
Thiosemicarbazide-Substituted Coumarins as Selective Inhibitors of the Tumor Associated Human Carbonic Anhydrases IX and XII

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
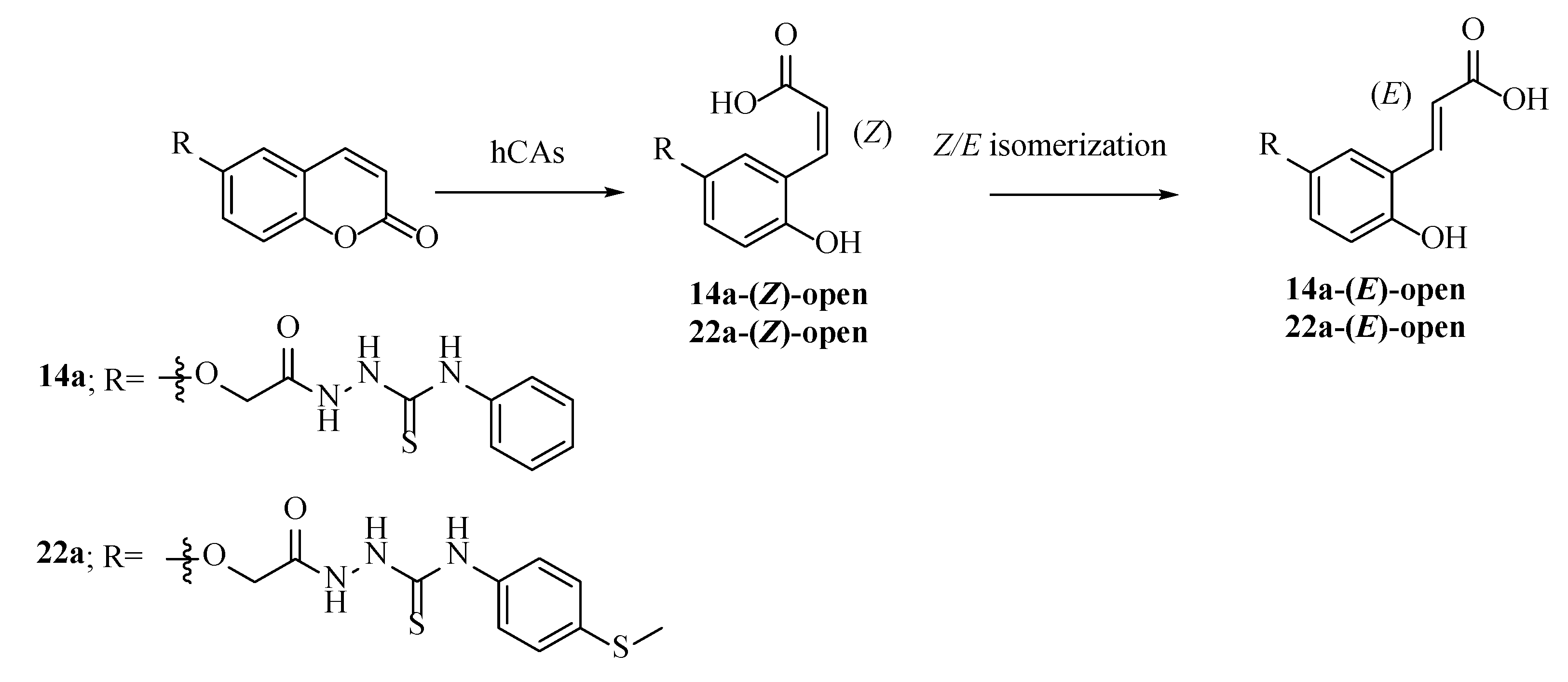
2.1. Synthesis of Coumarins 14a,b–22a,b and 23b
2.2. CA In Vitro Inhibition Assay
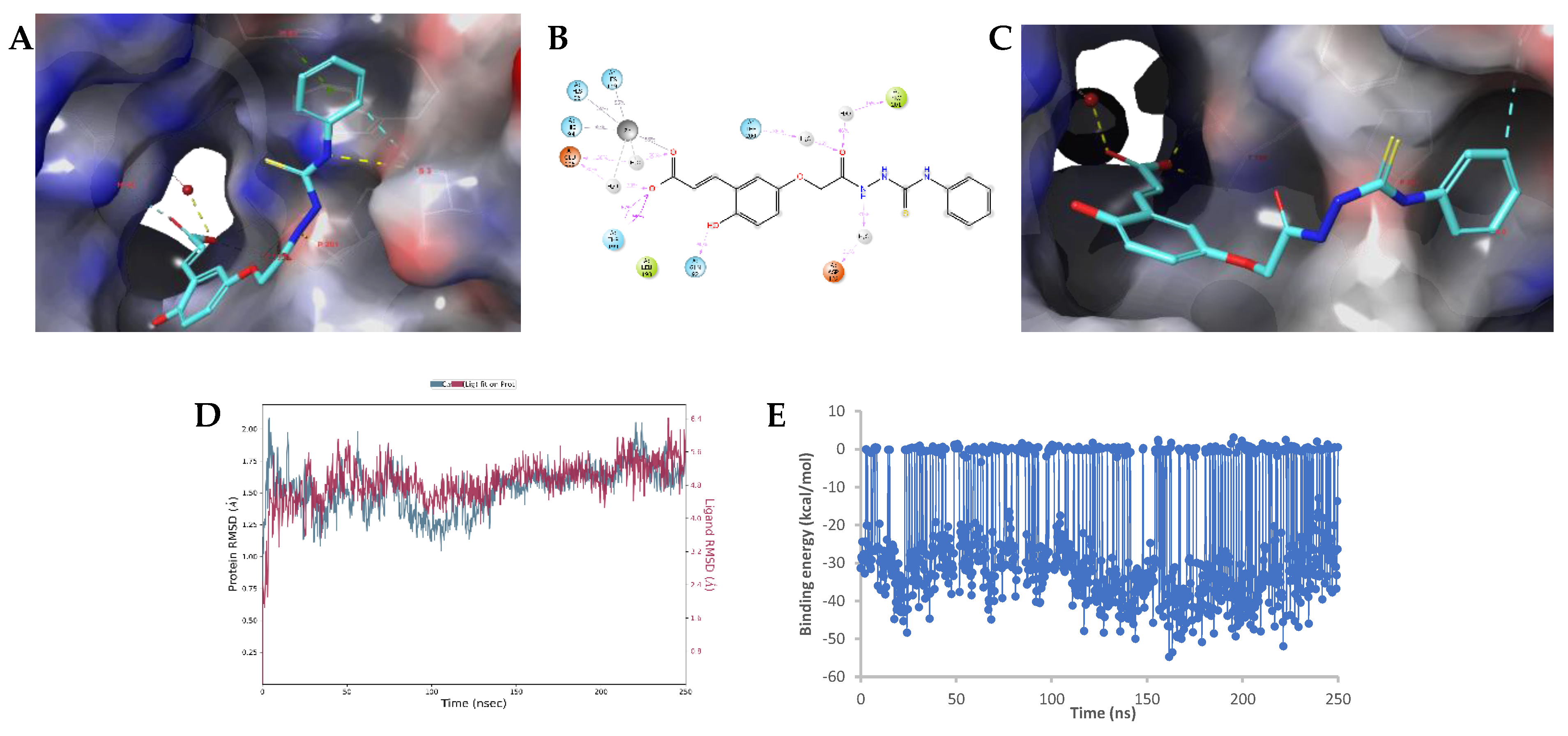
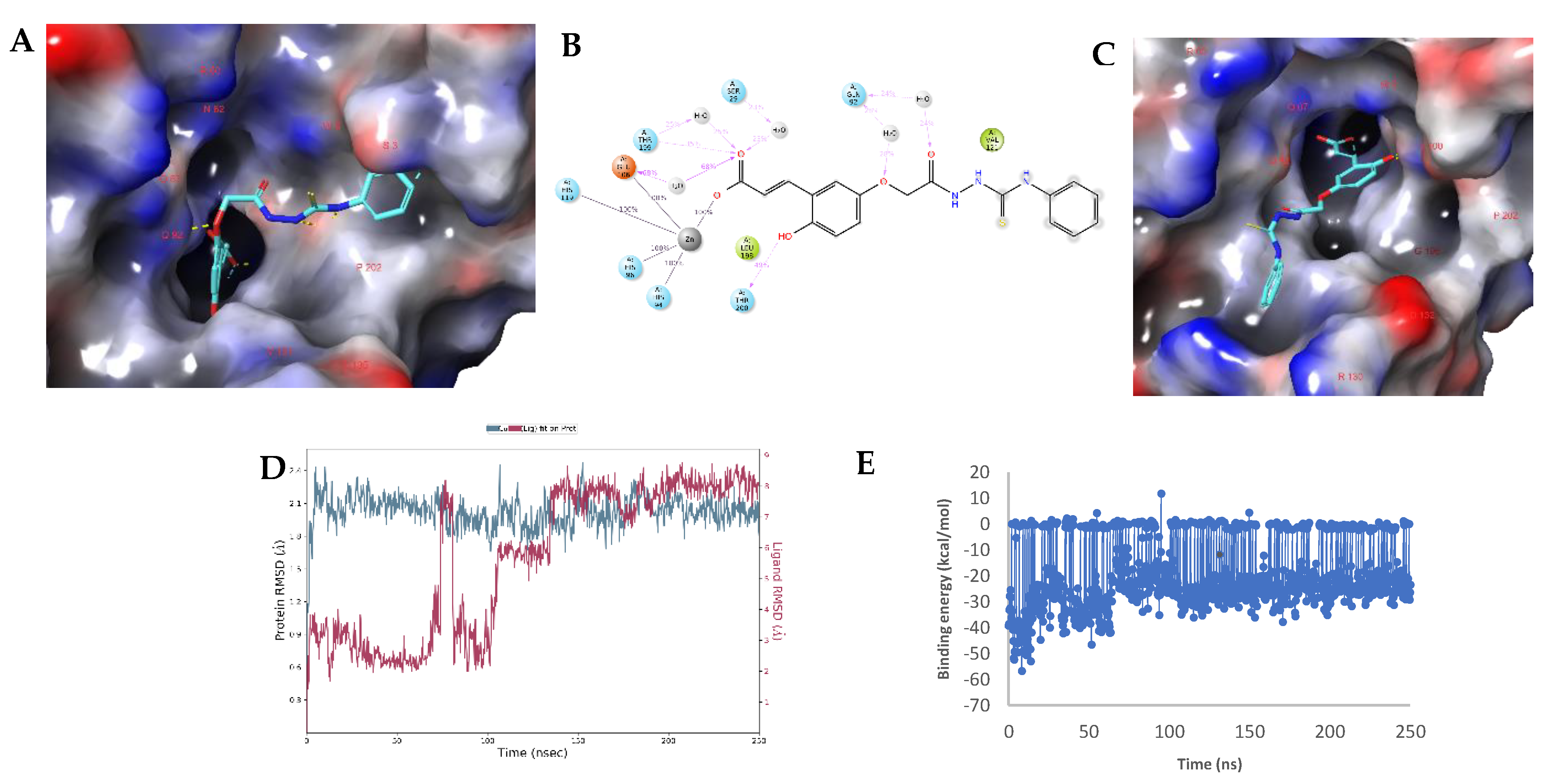
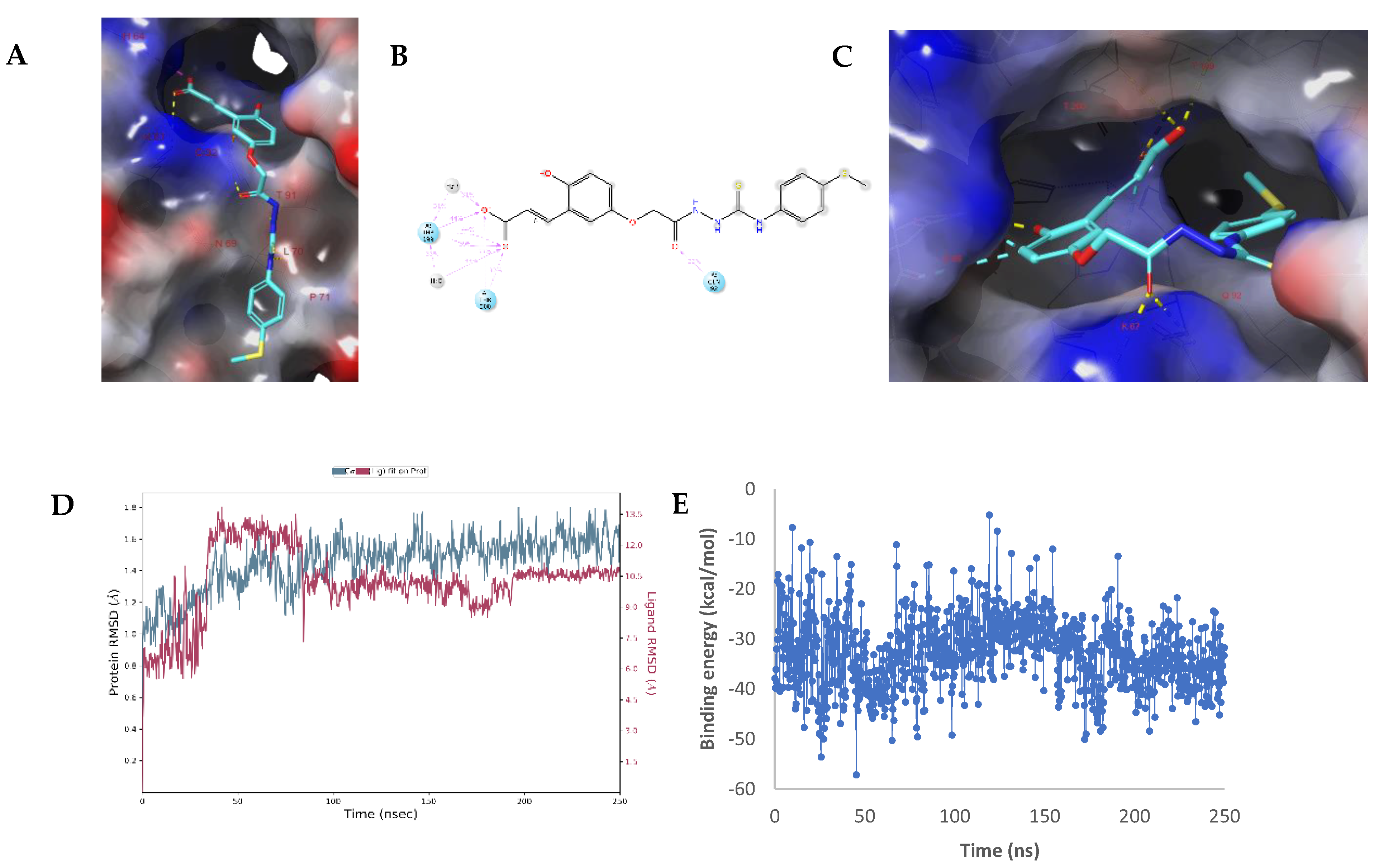
2.3. Molecular Modelling Studies
Modelling Studies of hCA IX– Hydrolyzed 14a-Open
2.4. Modelling Studies of hCA XII–Hydrolyzed 22a-Open
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure for Synthesis of 14a,b–22a,b and 23b
3.3. In Vitro Carbonic Anhydrase Inhibition
3.4. Preparation of Protein Structures
3.5. Docking Studies
3.6. Molecular Dynamics Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonelli, J.; Ortega-Forte, E.; Rovira, A.; Bosch, M.; Torres, O.; Cuscó, C.; Rocas, J.; Ruiz, J.; Marchán, V. Improving photodynamic therapy anticancer activity of a mitochondria-targeted coumarin photosensitizer using a polyurethane-polyurea hybrid nanocarrier. Biomacromolecules 2022, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Shi, W.; Wu, X.-F. Transition-Metal-Catalyzed carbonylative multifunctionalization of alkynes. J. Org. Chem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Akkol, E.K.; Genç, Y.; Karpuz, B.; Sobarzo-Sánchez, E.; Capasso, R. Coumarins and coumarin-related compounds in pharmacotherapy of cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.A.; Shawky, A.M. Thiosemicarbazides in heterocyclization. J. Heterocycl. Chem. 2011, 3, 495–516. [Google Scholar] [CrossRef]
- Magdy, G.; Al-Enna, A.A.; Belal, F.; El-Domany, R.A.; Abdel-Megied, A.M. Application of sulfur and nitrogen doped carbon quantum dots as sensitive fluorescent nanosensors for the determination of saxagliptin and gliclazide. R. Soc. Open Sci. 2022, 6, 220285. [Google Scholar] [CrossRef]
- Peperidou, A.; Bua, S.; Bozdag, M.; Hadjipavlou-Litina, D.; Supuran, C.T. Novel 6- and 7-substituted coumarins with inhibitory action against lipoxygenase and tumor-associated carbonic anhydrase IX. Molecules 2018, 23, 153. [Google Scholar] [CrossRef] [Green Version]
- Al-Amiery, A.A.; Musa, A.Y.; Kadhum, A.A.H.; Mohamad, A.B. The use of umbelliferone in the synthesis of new heterocyclic compounds. Molecules 2011, 16, 6833–6843. [Google Scholar] [CrossRef]
- Janowska, S.; Khylyuk, D.; Andrzejczuk, S.; Wujec, M. Design, synthesis, antibacterial evaluations and in silico studies of novel thiosemicarbazides and 1,3,4-thiadiazoles. Molecules 2022, 10, 3161. [Google Scholar] [CrossRef]
- Icharam Narkhede, H.; Shridhar Dhake, A.; Rikhabchand Surana, A. Synthesis and screening of thiosemicarbazide-dithiocarbamate conjugates for antioxidant and anticancer activities. Bioorg. Chem. 2022, 124, 105832. [Google Scholar] [CrossRef]
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-zinc mediated inhibition of carbonic anhydrases: Coumarins are a new class of suicide inhibitors. J. Am. Chem. Soc. 2009, 8, 3057–3062. [Google Scholar] [CrossRef] [Green Version]
- Cecchi, A.; Ciani, L.; Winum, J.-Y.; Montero, J.L.; Scozzafava, A.; Ristori, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Design of spin-labeled sulfonamides incorporating TEMPO moieties as probes for cytosolic or transmembrane isozymes. Bioorg. Med. Chem. Lett. 2008, 12, 3475–3480. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561. [Google Scholar] [CrossRef]
- Lazaridis, T. Inhomogeneous fluid approach to solvation thermodynamics. 1. Theory. J. Phys. Chem. B 1998, 102, 3531–3541. [Google Scholar] [CrossRef]
- Lazaridis, T. Solvent reorganization energy and entropy in hydrophobic hydration. J. Phys. Chem. B 2000, 104, 4964–4979. [Google Scholar] [CrossRef]
- Abel, R.; Young, T.; Farid, R.; Berne, B.J.; Friesner, R.A. Role of the active-site solvent in the thermodynamics of Factor Xa ligand binding. J. Am. Chem. Soc. 2008, 9, 2817–2831. [Google Scholar] [CrossRef] [Green Version]
- Young, T.; Abel, R.; Kim, B.; Berne, B.J.; Friesner, R.A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc. Natl. Acad. Sci. USA 2007, 3, 808–813. [Google Scholar] [CrossRef] [Green Version]
- Raman, K.; Singh, H.K.; Salzman, S.K.; Parmar, S.S. Substituted thiosemicarbazides and corresponding cyclized 1,3,4-oxadiazoles and their anti-inflammatory activity. J. Pharm. Sci. 1993, 2, 167–169. [Google Scholar] [CrossRef]
- Karioti, A.; Carta, F.; Supuran, C.T. Phenols and polyphenols as carbonic anhydrase inhibitors. Molecules 2016, 12, 1649. [Google Scholar] [CrossRef] [Green Version]
- Carta, F.; Maresca, A.; Scozzafava, A.; Supuran, C.T. 5- and 6-Membered (thio)lactones are prodrug type carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2012, 1, 267–270. [Google Scholar] [CrossRef]
- Carta, F.; Vullo, D.; Maresca, A.; Scozzafava, A.; Supuran, C.T. New chemotypes acting as isozyme-selective carbonic anhydrase inhibitors with low affinity for the offtarget cytosolic isoform II. Bioorg. Med. Chem. Lett. 2012, 6, 2182–2185. [Google Scholar] [CrossRef]
- Carta, F.; Vullo, D.; Maresca, A.; Scozzafava, A.; Supuran, C.T. Mono-/dihydroxybenzoic acid esters and phenol pyridinium derivatives as inhibitors of the mammalian carbonic anhydrase isoforms I, II, VII, IX, XII and XIV. Bioorg. Med. Chem. 2013, 6, 1564–1569. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
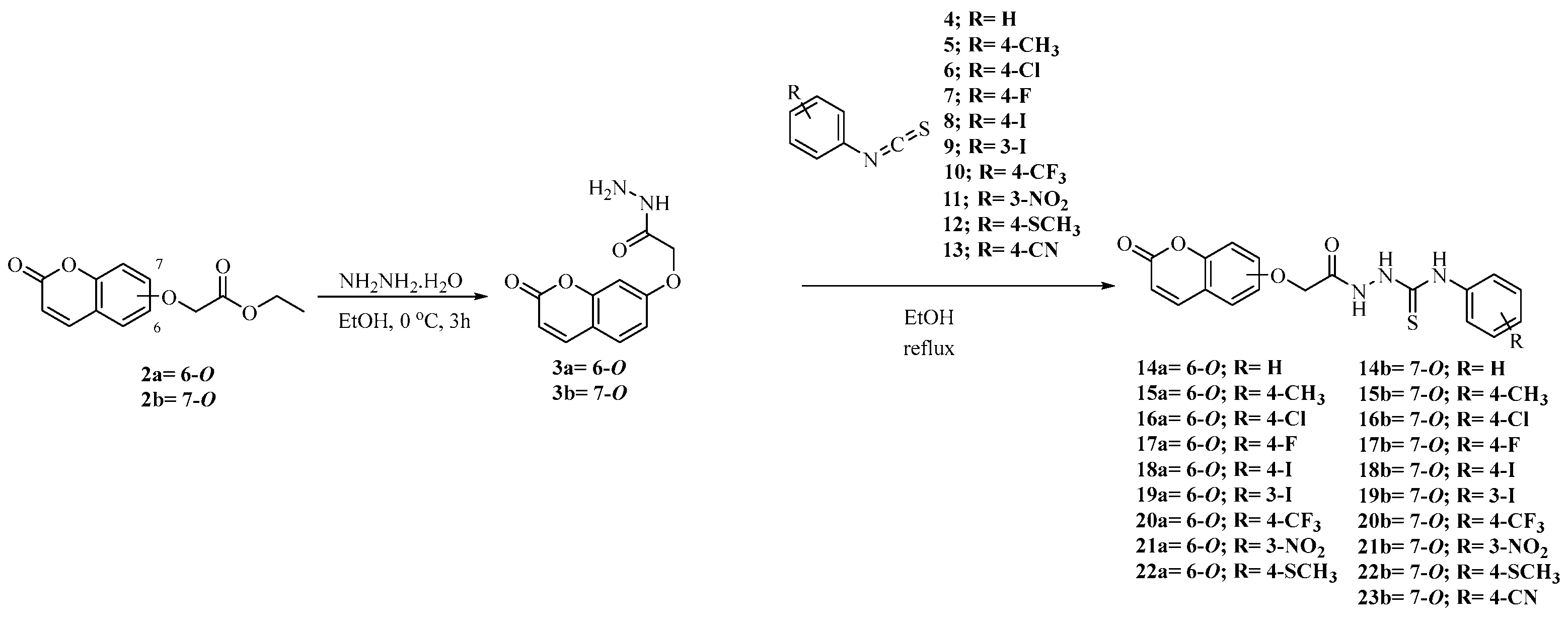{kind=link}
| Compound | hCA I (Ki; nM *) | hCA II (Ki; nM *)) | hCA IX (Ki; nM *)) | hCA XII (Ki; nM *)) |
|---|---|---|---|---|
| 14a | >10,000 | >10,000 | 8.8 | 739 |
| 14b | >10,000 | >10,000 | 46 | 743.5 |
| 15a | >10,000 | >10,000 | 234 | 709 |
| 15b | >10,000 | >10,000 | 78 | 684 |
| 16a | >10,000 | >10,000 | 48 | 608 |
| 16b | >10,000 | >10,000 | 55 | 866 |
| 17a | >10,000 | >10,000 | 39 | 494 |
| 17b | >10,000 | >10,000 | 64 | 745 |
| 18a | >10,000 | >10,000 | 9.6 | 8.4 |
| 18b | >10,000 | >10,000 | 721 | 7.5 |
| 19a | >10,000 | >10,000 | 9.6 | 79 |
| 19b | >10,000 | >10,000 | 9.4 | 573 |
| 20a | >10,000 | >10,000 | 92 | 306 |
| 20b | >10,000 | >10,000 | 70 | 56.6 |
| 21a | >10,000 | >10,000 | 71 | 328.5 |
| 21b | >10,000 | >10,000 | 9.1 | 8.6 |
| 22a | >10,000 | >10,000 | 57 | 4 |
| 22b | >10,000 | >10,000 | 33 | 4.6 |
| 23b | >10,000 | >10,000 | 437.5 | 473 |
| COU [10] | 3100 | 9200 | >10,000 | >10,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gumus, A.; Bozdag, M.; Akdemir, A.; Angeli, A.; Selleri, S.; Carta, F.; Supuran, C.T. Thiosemicarbazide-Substituted Coumarins as Selective Inhibitors of the Tumor Associated Human Carbonic Anhydrases IX and XII. Molecules 2022, 27, 4610. https://doi.org/10.3390/molecules27144610
Gumus A, Bozdag M, Akdemir A, Angeli A, Selleri S, Carta F, Supuran CT. Thiosemicarbazide-Substituted Coumarins as Selective Inhibitors of the Tumor Associated Human Carbonic Anhydrases IX and XII. Molecules. 2022; 27(14):4610. https://doi.org/10.3390/molecules27144610
Chicago/Turabian StyleGumus, Arzu, Murat Bozdag, Atilla Akdemir, Andrea Angeli, Silvia Selleri, Fabrizio Carta, and Claudiu T. Supuran. 2022. "Thiosemicarbazide-Substituted Coumarins as Selective Inhibitors of the Tumor Associated Human Carbonic Anhydrases IX and XII" Molecules 27, no. 14: 4610. https://doi.org/10.3390/molecules27144610
APA StyleGumus, A., Bozdag, M., Akdemir, A., Angeli, A., Selleri, S., Carta, F., & Supuran, C. T. (2022). Thiosemicarbazide-Substituted Coumarins as Selective Inhibitors of the Tumor Associated Human Carbonic Anhydrases IX and XII. Molecules, 27(14), 4610. https://doi.org/10.3390/molecules27144610