave Spectrum of Methyl Hexanoate. Molecules 2022, 27, 2639. [Google Scholar] [CrossRef]
- Andresen, M.; Schwell, M.; Nguyen, H.V.L. The Two-Top Molecule 3-Penten-2-one: Acetyl Methyl Torsion in α,β-Unsaturated Ketones. J. Mol. Struct. 2022, 1247, 131337. [Google Scholar] [CrossRef]
- Plusquellic, D.F.; Kleiner, I.; Demaison, J.; Suenram, R.D.; Lavrich, R.J.; Lovas, F.J.; Fraser, G.T.; Ilyushin, V.V. The Microwave Spectrum of a Two-Top Peptide Mimetic: The N-Acetyl Alanine Methyl Ester Molecule. J. Chem. Phys. 2006, 125, 104312. [Google Scholar] [CrossRef]
- Ohashi, N.; Hougen, J.T.; Suenram, R.D.; Lovas, F.J.; Kawashima, Y.; Fujitake, M.; Pyka, J. Analysis and Fit of the Fourier-Transform Microwave Spectrum of the Two-Top Molecule N-Methylacetamide. J. Mol. Spectrosc. 2004, 227, 28–42. [Google Scholar] [CrossRef]
- Fujitake, M.; Kubota, Y.; Ohashi, N. Fourier Transform Microwave Spectroscopy of N,N-Dimethylacetamide. J. Mol. Spectrosc. 2006, 236, 97–109. [Google Scholar] [CrossRef]
- Schnell, M.; Hougen, J.T.; Grabow, J.-U. Towards the Complete Analysis of the Rotational Spectrum of (CH3)3SnCl. J. Mol. Spectrosc. 2008, 251, 38–55. [Google Scholar] [CrossRef] [Green Version]
- Merke, I.; Lüchow, A.; Stahl, W. Internal Rotation, Quadrupole Coupling and Structure of (CH3)3SiI Studied by Microwave Spectroscopy and Ab-Initio Calculations. J. Mol. Struct. 2006, 780–781, 295–299. [Google Scholar] [CrossRef]
- Van, V. Structures and Internal Dynamics of Cyclic Molecules Studied by Microwave Spectrsocopy and Quantum Chemistry; RWTH Aachen University: Aachen, Germany, 2017. [Google Scholar]
- Dreizler, H. Gruppentheoretische Betrachtungen zu den Mikrowellenspektren von Molekülen mit zwei behindert drehbaren dreizählig-symmetrischen Molekülgruppen. Z. Naturforsch. 1961, 16, 1354–1367. [Google Scholar] [CrossRef]
- Gunther-Mohr, G.R.; White, R.L.; Schawlow, A.L.; Good, W.E.; Coles, D.K. Hyperfine Structure in the Spectrum of N14H3 I. Experimental Results. Phys. Rev. 1954, 94, 1184. [Google Scholar]
- Tsunekawa, S.; Kojima, T.; Hougen, J.T. Analysis of the Microwave Spectrum of Hydrazine. J. Mol. Spectrosc. 1982, 95, 133–152. [Google Scholar]
- Merke, I.; Coudert, L.H. Microwave Spectrum, Tunneling Motions, and Quadrupole Coupling Hyperfine Structure of Ethylene Diamine. J. Mol. Spectrosc. 2006, 237, 174–204. [Google Scholar] [CrossRef]
- Kręglewski, M. The Geometry and Inversion-Internal Rotation Potential Function of Methylamine. J. Mol. Spectrosc. 1989, 133, 10–21. [Google Scholar] [CrossRef]
- Kleibömer, B.; Sutter, D.H. The Vibrational State Dependence of the 14N Quadrupole Coupling Tensor in Aniline. A Microwave Fourier-Transform Study Combined with Semirigid Bender Calculations. Z. Naturforsch. 1988, 43, 561–571. [Google Scholar] [CrossRef]
- Kasuja, T. Scientific Papers of the Institute of Physical and Chemical Research; Institute of Physical and Chemical Research: Tokyo, Japan, 1962; Volume 56, p. 1. [Google Scholar]
- Marstokk, K.M.; Møllendal, H. Microwave Spectrum, Conformational Equilibrium, Intramolecular Hydrogen Bonding, Inversion Tunnelling, Dipole Moments and Centrifugal Distortion of Ethylenediamine. J. Mol. Struct. 1978, 49, 221–237. [Google Scholar] [CrossRef]
- Baraban, J.H.; Martin-Drumel, M.-A.; Changala, P.B.; Eibenberger, S.; Nava, M.; Patterson, D.; Stanton, J.F.; Ellison, G.B.; McCarthy, M.C. The Molecular Structure of gauche-1,3-Butadiene: Experimental Establishment of Non-planarity. Angew. Chem. Int. Ed. 2018, 57, 1821–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, S.; Lerch, P.; Prentner, R.; Quack, M. Tunneling and Tunneling Switching Dynamics in Phenol and Its Isotopomers from High-Resolution FTIR Spectroscopy with Synchrotron Radiation. Angew. Chem. Int. Ed. 2013, 52, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, P.; Lengsfeld, K.G.; Aydt, K.; Jahn, M.K.; Herbers, S.; Travers, M.J.; Nguyen, H.V.L.; Grabow, J.-U. Proton Inversion Tunneling in the Rotational Spectrum of Acetone Cyanohydrin. J. Mol. Spectrosc. 2020, 373, 111372. [Google Scholar] [CrossRef]
- Li, W.; Evangelisti, L.; Gou, Q.; Caminati, W.; Meyer, R. The Barrier to Proton Transfer in the Dimer of Formic Acid: A Pure Rotational Study. Angew. Chem. Int. Ed. 2019, 58, 859–865. [Google Scholar] [CrossRef]
- Schnell, M.; Erlekam, U.; Bunker, P.R.; von Helden, G.; Grabow, J.-U.; Meijer, G.; van der Avoird, A. Structure of the Benzene Dimer—Governed by Dynamics. Angew. Chem. Int. Ed. 2013, 52, 5180–5183. [Google Scholar] [CrossRef] [Green Version]
- Sanz, M.E.; Lesarri, A.; López, J.C.; Alonso, J.L. Hydrogen Bond in Molecules with Large-Amplitude Motions: A Rotational Study of Trimethylene Sulfide⋅⋅⋅HCl. Angew. Chem. Int. Ed. 2001, 40, 935–938. [Google Scholar] [CrossRef]
- Giuliano, B.M.; Caminati, W. Isotopomeric conformational change in anisole–water. Angew. Chem. Int. Ed. 2005, 117, 609–612. [Google Scholar] [CrossRef]
- Ghosh, S.; Thomas, J.; Huang, W.; Xu, Y.; Jäger, W. Rotational Spectra of Two Hydrogen-Bonded Methyl Salicylate Monohydrates: Relative Stability and Tunneling Motions. J. Phys. Chem. Lett. 2015, 6, 3126–3131. [Google Scholar] [CrossRef]
- Feng, G.; Gou, Q.; Evangelisti, L.; Caminati, W. Frontiers in Rotational Spectroscopy: Shapes and Tunneling Dynamics of the Four Conformers of the Acrylic Acid—Difluoroacetic Acid Adduct. Angew. Chem. Int. Ed. 2014, 53, 530–534. [Google Scholar] [CrossRef]
- Vogelsanger, B.; Caminati, W.; Bauder, A. The Pure Rotational Spectrum of Cyclobutane-d1 Observed by Microwave Fourier Transform Spectroscopy. Chem. Phys. Lett. 1987, 141, 245–250. [Google Scholar] [CrossRef]
- Caminati, W.; Vogelsanger, B.; Meyer, R.; Grassi, G.; Bauder, A. Rotational Spectrum, Dipole Moment, and Ring-Puckering Potential of Cyclobutane-1,1-d2. J. Mol. Spectrosc. 1988, 131, 172–184. [Google Scholar] [CrossRef]
- López, J.C.; Alonso, J.L.; Charro, M.E.; Wlodarczak, G.; Demaison, J. The Millimeter-Wave Spectrum of Cyclopentene. J. Mol. Spetrosc. 1992, 155, 143–157. [Google Scholar] [CrossRef]
- Kleiner, I.; Tarrago, G.; Brown, L.R. Positions and Intensities in the 3ν2/ν2 + ν4 Vibrational System of 14NH3 near 4 μm J. Mol. Spectrosc. 1995, 173, 120–145. [Google Scholar] [CrossRef]
- Finneran, I.A.; Shipman, S.T.; Weaver, S.L.W. Rotational Spectroscopy of 2-Methylfuran from 8.7 to 960 GHz. J. Mol. Spectrosc. 2012, 280, 27–33. [Google Scholar] [CrossRef]
- Maris, A.; Melandri, S.; Evangelisti, L.; Vigorito, A.; Sigismondi, S.; Calabrese, C.; Usabiaga, I. Structure and Dynamics of Methacrylamide, a Computational and Free-jet Rotational Spectroscopic Study. J. Mol. Struct. 2022, 1248, 131391. [Google Scholar] [CrossRef]
- Bruckhuisen, J.; Dhont, G.; Roucou, A.; Jabri, A.; Bayoudh, H.; Tran, T.T.; Goubet, M.; Martin-Drumel, M.-A.; Cuisset, A. Intramolecular H-Bond Dynamics of Catechol Investigated by THz High-Resolution Spectroscopy of Its Low-Frequency Modes. Molecules 2021, 26, 3645. [Google Scholar] [CrossRef]
- Ilyushin, V.V.; Zakharenko, O.; Lewen, F.; Schlemmer, S.; Alekseev, E.A.; Pogrebnyak, M.; Lees, R.M.; Xu, L.-H.; Belloche, A.; Menten, K.M.; et al. Rotational Spectrum of Isotopic Methyl Mercaptan, 13CH3SH, in the Laboratory and towards Sagittarius B2(N2). Can. J. Phys. 2020, 98, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, N.; Takagi, K.; Hougen, J.T.; Olson, W.B.; Lafferty, W.L. Far-Infrared Spectrum of Methyl Amine: Assignment and Analysis of the First Torsional State. J. Mol. Spectrosc. 1988, 132, 242–260. [Google Scholar] [CrossRef]
- Kalkman, I.; Vu, C.; Schmitt, M.; Meerts, W.L. Structure and Internal Rotation in the S0 and S1 States of o-Toluidine Studied by High Resolution UV Spectroscopy. Phys. Chem. Chem. Phys. 2009, 11, 4311–4318. [Google Scholar] [CrossRef]
- Belorgeot, C.; Stern, V.; Goff, N.; Kachmarsky, J.; Möller, K.D. Far-Infrared Spectrum of the Internal Rotation in Methylamine. J. Mol. Spectrosc. 1982, 92, 91–100. [Google Scholar] [CrossRef]
- Durig, J.R.; Lin, J.; Guirgist, G.A.; Bell, S. Far-Infrared Spectrum, Barrier to Internal Rotation, r0 Structure, and Ab Initio Calculations for Acetylacetylene. Struct. Chem. 1990, 1, 547–559. [Google Scholar] [CrossRef]
- Stern, V.; Goff, N.; Kachmarsky, J.; Möller, K.D. Far-Infrared Internal Rotation Spectrum of CH3OD and CD3OD. J. Mol. Spectrosc. 1980, 79, 345–362. [Google Scholar] [CrossRef]
- Fateley, W.G.; Miller, F.A. Torsional Frequencies in the Far Infrared—III: The Form of the Potential Curve for Hindered Internal Rotation of a Methyl Group. Spectrochim. Acta 1963, 19, 611–628. [Google Scholar] [CrossRef]
- Xu, L.-H.; Lees, R.M.; Crabbe, G.T.; Myshrall, J.A.; Mueller, H.S.P.; Endres, C.P.; Baum, O.; Lewen, F.; Schlemmer, S.; Menten, K.M.; et al. Terahertz and Far-Infrared Synchrotron Spectroscopy and Global Modeling of Methyl Mercaptan, CH332SH J. Chem. Phys. 2012, 137, 104313. [Google Scholar] [CrossRef]
- Smithson, T.L.; Duckett, J.A.; Wieser, H. Far-Infrared Spectra and Skeletal Out-of-Plane Deformations of Indan, Phthalan, and Indoline. J. Phys. Chem. 1984, 88, 1102–1109. [Google Scholar] [CrossRef]
- Duckett, J.A.; Smithson, T.L.; Wieser, H. 1,3-Benzodioxole: Far-Infrared Spectrum 50–500 cm−1. Chem. Phys. Lett. 1979, 64, 261–265. [Google Scholar] [CrossRef]
- McCarthy, M.C.; McGuire, B.A. Aromatics and Cyclic Molecules in Molecular Clouds: A New Dimension of Interstellar Organic Chemistry. J. Phys. Chem. A 2021, 125, 3231–3243. [Google Scholar] [CrossRef]
- Hollis, J.M.; Remijan, A.J.; Jewell, P.R.; Lovas, F.J. Cyclopropenone (c-H2C3O): A New Interstellar Ring Molecule. Astrophys. J. 2006, 642, 933. [Google Scholar] [CrossRef] [Green Version]
- Guelin, M.; Cernicharo, J. Organic Molecules in Insterstellar Space: Latest advances. arXiv 2022, arXiv:2201.06106. [Google Scholar]
- Schnitzler, E.G.; Seifert, N.A.; Kusuma, I.; Jäger, W. Rotational Spectroscopy of p-Toluic Acid and Its 1:1 Complex with Water. J. Phys. Chem. A 2017, 121, 8625–8631. [Google Scholar] [CrossRef]
- Murugachandran, S.I.; Tang, J.; Peña, I.; Loru, D.; Sanz, M.E. New Insights into Secondary Organic Aerosol Formation: Water Binding to Limonene. J. Phys. Chem. Lett. 2021, 12, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.R. New Opportunities and Emerging Themes of Research in Microwave Spectroscopy. Phil. Trans. R. Soc. A 2007, 365, 2813. [Google Scholar] [CrossRef] [PubMed]
- Kannengießer, R.; Klahm, S.; Nguyen, H.V.L.; Lüchow, A.; Stahl, W. The Effects of Methyl Internal Rotation and 14N Quadrupole Coupling in the Microwave Spectra of Two Conformers of N,N-Diethylacetamide. J. Chem. Phys. 2014, 141, 204308. [Google Scholar] [CrossRef] [PubMed]
- Caminati, W.; Wilson, E.B. Internal Hydrogen Bond, Torsional Motion, and Molecular Properties of 2-Methoxyethylamine by Microwave Spectroscopy: Methyl Barrier to Internal Rotation for 2-Methoxyethanol. J. Mol. Spectrosc. 1980, 81, 356–372. [Google Scholar] [CrossRef]
- Durig, J.R.; Groner, P.; Lin, J.; van der Veken, B.J. Structure of Methyl Cyanoformate from Microwave Spectroscopy and Ab Initio Calculations. J. Chem. Phys. 1992, 96, 8062. [Google Scholar] [CrossRef]
- Marstokk, K.-M.; Møllendal, H. Microwave Spectrum and Dipole Moment of Glycolaldehyde. J. Mol. Struct. 1970, 5, 205–213. [Google Scholar] [CrossRef]
- Balle, T.J.; Flygare, W.H. Fabry–Perot Cavity Pulsed Fourier Transform Microwave Spectrometer with a Pulsed Nozzle Particle Source. Rev. Sci. Instrum. 1981, 52, 33. [Google Scholar] [CrossRef]
- Grabow, J.-U. Fourier Transform Microwave Spectroscopy Measurement and Instrumentation. In Handbook of High-Resolution Spectroscopy; Wiley: New York, NY, USA, 2011. [Google Scholar] [CrossRef]
- Grabow, J.-U.; Stahl, W.; Dreizler, H. A Multioctave Coaxially Oriented Beam-Resonator Arrangement Fourier-Transform Microwave Spectrometer. Rev. Sci. Instrum. 1996, 67, 4072. [Google Scholar] [CrossRef]
- Grabow, J.-U.; Stahl, W. A Pulsed Molecular Beam Microwave Fourier Transform Spectrometer with Parallel Molecular Beam and Resonator Axes. Z. Naturforsch. 1990, 45, 1043–1044. [Google Scholar] [CrossRef]
- Pajski, J.J.; Logan, M.D.; Douglass, K.O.; Brown, G.G.; Suenram, R.D.; Dian, B.C.; Pate, B.H. Chirped-Pulse Fourier Transform Microwave Spectroscopy: A New Technique for Rapid Identification of Chemical Agents. Int. J. High Speed Electron. 2008, 18, 31–45. [Google Scholar] [CrossRef]
- Herbers, S.; Fritz, S.M.; Mishra, P.; Nguyen, H.V.L.; Zwier, T.S. Local and Global Approaches to Treat the Torsional Barriers of 4-Methylacetophenone Using Microwave Spectroscopy. J. Chem. Phys. 2020, 152, 074301. [Google Scholar] [CrossRef]
- Khemissi, S.; Pérez Salvador, A.; Nguyen, H.V.L. Large Amplitude Motions in 2,3-Dimethylfluorobenzene: Steric Effects Failing to Interpret Hindered Methyl Torsion. J. Phys. Chem. A 2021, 125, 8542–8548. [Google Scholar] [CrossRef]
- Mélan, J.; Khemissi, S.; Nguyen, H.V.L. Steric Effects on Two Inequivalent Methyl Internal Rotations of 3,4-Dimethylfluorobenzene. Spectrochim. Acta A 2021, 253, 119564. [Google Scholar] [CrossRef]
- Khemissi, S.; Nguyen, H.V.L. Two Equivalent Internal Rotations in the Microwave Spectrum of 2,6-Dimethylfluorobenzene. ChemPhysChem 2020, 21, 1682–1687. [Google Scholar] [CrossRef]
- Tudorie, M.; Kleiner, I.; Jahn, M.; Grabow, J.-U.; Goubet, M.; Pirali, O. Coupled Large Amplitude Motions: A Case Study of the Dimethylbenzaldehyde Isomers. J. Phys. Chem. A 2013, 117, 13636–13647. [Google Scholar] [CrossRef]
- Ferres, L.; Stahl, W.; Nguyen, H.V.L. Low torsional barrier challenges in the microwave spectrum of 2,4-dimethylanisole. J. Chem. Phys. 2019, 151, 104310. [Google Scholar] [CrossRef]
- Ferres, L.; Cheung, J.; Stahl, W.; Nguyen, H.V.L. Conformational Effect on the Large Amplitude Motions of 3,4-Dimethylanisole Explored by Microwave Spectroscopy. J. Phys. Chem. A 2019, 123, 3497–3503. [Google Scholar] [CrossRef]
- Ferres, L.; Truong, K.-N.; Stahl, W.; Nguyen, H.V.L. Interplay Between Microwave Spectroscopy and X-ray Diffraction: The Molecular Structure and Large Amplitude Motions of 2,3-Dimethylanisole. ChemPhysChem 2018, 19, 1781–1788. [Google Scholar] [CrossRef]
- Van, V.; Nguyen, T.; Stahl, W.; Nguyen, H.V.L.; Kleiner, I. Coupled Large Amplitude Motions: The Effects of Two Methyl Internal Rotations and 14N Quadrupole Coupling in 4,5-Dimethylthiazole Investigated by Microwave Spectroscopy. J. Mol. Struct. 2020, 1207, 127787. [Google Scholar] [CrossRef]
- Van, V.; Stahl, W.; Nguyen, H.V.L. The Structure and Torsional Dynamics of Two Methyl Groups in 2-Acetyl-5-methylfuran as Observed by Microwave Spectroscopy. ChemPhysChem 2016, 17, 3223–3228. [Google Scholar] [CrossRef]
- Nguyen, T.; Stahl, W.; Nguyen, H.V.L.; Kleiner, I. Local versus Global Approaches to Treat Two Equivalent Methyl Internal Rotations and 14N Nuclear Quadrupole Coupling of 2,5-Dimethylpyrrole. J. Chem. Phys. 2021, 154, 204304. [Google Scholar] [CrossRef]
- Van, V.; Bruckhuisen, J.; Stahl, W.; Ilyushin, V.; Nguyen, H.V.L. The Torsional Barriers of Two Equivalent Methyl Internal Rotations in 2,5-Dimethylfuran Investigated by Microwave Spectroscopy. J. Mol. Spectrosc. 2018, 343, 121–125. [Google Scholar] [CrossRef]
- Van, V.; Stahl, W.; Nguyen, H.V.L. Two Equivalent Methyl Internal Rotations in 2,5-Dimethylthiophene Investigated by Microwave Spectroscopy. Phys. Chem. Chem. Phys. 2015, 17, 32111–32114. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T. Far-Infrared Spectrum and Barrier to Internal Rotation in N-Methylaziridine. J. Mol. Spectrosc. 1970, 36, 268–283. [Google Scholar] [CrossRef]
- Fewster, S. Internal Rotation Studies in Some Small Aromatic Molecules; University of Manchester: Manchester, UK, 1970. [Google Scholar]
- Durig, J.R.; Bist, H.D.; Furic, K.; Qiu, J.; Little, T.S. Far Infrared Spectra and Barriers to Internal Rotation of Benzaldehyde, Benzoyl Fluoride, Benzoyl Chloride and Acetophenone. J. Mol. Struct. 1985, 129, 45–56. [Google Scholar] [CrossRef]
- Zachariou, A.; Hawkins, A.P.; Collier, P.; Howe, R.F.; Lennon, D.; Parker, S.F. The Methyl Torsion in Unsaturated Compounds. ACS Omega 2020, 5, 2755–2765. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.V.L.; Kleiner, I. Understanding (Coupled) Large Amplitude Motions: The Interplay of Microwave Spectroscopy, Spectral Modeling, and Quantum Chemistry. Phys. Sci. Rev. 2020, 7, 679–726. [Google Scholar] [CrossRef]
- Townes, C.H.; Schawlow, A.L. Microwave Spectroscopy; McGraw-Hill: New York, NY, USA, 1955. [Google Scholar]
- Pozdeev, N.M.; Gunderova, L.N.; Shapkin, A.A. Microwave Spectrum, Internal Rotation and Dipole Moment of 2-Methylthiophene. Opt. Spektrosk. 1970, 28, 254–259. [Google Scholar]
- Ogata, T.; Kozima, K. Microwave Spectrum, Barrier Height to Internal Rotation of Methyl Group of 3-Methylthiophene, and Dipole Moments of 3-Methylthiophene and Thiophene. J. Mol. Spectrosc. 1972, 42, 38–46. [Google Scholar] [CrossRef]
- Grabow, J.-U.; Hartwig, H.; Heineking, N.; Jäger, W.; Mäder, H.; Nicolaisen, H.W.; Stahl, W. The Microwave Spectrum of 2-Methylthiazole: Methyl Internal Rotation and 14N Nuclear Quadrupole Coupling. J. Mol. Struct. 2002, 612, 349–356. [Google Scholar] [CrossRef]
- Nguyen, T.; Van, V.; Gutlé, C.; Stahl, W.; Schwell, M.; Kleiner, I.; Nguyen, H.V.L. The Microwave Spectrum of 2-Methylthiazole: 14N Nuclear Quadrupole Coupling and Methyl Internal Rotation. J. Chem. Phys. 2020, 152, 134306. [Google Scholar] [CrossRef]
- Jäger, W.; Mäder, H. The Microwave Spectrum of 4-Methylthiazole: Methyl Internal Rotation, 14N Nuclear Quadrupole Coupling and Electric Dipole Moment. Z. Naturforsch. 1987, 42, 1405–1409. [Google Scholar] [CrossRef]
- Jäger, W.; Mäder, H. The Microwave Spectrum of 5-Methylthiazole: Methyl Internal Rotation, 14N Nuclear Quadrupole Coupling and Electric Dipole Moment. J. Mol. Struct. 1988, 190, 295–305. [Google Scholar] [CrossRef]
- Khemissi, S.; Schwell, M.; Kleiner, I.; Nguyen, H.V.L. Influence of π-Electron Conjugation Outside the Aromatic Ring on the Methyl Internal Rotation of 4-Methyl-5-vinylthiazole. Mol. Phys. 2022, 2022, e2052372. [Google Scholar] [CrossRef]
- Nicolaisen, H.-W.; Grabow, J.-U.; Heineking, N.; Stahl, W. The Microwave Spectrum of 4-Methylisothiazole. Z. Naturforsch. A 1991, 46, 635–638. [Google Scholar] [CrossRef]
- Van, V.; Nicolaisen, H.W.; Grabow, J.-U.; Heineking, N.; Stahl, W.; Nguyen, H.V.L. The Microwave Spectrum of 5-Methylisothiazole. 2022; Manuscript in preparation. [Google Scholar]
- Norris, W.G.; Krisher, L.C. Microwave Spectrum of 2-Methylfuran. J. Chem. Phys. 1969, 51, 403. [Google Scholar] [CrossRef]
- Ogata, T.; Kozima, K. Microwave Spectrum, Barrier Height to Internal Rotation of Methyl Group, and Dipole Moment of 3-Methylfuran. Bull. Chem. Soc. Jpn. 1971, 44, 2344–2346. [Google Scholar] [CrossRef] [Green Version]
- Fliege, E.R.L. A Reanalysis of 14N Nuclear Quadrupole Coupling and Methyl Internal Rotation in the Rotational Spectra of Monomethyl Oxazoles and Isoxazoles. Z. Naturforsch. A 1990, 45, 911–922. [Google Scholar] [CrossRef]
- Fliege, E.; Dreizler, H.; Meyer, M.; Iqbal, K.; Sheridan, J. 14N Nuclear Quadrupole Coupling and Methyl Internal Rotation of 2-, 4-, and 5-Methyl Oxazole. Z. Naturforsch. A 1986, 41, 623–636. [Google Scholar] [CrossRef]
- Fliege, E.; Dreizler, H.; Sheridan, J.; Walls, C.T. Internal Rotation and 14N Quadrupole Coupling of 3- and 5-Methylisoxazole. J. Mol. Spectrosc. 1985, 113, 362–372. [Google Scholar] [CrossRef]
- Jäger, W.; Dreizler, H.; Mäder, H.; Sheridan, J.; Walls, C.T. The Microwave Spectrum of 4-Methylisoxazole: 14N Nuclear Quadrupole Coupling, Methyl Internal Rotation and Electric Dipole Moment. Z. Naturforsch. A 1987, 42, 501–506. [Google Scholar] [CrossRef]
- Makarewicz, J.; Huber, S.; Brupbacher-Gatehouse, B.; Bauder, A. Internal Rotation Dependent Quadrupole Hyperfine Splittings of Rotational Transitions of N-Methylpyrrole. J. Mol. Struct. 2002, 612, 117–123. [Google Scholar] [CrossRef]
- Nguyen, T.; Dindic, C.; Stahl, W.; Nguyen, H.V.L.; Kleiner, I. 14N Nuclear Quadrupole Coupling and Methyl Internal Rotation in the Microwave Spectrum of 2-Methylpyrrole. Mol. Phys. 2020, 118, 1668572. [Google Scholar] [CrossRef]
- Nguyen, T.; Stahl, W.; Nguyen, H.V.L.; Kleiner, I. 14N Nuclear Quadrupole Coupling and Methyl Internal Rotation in 3-Methylpyrrole Investigated by Microwave Spectroscopy. J. Mol. Spectrosc. 2020, 372, 111351. [Google Scholar] [CrossRef]
- Gougoula, E.; Medcraft, C.; Heitkämper, M.; Walker, N.R. Barriers to Internal Rotation in Methylimidazole Isomers Determined by Rotational Spectroscopy. J. Chem. Phys. 2019, 151, 144301. [Google Scholar] [CrossRef]
- Saegebarth, E. Microwave Spectrum of Monomethylfurazan. J. Chem. Phys. 1970, 52, 1476. [Google Scholar] [CrossRef]
- Srivastava, K.S.L.; Narain, N.K. Microwave Spectrum and Barrier to Internal Rotation in N-Methyl Pyrazole. Indian J. Phys. B 1977, 51, 8–16. [Google Scholar]
- Jabri, A.; Van, V.; Nguyen, H.V.L.; Mouhib, H.; Tchana, F.K.; Manceron, L.; Stahl, W.; Kleiner, I. Laboratory Microwave, Millimeter Wave and Far-Infrared Spectra of Dimethyl Sulfide. Astron. Astrophys. 2016, 589, A127. [Google Scholar] [CrossRef] [Green Version]
- Neustock, W.; Guarnieri, A.; Demaison, J.; Wlodarczak, G. The Millimeter and Submillimeter-Wave Spectrum of Dimethylether. Z. Naturforsch. A 1990, 45, 702–706. [Google Scholar] [CrossRef]
- Hayashi, M.; Adachi, M.; Nakagawa, J. Microwave Spectrum, Structure, Dipole Moment, and Internal Rotation of the Trans Isomer of Ethyl Methyl Sulfide. J. Mol. Spectrosc. 1981, 86, 129–135. [Google Scholar] [CrossRef]
- Hayashi, M.; Kuwada, K. Microwave Spectrum, Structure, Dipole Moment and Internal Rotation of Trans-Ethylmethylether. J. Mol. Struct. 1975, 28, 147–161. [Google Scholar] [CrossRef]
- Tulimat, L.; Mouhib, H.; Nguyen, H.V.L.; Stahl, W. Laboratory Rotational Spectroscopy of Methyl n-Propyl Sulfide: Conformational Analysis and Methyl Internal Rotations. J. Mol. Spectrosc. 2020, 373, 111356. [Google Scholar] [CrossRef]
- Kato, H.; Nakagawa, J.; Hayashi, M. Microwave Spectrum, Structure, and Dipole Moment of the Trans-Trans Isomer of Methylpropylether. J. Mol. Spectrosc. 1980, 80, 272–278. [Google Scholar] [CrossRef]
- Hensel, K.D.; Gerry, M.C.L. Microwave Spectrum of Tetrolyl Fluoride. J. Chem. Soc. Faraday Trans. 1994, 90, 3023–3027. [Google Scholar] [CrossRef]
- Stolwijk, V.M.; van Eijck, B.P. Microwave Spectrum and Barrier to Internal Rotation of 1-Chloro-2-butyne. J. Mol. Spectrosc. 1987, 124, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Eibl, K.; Kannengießer, R.; Stahl, W.; Nguyen, H.V.L.; Kleiner, I. Low Barrier Methyl Rotation in 3-Pentyn-1-ol as Observed by Microwave Spectroscopy. Mol. Phys. 2016, 114, 3483–3489. [Google Scholar] [CrossRef]
- Eibl, K.; Stahl, W.; Kleiner, I.; Nguyen, H.V.L. Conformational Effect on the Almost Free Internal Rotation in 4-Hexyn-3-ol Studied by Microwave Spectroscopy and Quantum Chemistry. J. Chem. Phys. 2018, 149, 144306. [Google Scholar] [CrossRef]
- Ferres, L.; Mouhib, H.; Stahl, W.; Nguyen, H.V.L. Methyl Internal Rotation in the Microwave Spectrum of o-Methyl Anisole. ChemPhysChem 2017, 18, 1855–1859. [Google Scholar] [CrossRef]
- Herbers, S.; Buschmann, P.; Wang, J.; Lengsfeld, K.G.; Nair, K.P.R.; Grabow, J.-U. Reactivity and Rotational Spectra: The Old Concept of Substitution Effects. Phys. Chem. Chem. Phys. 2020, 22, 11490–11497. [Google Scholar] [CrossRef]
- Nair, K.P.R.; Herbers, S.; Grabow, J.-U.; Nguyen, H.V.L. Neighborhood Matters: Steric Effects on Methyl Internal Rotation and Chlorine Nuclear Quadrupole Coupling in 2-Fluoro-4-chlorotoluene. J. Mol. Struct. 2021, 1246, 131096. [Google Scholar] [CrossRef]
- Nair, K.P.R.; Herbers, S.; Bailey, W.C.; Obenchain, D.A.; Lesarri, A.; Grabow, J.-U.; Nguyen, H.V.L. Internal Rotation and Chlorine Nuclear Quadrupole Coupling in 2-Chloro-4-fluorotoluene Explored by Microwave Spectroscopy and Quantum Chemistry. Spectrochim. Acta A 2021, 247, 119120. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, E.G.; Zenchyzen, B.L.M.; Jäger, W. High-Resolution Fourier-Transform Microwave Spectroscopy of Methyl-and Dimethylnapthalenes. Astrophys. J. 2015, 805, 141. [Google Scholar] [CrossRef] [Green Version]
- Dindić, C.; Ludovicy, J.; Terzi, V.; Lüchow, A.; Vogt, N.; Demaison, J.; Nguyen, H.V.L. Determination of the Semiexperimental Equilibrium Structure of 2-Acetylthiophene in the Presence of Methyl Internal Rotation and Substituent Effects Compared to Thiophene. Phys. Chem. Chem. Phys. 2022, 24, 3804–3815. [Google Scholar] [CrossRef]
- Dindić, C.; Lüchow, A.; Vogt, N.; Demaison, J.; Nguyen, H.V.L. Equilibrium Structure in the Presence of Methyl Internal Rotation: Microwave Spectroscopy and Quantum Chemistry Study of the Two Conformers of 2-Acetylfuran. J. Phys. Chem. A 2021, 125, 4986–4997. [Google Scholar] [CrossRef]
- Hakiri, R.; Derbel, N.; Nguyen, H.V.L.; Mouhib, H. Communication Through the Furan Ring: Conformational Effect on the Internal Rotation of 5-Methyl Furfural Studied by Microwave Spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 25577–25582. [Google Scholar] [CrossRef]
- Andresen, M.; Schöngen, D.; Kleiner, I.; Schwell, M.; Stahl, W.; Nguyen, H.V.L. Internal Rotation of the Acetyl Methyl Group in Methyl Alkyl Ketones: The Microwave Spectrum of Octan-2-one. ChemPhysChem 2020, 21, 2206. [Google Scholar] [CrossRef]
- Kao, J.; Radom, L. Conformations, Stabilities, and Charge Distributions in 2-and 3-Monosubstituted Thiophenes. An Ab Initio Molecular Orbital Study. J. Am. Chem. Soc. 1979, 101, 311–318. [Google Scholar] [CrossRef]
- Rudolph, H.D.; Dreizler, H.; Jaeschke, A.; Wendling, P. Mikrowellenspektrum, Hinderungspotential der internen Rotation und Dipolmoment des Toluols. Z. Naturforsch. A 1967, 22, 940–944. [Google Scholar] [CrossRef]
- Kreiner, W.A.; Rudolph, H.D.; Tan, B.T. Microwave Spectra of Several Molecular Isotopes of Toluene. J. Mol. Spectrosc. 1973, 48, 86–99. [Google Scholar] [CrossRef]
- Amir-Ebrahimi, V.; Choplin, A.; Demaison, J.; Roussy, G. Microwave Spectrum of the 13C-Ring-Monosubstituted Toluenes and Structure of Toluene. J. Mol. Spectrosc. 1981, 89, 42–52. [Google Scholar] [CrossRef]
- Kisiel, Z.; Bialkowska-Jaworska, E.; Pszczólkowski, L.; Mäder, H. Ground State Rotational Spectrum of Toluene. J. Mol. Spectrosc. 2004, 227, 109–113. [Google Scholar] [CrossRef]
- Ilyushin, V.V.; Kisiel, Z.; Pszczółkowski, L.; Mäder, H.; Hougen, J.T. A New Torsion–Rotation Fitting Program for Molecules with a Sixfold Barrier: Application to the Microwave Spectrum of Toluene. J. Mol. Spectrosc. 2010, 259, 26–38. [Google Scholar] [CrossRef]
- Ilyushin, V.V.; Alekseev, E.A.; Kisiel, Z.; Pszczółkowski, L. High-J Rotational Spectrum of Toluene in |m| ≤ 3 Torsional States. J. Mol. Spectrosc. 2017, 339, 31–39. [Google Scholar] [CrossRef]
- Welzel, A.; Hellweg, A.; Merke, I.; Stahl, W. Structural and Torsional Properties of o-Cresol and o-Cresol-OD as Obtained from Microwave Spectroscopy and ab Initio Calculations. J. Mol. Spectrosc. 2002, 215, 58–65. [Google Scholar] [CrossRef]
- Hellweg, A.; Hättig, C.; Merke, I.; Stahl, W. Microwave and Theoretical Investigation of the Internal Rotation in m-Cresol. J. Chem. Phys. 2006, 124, 204305. [Google Scholar] [CrossRef]
- Hellweg, A.; Hättig, C. On the Internal Rotations in p-Cresol in its Ground and First Electronically Excited States. J. Chem. Phys. 2007, 127, 024307. [Google Scholar] [CrossRef]
- Hellweg, A. Determining the Internal Rotations of p-Thiocresol. Chem. Phys. Lett. 2009, 475, 198–201. [Google Scholar] [CrossRef]
- Schmitz, D.; Shubert, V.A.; Giuliano, B.M.; Schnell, M. The Broadband Microwave Spectra of the Monoterpenoids Thymol and Carvacrol: Conformational Landscape and Internal Dynamics. J. Chem. Phys. 2014, 141, 034304. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Castillo, A.O.; Calabrese, C.; Fritz, S.M.; Uriarte, I.; Cocinero, E.J.; Zwier, T.S. Bond Length Alternation and Internal Dynamics in Model Aromatic Substituents of Lignin. ChemPhysChem 2022, 23, e202100808. [Google Scholar] [CrossRef]
- Ferres, L.; Stahl, W.; Nguyen, H.V.L. Conformational Effects on the Torsional Barriers in m-Methylanisole Studied by Microwave Spectroscopy. J. Chem. Phys. 2018, 148, 124304. [Google Scholar] [CrossRef] [PubMed]
- Herbers, S.; Nguyen, H.V.L. Next Level Achievement of the XIAM Code in Modeling the Microwave Spectrum of m-Methylanisole. J. Mol. Spectrosc. 2020, 370, 111289. [Google Scholar] [CrossRef]
- Ferres, L.; Stahl, W.; Kleiner, I.; Nguyen, H.V.L. The Effect of Internal Rotation in p-Methyl Anisole Studied by Microwave Spectroscopy. J. Mol. Spectrosc. 2018, 343, 44–49. [Google Scholar] [CrossRef]
- Écija, P.; Evangelisti, L.; Vallejo, M.; Basterretxea, F.J.; Lesarri, A.; Castanño, F.; Caminati, W.; Cocinero, E.J. Conformational Flexibility of Mephenesin. J. Phys. Chem. B 2014, 118, 5357–5364. [Google Scholar] [CrossRef]
- Obenchain, D.A.; Pinacho, P.; Zinn, S.; Schnell, M. The Low-Barrier Methyl Internal Rotation in the Rotational Spectrum of 3-Methylphenylacetylene. J. Mol. Struct. 2020, 1213, 128109. [Google Scholar] [CrossRef]
- Shirar, A.J.; Wilcox, D.S.; Hotopp, K.M.; Storck, G.L.; Kleiner, I.; Dian, B.C. Impact of Molecular Conformation on Barriers to Internal Methyl Rotation: The Rotational Spectrum of m-Methylbenzaldehyde. J. Phys. Chem. A 2010, 114, 12187–12194. [Google Scholar] [CrossRef]
- Saal, H.; Grabow, J.-U.; Walker, A.H.; Hougen, J.; Kleiner, I.; Caminati, W. Microwave Study of Internal Rotation in para-Tolualdehyde: Local versus Global Symmetry Effects at the Methyl-Rotor Site. J. Mol. Spectrosc. 2018, 351, 55–61. [Google Scholar] [CrossRef]
- Schnitzler, E.G.; Zenchyzen, B.L.M.; Jäger, W. Rotational Spectroscopy of the Atmospheric Photo-Oxidation Product o-Toluic Acid and its Monohydrate. Phys. Chem. Chem. Phys. 2016, 18, 448–457. [Google Scholar] [CrossRef]
- Al-Jabiri, M.; Schnitzler, E.G.; Seifert, N.A.; Jäger, W. Microwave Spectroscopic Study of the Atmospheric Oxidation Product m-Toluic Acid and its Monohydrate. RH05. In Proceedings of the 72nd International Symposium on Molecular Spectroscopy (ISMS), Urbana-Champaign, IL, USA, 19–23 June 2017. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Zhang, J.; Feng, G.; Grabow, J.-U.; Gou, Q. Conformational Preference Determined by Inequivalent n-Pairs: Rotational Studies on Acetophenone and its Monohydrate. Phys. Chem. Chem. Phys. 2019, 21, 22888–22894. [Google Scholar] [CrossRef]
- Cocinero, E.J.; Basterretxea, F.J.; Écija, P.; Lesarri, A.; Fernández, J.A.; Castaño, F. Conformational Behaviour, Hydrogen Bond Competition and Intramolecular Dynamics in Vanillin Derivatives: Acetovanillone and 6-Hydroxy-3-methoxyacetophenone. Phys. Chem. Chem. Phys. 2011, 13, 13310–13318. [Google Scholar] [CrossRef]
- Herbers, S.; Zingsheim, O.; Nguyen, H.V.L.; Bonah, L.; Heyne, B.; Wehres, N.; Schlemmer, S. Internal Rotation Arena: Program Performances on the Low Barrier Problem of 4-Methylacetophenone. J. Chem. Phys. 2021, 155, 224302. [Google Scholar] [CrossRef]
- Jacobsen, S.; Andresen, U.; Mäder, H. Microwave Spectra of o-Fluorotoluene and Its 13C Isotopic Species: Methyl Internal Rotation and Molecular Structure. Struct. Chem. 2003, 14, 217–225. [Google Scholar] [CrossRef]
- Rudolph, H.D.; Trinkaus, A. Mikrowellenspektrum, Hinderungspotential der internen Rotation und Dipolmoment des meta-Fluortoluols. Z. Naturforsch. A 1968, 23, 68–76. [Google Scholar] [CrossRef]
- Nair, K.P.R.; Herbers, S.; Nguyen, H.V.L.; Grabow, J.-U. The Structure and Low-Barrier Methyl Torsion of 3-Fluorotoluene. Spectrochim. Acta. A 2020, 242, 118709. [Google Scholar] [CrossRef]
- Rottstegge, J.; Hartwig, H.; Dreizler, H. The Rotational Spectrum, Structure and Barrier V6 to Internal Rotation of p-Fluorotoluene. J. Mol. Struct. 1999, 478, 37–47. [Google Scholar] [CrossRef]
- Nair, K.P.R.; Herbers, S.; Grabow, J.-U.; Lesarri, A. Internal Rotation in Halogenated Toluenes: Rotational Spectrum of 2,3-Difluorotoluene. J. Mol. Spectrosc. 2018, 349, 37–42. [Google Scholar] [CrossRef]
- Nair, K.P.R.; Herbers, S.; Obenchain, D.A.; Grabow, J.-U.; Lesarri, A. The Low Internal Rotation Barriers of Halogenated Toluenes: Rotational Spectrum of 2,4-Difluorotoluene. J. Mol. Spectrosc. 2018, 344, 21–26. [Google Scholar] [CrossRef]
- Nair, K.P.R.; Wachsmuth, D.; Grabow, J.-U.; Lesarri, A. Internal Rotation in Halogenated Toluenes: Rotational Spectrum of 2,5-Difluorotoluene. J. Mol. Spectrosc. 2017, 337, 46–50. [Google Scholar] [CrossRef]
- Nair, K.P.R.; Jahn, M.K.; Lesarri, A.; Ilyushin, V.V.; Grabow, J.-U. Six-Fold-Symmetry Internal Rotation in Toluenes: The Low Barrier Challenge of 2,6- and 3,5-Difluorotoluene. Phys. Chem. Chem. Phys. 2015, 17, 26463–26470. [Google Scholar] [CrossRef] [Green Version]
- Nair, K.P.R.; Herbers, S.; Grabow, J.-U. Structure and Methyl Torsion of Halogenated Toluenes: Rotational Spectrum of 3,4-Difluorotoluene. J. Mol. Spectrosc. 2019, 355, 19–25. [Google Scholar] [CrossRef]
- Nair, K.P.R.; Herbers, S.; Obenchain, D.A.; Grabow, J.-U. Internal Methyl Rotation and Molecular Structure of Trifluorotoluenes: Microwave Rotational Spectra of 2,3,4- and 2,4,5-Trifluorotoluene. Can. J. Phys. 2020, 98, 543. [Google Scholar] [CrossRef]
- Nair, K.P.R.; Herbers, S.; Grabow, J.-U.; Lesarri, A. Molecular Systems with Nearly-Free Internal Rotation and Nuclear Quadrupole Coupling: Meta-Chlorotoluene. J. Mol. Spectrosc. 2019, 361, 1–7. [Google Scholar] [CrossRef]
- Schubert, V.A.; Schmitz, D.; Schnell, M. Communication Through the Phenyl Ring: Internal Rotation and Nuclear Quadrupole Splitting in p-Halotoluenes. Mol. Phys. 2013, 111, 2189–2197. [Google Scholar] [CrossRef]
- Dreizler, H.; Rudolph, H.D.; Mäder, H. Mikrowellenspektrum, Hinderungspotential der internen Rotation, Quadrupolkopplungskonstanten und Dipolmoment des 2-Methyl-Pyridins. Z. Naturforsch. A 1970, 25, 25–35. [Google Scholar] [CrossRef]
- Wörmke, S.; Brendel, K.; Andresen, U.; Mäder, H. A Molecular Beam Fourier Transform Microwave Study of 2-Methylpyridine and its Complex with Argon: Structure, Methyl Internal Rotation and 14N Nuclear Quadrupole Coupling. Mol. Phys. 2004, 102, 1625–1639. [Google Scholar] [CrossRef]
- Heineking, N.; Dreizler, H.; Endo, K.; Kamura, Y. High Resolution Microwave Spectra of Pyridine-N-oxideanda-Picoline-N-oxide. Z. Naturforsch. A 1989, 44, 1196–1200. [Google Scholar] [CrossRef]
- Rudolph, H.D.; Dreizler, H.; Seiler, H. Hinderungspotential der internen Rotation, Dipolmoment und Quadrupolkopplungskonstanten aus dem Mikrowellenspektrum des 4-Methyl-Pyridins. Z. Naturforsch. A 1967, 22, 1738–1743. [Google Scholar] [CrossRef]
- Jaman, A.I.; Maiti, S.; Nandi, R.N. Microwave Spectrum and Barrier to Internal Rotation in ortho-Tolunitrile. J. Mol. Spectrosc. 1998, 192, 148–151. [Google Scholar] [CrossRef]
- Hansen, N.; Mäder, H.; Bruhn, T. A Molecular Beam Fourier Transform Microwave Study of o-Tolunitrile: 14N Nuclear Quadrupole Coupling and Methyl Internal Rotation Effects. Mol. Phys. 1999, 97, 587–595. [Google Scholar] [CrossRef]
- Bruhn, T.; Mäder, H. The Microwave Spectrum of m-Tolunitrile: Methyl Internal Rotation and 14N Nuclear Quadrupole Coupling. J. Mol. Spectrosc. 2000, 200, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Roucou, A.; Goubet, M.; Kleiner, I.; Bteich, S.; Cuisset, A. Large Amplitude Torsions in Nitrotoluene Isomers Studied by Rotational Spectroscopy and Quantum Chemistry Calculations. ChemPhysChem 2020, 21, 2523–2538. [Google Scholar] [CrossRef]
- Roucou, A.; Kleiner, I.; Goubet, M.; Bteich, S.; Mouret, G.; Bocquet, R.; Hindle, F.; Meerts, W.L.; Cuisset, A. Towards the Detection of Explosive Taggants: Microwave and Millimetre-Wave Gas-Phase Spectroscopies of 3-Nitrotoluene. ChemPhysChem 2018, 19, 1056–1067. [Google Scholar] [CrossRef]
- Bird, R.G.; Pratt, D.W. Methyl rotors in the gas phase: A Study of o- and m-Toluidine by Chirped-Pulse Fourier Transform Microwave Spectroscopy. J. Mol. Spectrosc. 2011, 266, 81–85. [Google Scholar] [CrossRef]
- Hellweg, A. Inversion, Internal Rotation, and Nitrogen Nuclear Quadrupole Coupling of p-Toluidine as Obtained from Microwave Spectroscopy and Ab Initio Calculations. Chem. Phys. 2008, 344, 281–290. [Google Scholar] [CrossRef]
- Caminanti, W.; Cazzoli, G.; Troiano, D. 2-Methyl-Pyrimidine: Determination of V6 Barrier and Dipole Moment by Microwave Spectroscopy and Comparison with 5-Methyl-Pyrimidine. Chem. Phys. Lett. 1976, 43, 65–68. [Google Scholar] [CrossRef]
- Nguyen, T. Structures and Internal Dynamics of Methylated Nitrogen Containing Aromatic Rings Probed by Means of Microwave Spectroscopy, Quantum Chemical Calculation and Spectral Modelling; Université de Paris: Paris, France, 2021. [Google Scholar]
- Caminati, W.; Cazzoli, G.; Mirri, A.M. Microwave Spectrum, Barrier to Internal Rotation and Dipole Moment in 5-Methyl-pyrimidine. Chem. Phys. Lett. 1975, 31, 104–107. [Google Scholar] [CrossRef]
- López, J.C.; Peña, M.I.; Sanz, M.E.; Alonso, J.L. Probing Thymine with Laser Ablation Molecular Beam Fourier Transform Microwave Spectroscopy. J. Chem. Phys. 2007, 126, 191103. [Google Scholar] [CrossRef]
- Gurusinghe, R.M.; Tubergen, M.J. Probing the Electronic Environment of Methylindoles using Internal Rotation and 14N Nuclear Quadrupole Coupling. J. Phys. Chem. A 2016, 120, 3491–3496. [Google Scholar] [CrossRef]
- Tan, X.-Q.; Majewski, W.A.; Plusquellic, D.F.; Pratt, D.W. Methyl group torsional dynamics from rotationally resolved electronic spectra. 1- and 2-methylnaphthalene. J. Chem. Phys. 1991, 94, 7721. [Google Scholar] [CrossRef]
- Brannys, G. Internal Rotation of Two Inequivalent Methyl Groups in 2,3-Dimethylfurane Studied by Microwave Spectroscopy and Quantum Chemistry. Bachelor’s Thesis, RWTH Aachen University, Aachen, Germany, 2021. [Google Scholar]
- Barth, M.; Nguyen, H.V.L. Microwave Spectroscopic and Quantum Chemical Investigations on 2,4-Dimethylpyrrole. Poster A70. In Proceedings of the 27th Colloquium on High-Resolution Molecular Spectroscopy (HRMS), Cologne, Germany, 30 August–3 September 2021. [Google Scholar]
- Khemissi, S.; Van, V.; Kleiner, I.; Schwell, M.; Nguyen, H.V.L. Two Inequivalent Methyl Internal Rotations and 14N Quadrupole Coupling in the Microwave Spectrum of 2,4-Dimethylthiazole. 2022; Manuscript in preparation. [Google Scholar]
- Dindić, C.; Nguyen, H.V.L. Microwave Spectrum of Two-Top Molecule: 2-Acetyl-3-methylthiophene. ChemPhysChem 2021, 22, 2420–2428. [Google Scholar] [CrossRef]
- Dindić, C.; Barth, M.; Nguyen, H.V.L. Two Methyl Internal Rotations of 2-Acetyl-4-methylthiophene Explored by Microwave Spectroscopy and Quantum Chemistry. Spectrochim. Acta A 2022, 121505. [Google Scholar] [CrossRef]
- Dindić, C.; Nguyen, H.V.L. Benchmarking Acetylthiophene Derivatives: Methyl Internal Rotations in the Microwave Spectrum of 2-Acetyl-5-Methylthiophene. Phys. Chem. Chem. Phys. 2022; Manuscript to be submitted. [Google Scholar]
- Rudolph, H.D.; Walzer, K.; Krutzik, I. Microwave spectrum, barrier for methyl rotation, methyl conformation, and dipole moment of ortho-xylene. J. Mol. Spectrosc. 1973, 47, 314–339. [Google Scholar] [CrossRef]
- Thomsen, C.; Dreizler, H. The Microwave Spectra of m-Xylene and m-Xylene-d10. Determination of the Low Methyl Internal Rotation Barrier. Z. Naturforsch. A 2001, 56, 635–640. [Google Scholar] [CrossRef]
- Ferres, L. Quantum Chemical and Microwave Spectroscopic Investigations on Phenyl Ring Containing Molecules; RWTH Aachen University: Aachen, Germany, 2019. [Google Scholar]
- Sørensen, G.O.; Pedersen, T.; Dreizler, H.; Guarnieri, A.; Cox, A.P. Microwave Spectra of Nitromethane and D3-Nitromethane. J. Mol. Struct. 1983, 97, 77–82. [Google Scholar] [CrossRef]
- Ferres, L.; Spautz, J.; Stahl, W.; Nguyen, H.V.L. Lowering the Torsional Barriers by Sterical Hindrance: Microwave Spectrum of the Three-Top Molecule 2,6-Dimethylanisole. TA07. In Proceedings of the Virtual International Symposium on Molecular Spectroscopy (ISMS), Virtual, 21–25 June 2021. [Google Scholar] [CrossRef]
- Khemissi, S.; Kleiner, I.; Schwell, M.; Nguyen, H.V.L. The Microwave Spectrum of 2,4-Dimethylfluorobenzene. 2022; Manuscript in preparation. [Google Scholar]
- Sun, H.; Khemissi, S.; Kleiner, I.; Schwell, M.; Nguyen, H.V.L. The Microwave Spectrum of 2,5-Dimethylfluorobenzene. 2022; Manuscript in preparation. [Google Scholar]
- Onda, M.; Toda, A.; Mori, S.; Yamaguchi, I. Microwave Spectrum of Anisole. J. Mol. Struct. 1986, 144, 47–51. [Google Scholar] [CrossRef]
- Ferres, L.; Stahl, W.; Nguyen, H.V.L. The Molecular Structure of Phenetole Studied by Microwave Spectroscopy and Quantum Chemical Calculations. Mol. Phys. 2016, 114, 2788–2793. [Google Scholar] [CrossRef] [Green Version]
- Kakar, R.K.; Rinehart, E.A.; Quade, C.R.; Kojima, T. Microwave Spectrum of Benzaldehyde. J. Chem. Phys. 1970, 52, 3803. [Google Scholar] [CrossRef]
- Utzat, K.A.; Bohn, R.K.; Montgomery, J.A., Jr.; Michels, H.H.; Caminati, W. Rotational Spectrum, Tunneling Motions, and Potential Barriers of Benzyl Alcohol. J. Phys. Chem. A 2010, 114, 6913–6916. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, L.; Caminati, W. Modeling the Internal Rotation Tunnelling in Benzyl Alcohol by Ring Fluorination: The Rotational Spectrum of 3,5-Difluorobenzyl Alcohol. Chem. Phys. Lett. 2019, 737S, 100004. [Google Scholar] [CrossRef]
- Aviles Moreno, J.-R.; Petitprez, D.; Huet, T.R. The Conformational Flexibility in N-Phenylformamide: An Ab Initio Approach Supported by Microwave Spectroscopy. Chem. Phys. Lett. 2006, 419, 411–416. [Google Scholar] [CrossRef]
- Cabezas, C.; Varela, M.; Caminati, W.; Mata, S.; López, J.C.; Alonso, J.L. The Two Conformers of Acetanilide Unraveled Using LA-MB-FTMW Spectroscopy. J. Mol. Spectrosc. 2011, 268, 42–46. [Google Scholar] [CrossRef]
- Ferres, L.; Mouhib, H.; Stahl, W.; Schwell, M.; Nguyen, H.V.L. Molecular Structure and Ring Tunneling of Phenyl Formate as Observed by Microwave Spectroscopy and Quantum Chemistry. J. Mol. Spectrosc. 2017, 337, 59–64. [Google Scholar] [CrossRef]
- Evangelisti, L.; Maris, A.; Melandri, S.; Caminati, W. Internal Dynamics in Phenylacetate. Poster D42. In Proceedings of the 22nd International Conference on High Resolution Molecular Spectroscopy (HRMS), Praha, Czech Republic, 4–9 September 2012. [Google Scholar]
- Ferres, L.; Evangelisti, L.; Maris, A.; Melandri, S.; Stahl, W.; Caminati, W.; Nguyen, H.V.L. Skeletal Torsion Tunneling and Methyl Internal Rotation: The Coupled Large Amplitude Motions in Phenyl Acetate. Molecules 2022, 27, 2730. [Google Scholar] [CrossRef]
- Nguyen, H.V.L.; Stahl, W. The Microwave Spectrum of Isopropenyl Acetate: An Asymmetric Molecule with Two Internal Rotors. J. Mol. Spectrosc. 2010, 264, 120–124. [Google Scholar] [CrossRef]
- Ferres, L.; Cheung, J.; Stahl, W.; Nguyen, H.V.L. Microwave Spectroscopic and Quantum Chemical Studies of the Coupled Large Amplitude Motions in S-Phenyl Thioacetate. A3.4. In Proceedings of the 25th International Conference on High Resolution Molecular Spectroscopy (HRMS), Bilbao, Spain, 3–7 September 2018. [Google Scholar]
- Ilyushin, V.V.; Cloessner, E.A.; Chou, Y.-C.; Picraux, L.B.; Hougen, J.T.; Lavrich, R. A Microwave Study of Hydrogen-Transfer-Triggered Methyl-Group Rotation in 5-Methyltropolone. J. Chem. Phys. 2010, 133, 184307. [Google Scholar] [CrossRef] [Green Version]
- Ilyushin, V.V.; Alekseev, E.A.; Chou, Y.-C.; Hsu, Y.-C.; Hougen, J.T.; Lovas, F.J.; Picraux, L.B. Reinvestigation of the Microwave Spectrum of 2-Methylmalonaldehyde. J. Mol. Spectrosc. 2008, 251, 56–63. [Google Scholar] [CrossRef]
- Ilyushin, V.V.; Johnson, A.M.; Hohl, J.; Cloessner, E.A.; Lovas, F.J.; Lavrich, R.J. Isotopic Dependence of the Hydrogen-Transfer-Triggered Methyl-Group Rotation in Deuterated 5-Methyltropolone. J. Mol. Spectrosc. 2018, 343, 76–80. [Google Scholar] [CrossRef]
- Caminati, W.; Damiani, D.; Corbelli, G.; Favero, L.B. Ring Puckering Motion in Indan: A Microwave Spectroscopy Study. Mol. Phys. 1992, 75, 857–865. [Google Scholar] [CrossRef]
- Meyer, R. Flexible Models for Intramolecular Motion, a Versatile Treatment and its Application to Glyoxal. J. Mol. Spectrosc. 1979, 76, 266–300. [Google Scholar] [CrossRef]
- Caminati, W.; Favero, L.B.; Velino, B.; Zerbetto, F. A Study of the Large Amplitude Motions of Indoline through Microwave Spectroscopy and Ab Initio Calculations. Mol. Phys. 1993, 78, 1561–1574. [Google Scholar] [CrossRef]
- Fantoni, A.C.; Caminati, W. Large Amplitude Motions in 2,3-Cyclopentenopyridine. J. Mol. Spectrosc. 1997, 186, 105–112. [Google Scholar] [CrossRef]
- Ottaviani, P.; Caminati, W. Ring-Puckering and Anomeric Effect in Coumaran. Chem. Phys. Lett. 2005, 405, 68–72. [Google Scholar] [CrossRef]
- Caminati, W.; Melandri, S.; Corbelli, G.; Favero, L.B.; Meyer, R. Chair Conformation and Barrier to Ring Puckering in 1,3-Benzodioxole. Mol. Phys. 1993, 80, 1297–1315. [Google Scholar] [CrossRef]
- Caminati, W.; Damiani, D.; Favero, L.B. Non-Planarity and Barrier to Ring Puckering in Phthalan. Mol. Phys. 1993, 79, 699–708. [Google Scholar] [CrossRef]
- Caminati, W.; Corbelli, G.; Favero, L.B. Planarity and Low-Energy Vibrations of Catecholborane: A Microwave Spectroscopic Study. J. Chem. Soc. Faraday Trans. 1993, 89, 1631–1636. [Google Scholar] [CrossRef]
























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, H.V.L.; Caminati, W.; Grabow, J.-U. The LAM of the Rings: Large Amplitude Motions in Aromatic Molecules Studied by Microwave Spectroscopy. Molecules 2022, 27, 3948. https://doi.org/10.3390/molecules27123948
Nguyen HVL, Caminati W, Grabow J-U. The LAM of the Rings: Large Amplitude Motions in Aromatic Molecules Studied by Microwave Spectroscopy. Molecules. 2022; 27(12):3948. https://doi.org/10.3390/molecules27123948
Chicago/Turabian StyleNguyen, Ha Vinh Lam, Walther Caminati, and Jens-Uwe Grabow. 2022. "The LAM of the Rings: Large Amplitude Motions in Aromatic Molecules Studied by Microwave Spectroscopy" Molecules 27, no. 12: 3948. https://doi.org/10.3390/molecules27123948
APA StyleNguyen, H. V. L., Caminati, W., & Grabow, J. -U. (2022). The LAM of the Rings: Large Amplitude Motions in Aromatic Molecules Studied by Microwave Spectroscopy. Molecules, 27(12), 3948. https://doi.org/10.3390/molecules27123948