

Investigation of Water Interaction with Polymer Matrices by Near-Infrared (NIR) Spectroscopy




Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Data Aquisition
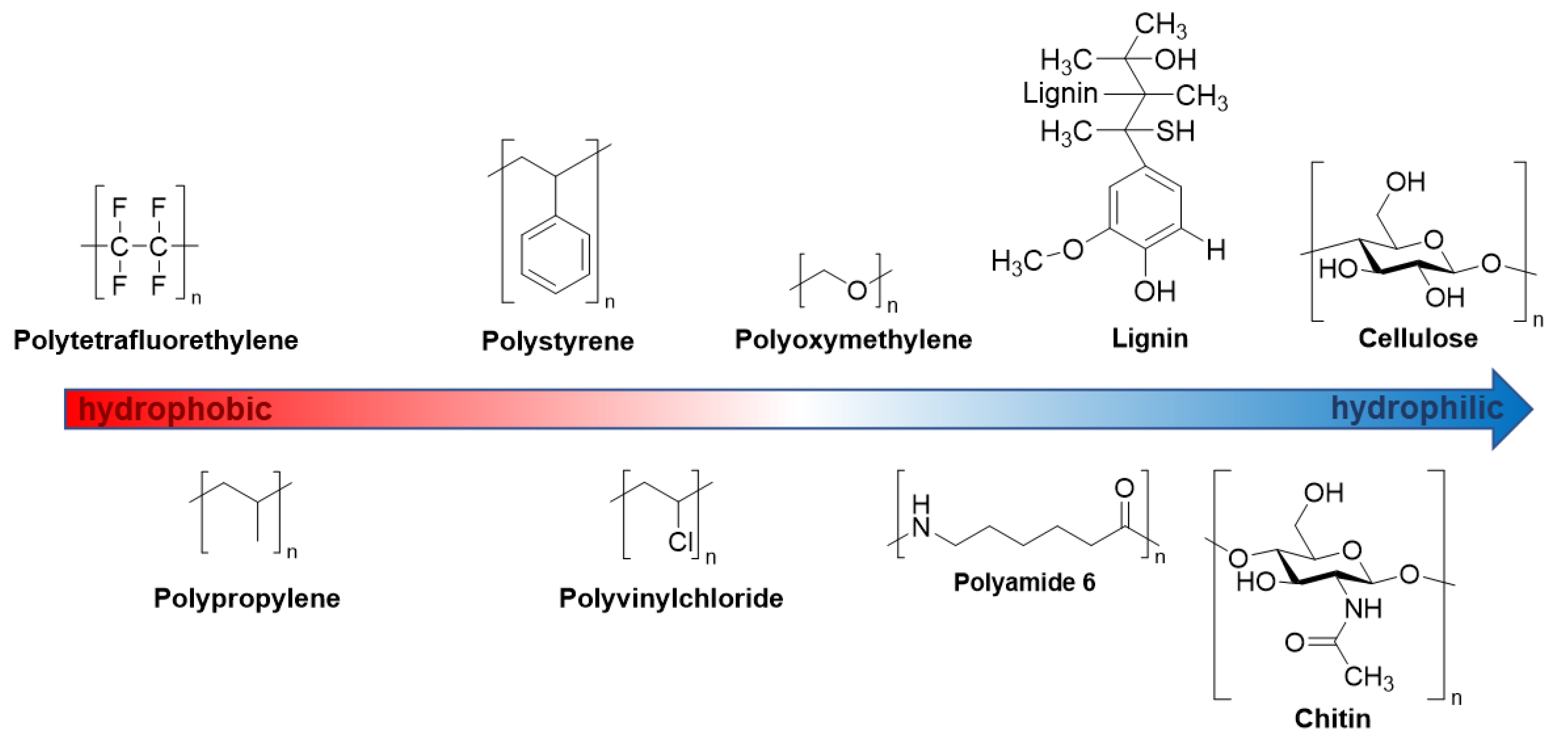
2.1.1. Polymer Samples
2.1.2. NIRFlex N-500 FT-NIR Spectrometer
2.1.3. Data Acquisition
2.2. Chemometric Methods–Spectra Processing and Analysis
2.2.1. Principal Component Analysis (PCA) and Median Linkage Clustering (MLC)
2.2.2. Partial Least Squares Regression (PLSR)
2.2.3. Difference Spectroscopy
2.2.4. Multivariate Curve Resolution Alternating Least Squares (MCR-ALS)
2.2.5. Two-Dimensional Correlation Spectroscopy (2D-COS)
2.2.6. Aquagrams
3. Results
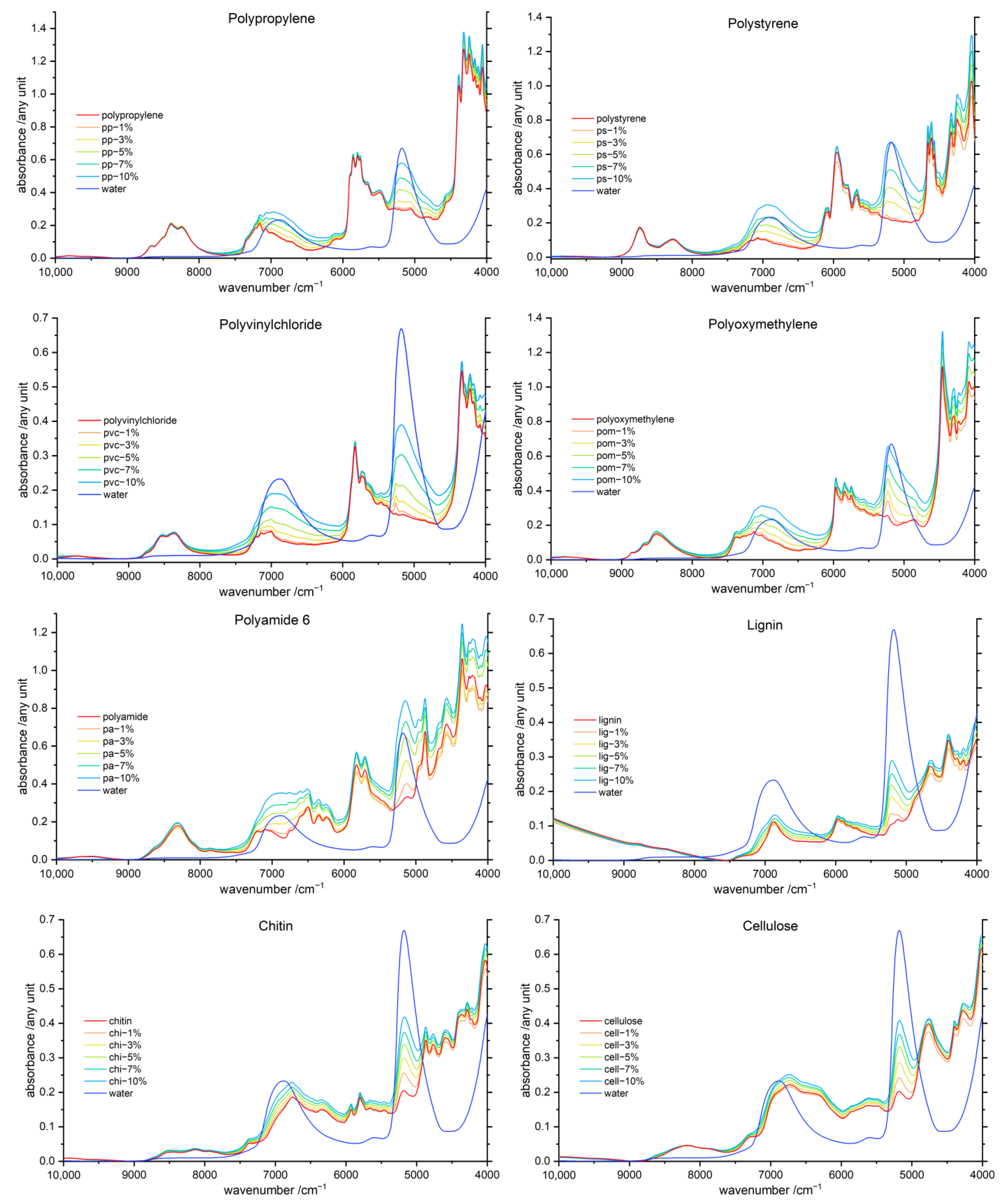
3.1. General Features of the NIR Spectra of Polymer–Water Systems
3.2. Band Assignment
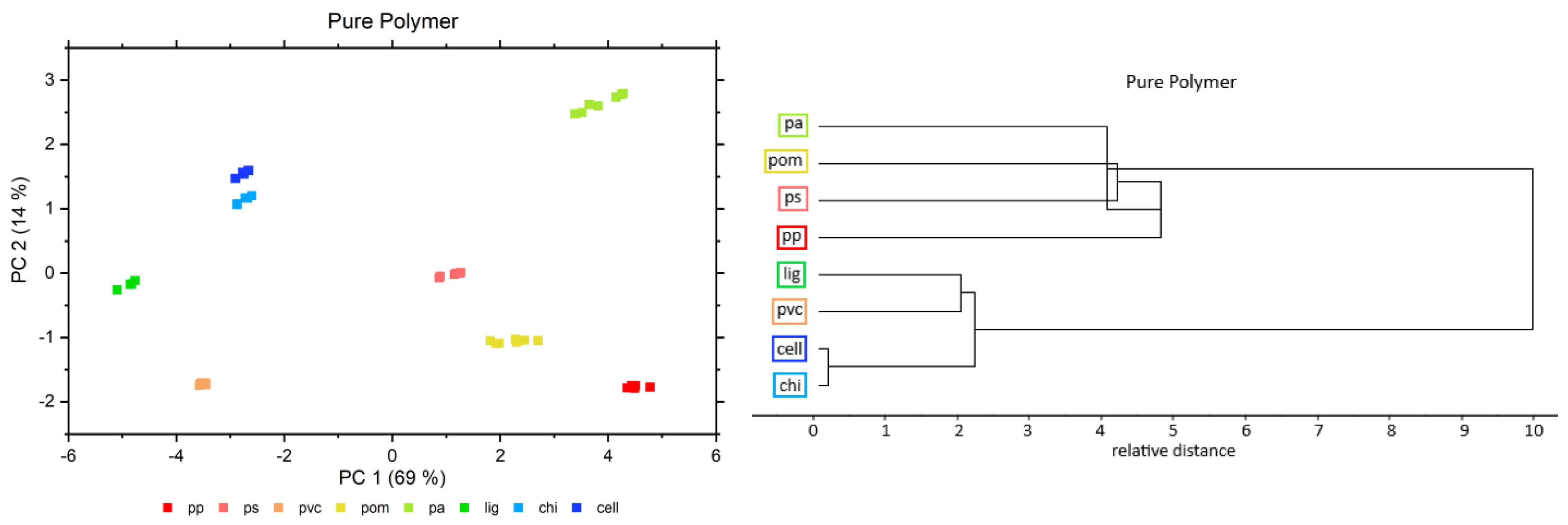
3.3. Principal Component Analysis (PCA) and Median Linkage Clustering (MLC)
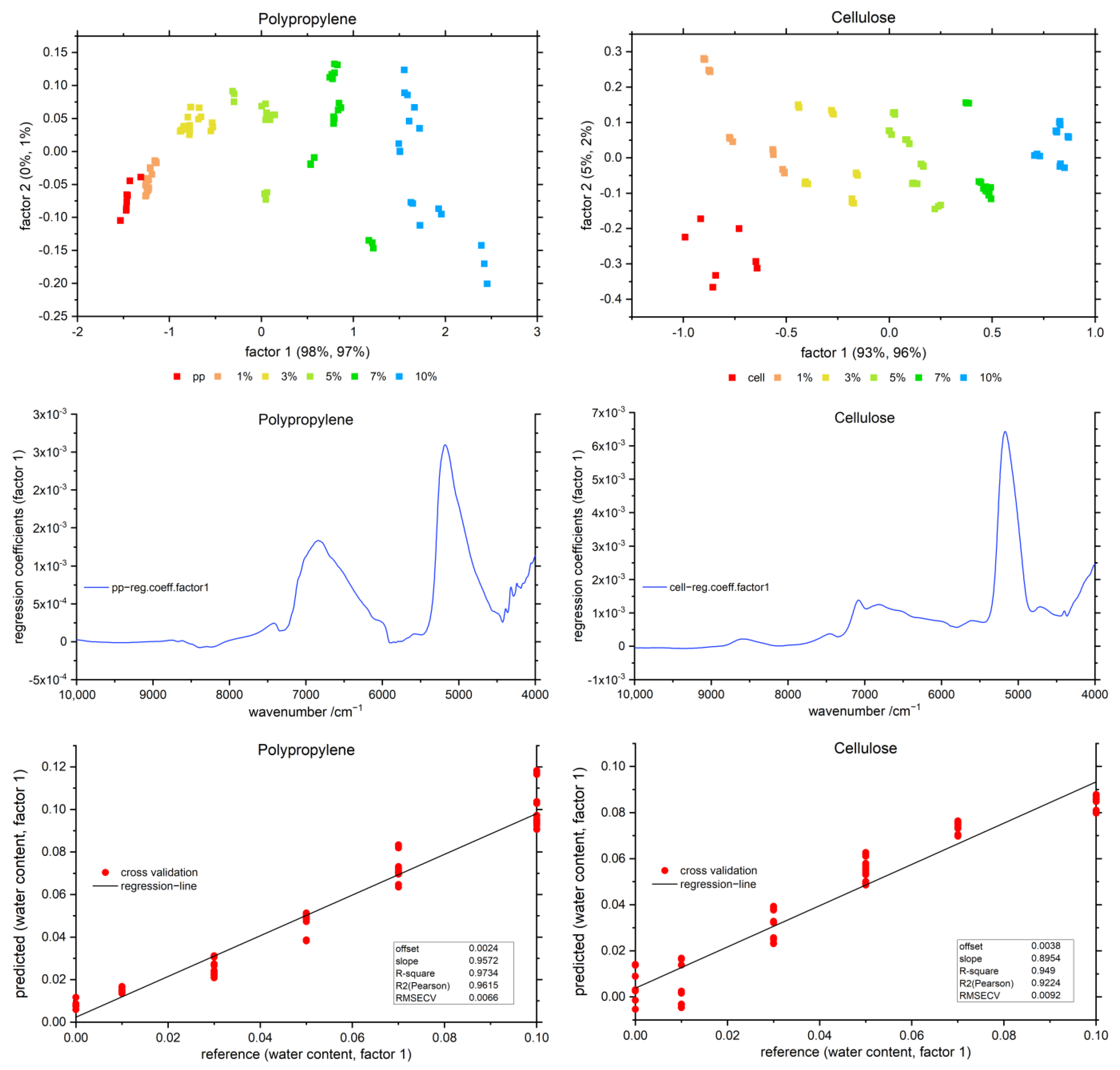
3.4. Partial Least Squares Regression (PLSR)
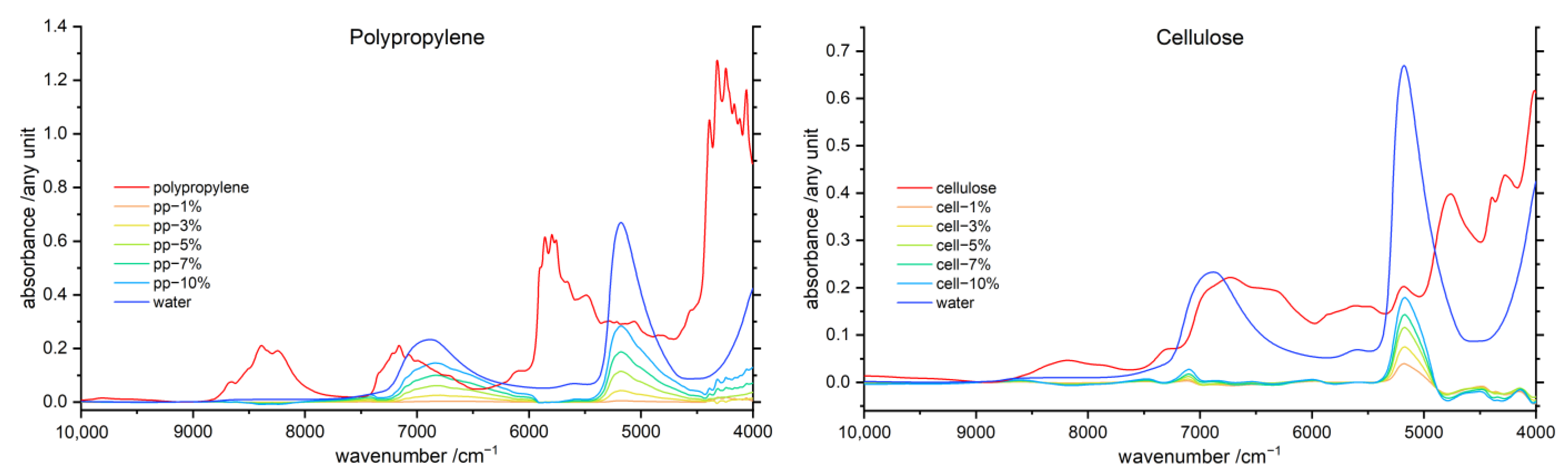
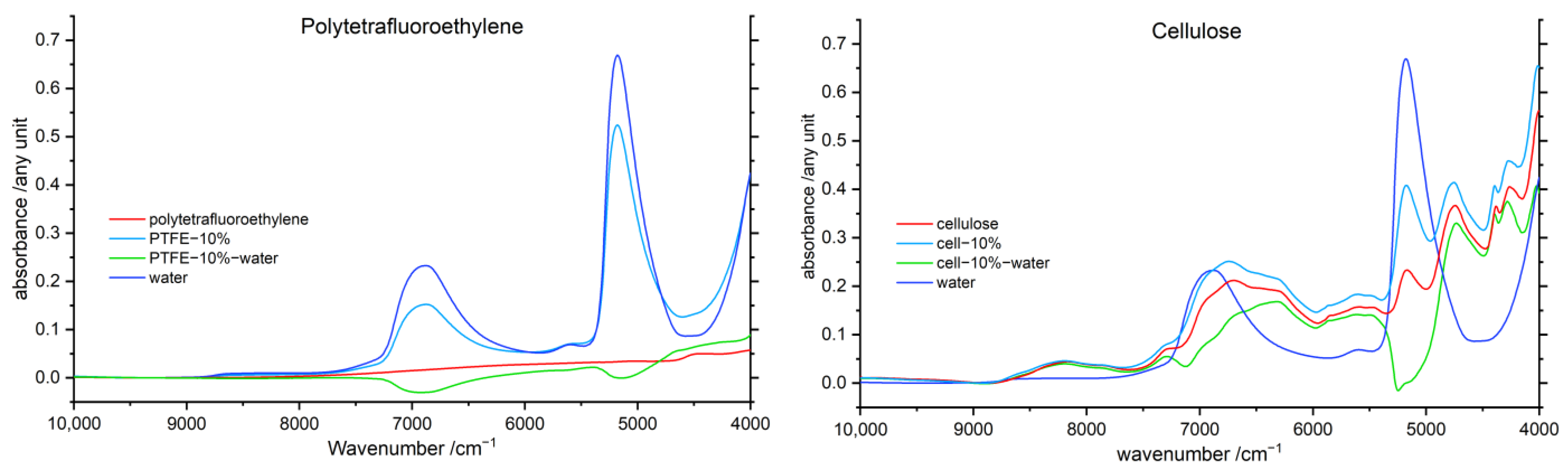
3.5. Difference Spectroscopy
3.5.1. Water difference Spectra
3.5.2. Polymer Difference Spectra
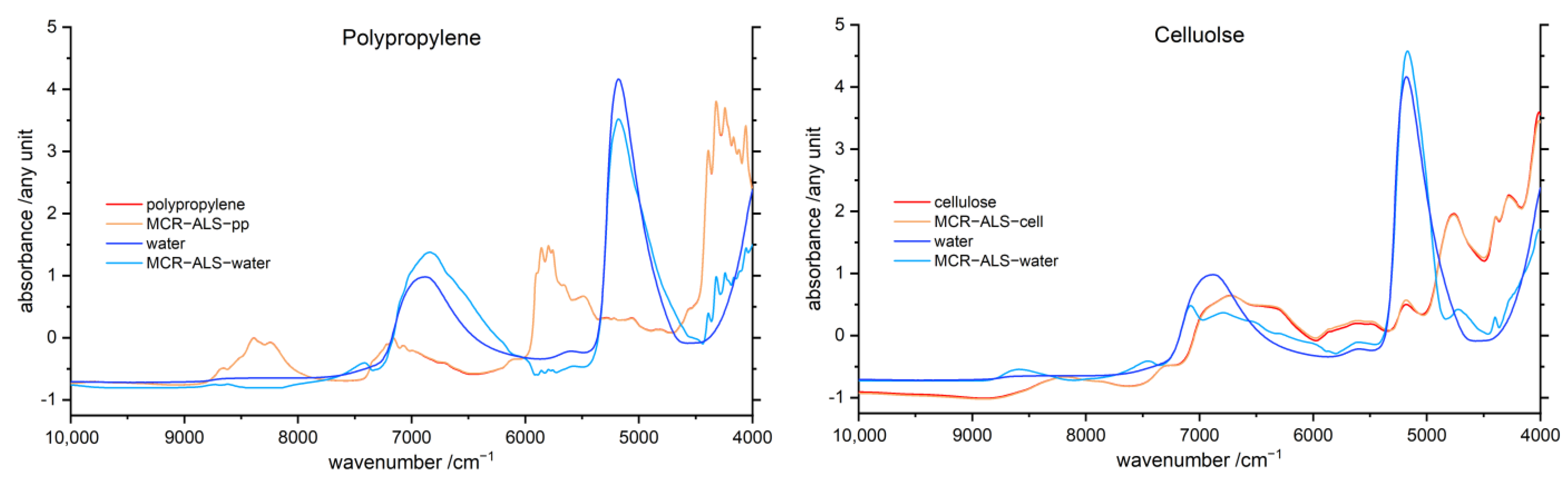
3.6. Multivariate Curve Resolution Alternating Least Squares (MCR-ALS)
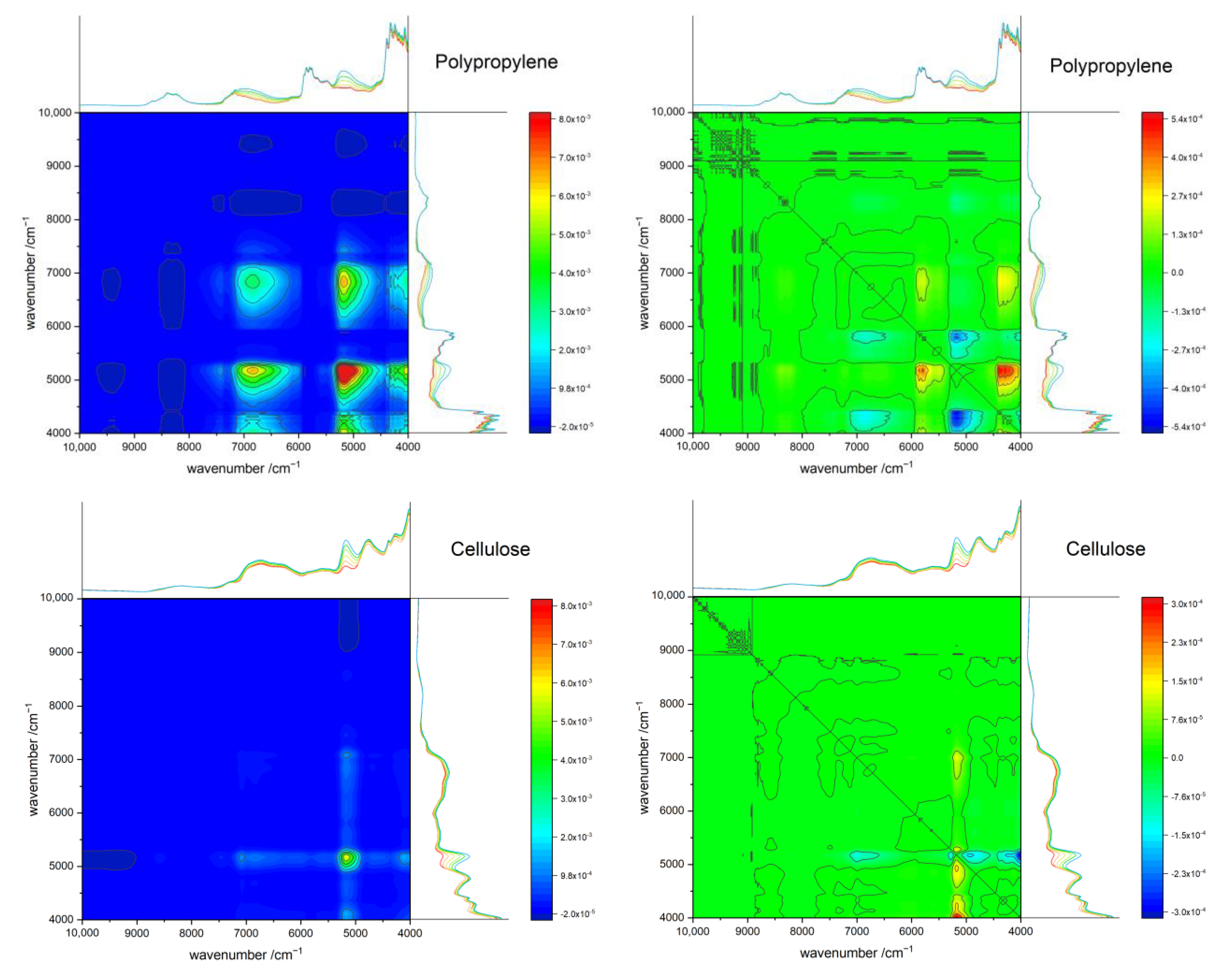
3.7. Two-Dimensional Correlation Spectroscopy (2D-COS)
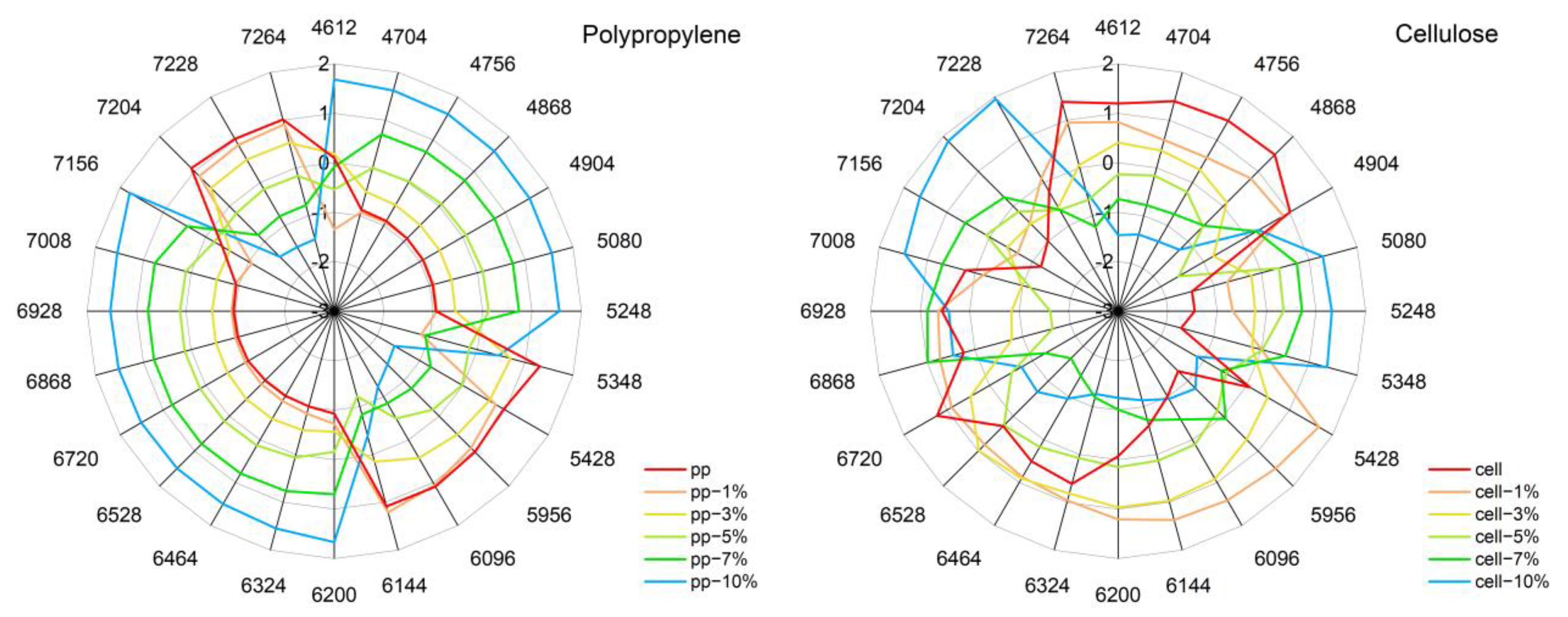
3.8. Aquagrams
4. Discussion
4.1. Polymer Hydrophilicity as the Background for the NIR Spectral Trend in Polymer–Water Systems
4.2. General Discussion and Comparison of the Information Derived from Synergistic Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hatakeyama, H.; Hatakeyama, T. Interaction between water and hydrophilic polymers. Thermochim. Acta 1998, 308, 3–22. [Google Scholar] [CrossRef]
- Hsu, S.L.; Patel, J.; Zhao, W. Vibrational spectroscopy of polymers. In Molecular Characterization of Polymers; Elsevier: Amsterdam, The Netherlands, 2021; pp. 369–407. ISBN 9780128197684. [Google Scholar]
- Jellinek, H.H.G. (Ed.) Water Structure at the Water-Polymer Interface: Proceedings of a Symposium held on March 30 and April 1, 1971, at the 161st National Meeting of the American Chemical Society; Springer US: Boston, MA, USA, 1972; ISBN 978-1-4615-8683-8. [Google Scholar]
- Yasoshima, N.; Ishiyama, T.; Gemmei-Ide, M.; Matubayasi, N. Molecular Structure and Vibrational Spectra of Water Molecules Sorbed in Poly(2-methoxyethylacrylate) Revealed by Molecular Dynamics Simulation. J. Phys. Chem. B 2021, 125, 12095–12103. [Google Scholar] [CrossRef] [PubMed]
- Arif, U.; Haider, S.; Haider, A.; Khan, N.; Alghyamah, A.A.; Jamila, N.; Khan, M.I.; Almasry, W.A.; Kang, I.-K. Biocompatible Polymers and their Potential Biomedical Applications: A Review. Curr. Pharm. Des. 2019, 25, 3608–3619. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Motomura, T.; Ishii, N.; Shimura, K.; Onishi, M.; Mochizuki, A.; Hatakeyama, T. Cold crystallization of water in hydrated poly(2-methoxyethyl acrylate) (PMEA). Polym. Int. 2000, 49, 1709–1713. [Google Scholar] [CrossRef]
- Koguchi, R.; Jankova, K.; Hayasaka, Y.; Kobayashi, D.; Amino, Y.; Miyajima, T.; Kobayashi, S.; Murakami, D.; Yamamoto, K.; Tanaka, M. Understanding the Effect of Hydration on the Bio-inert Properties of 2-Hydroxyethyl Methacrylate Copolymers with Small Amounts of Amino- or/and Fluorine-Containing Monomers. ACS Biomater. Sci. Eng. 2020, 6, 2855–2866. [Google Scholar] [CrossRef]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A View of the Hydrophobic Effect. J. Phys. Chem. B 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Foster, J.C.; Akar, I.; Grocott, M.C.; Pearce, A.K.; Mathers, R.T.; O’Reilly, R.K. 100th Anniversary of Macromolecular Science Viewpoint: The Role of Hydrophobicity in Polymer Phenomena. ACS Macro Lett. 2020, 9, 1700–1707. [Google Scholar] [CrossRef]
- Sato, K.; Kobayashi, S.; Kusakari, M.; Watahiki, S.; Oikawa, M.; Hoshiba, T.; Tanaka, M. The Relationship Between Water Structure and Blood Compatibility in Poly(2-methoxyethyl Acrylate) (PMEA) Analogues. Macromol. Biosci. 2015, 15, 1296–1303. [Google Scholar] [CrossRef]
- Christy, A.A. Chemistry of Desiccant Properties of Carbohydrate Polymers as Studied by Near-Infrared Spectroscopy. Ind. Eng. Chem. Res. 2013, 52, 4510–4516. [Google Scholar] [CrossRef]
- Tanaka, M.; Hayashi, T.; Morita, S. The roles of water molecules at the biointerface of medical polymers. Polym. J. 2013, 45, 701–710. [Google Scholar] [CrossRef]
- Ryabov, Y.E.; Feldman, Y.; Shinyashiki, N.; Yagihara, S. The symmetric broadening of the water relaxation peak in polymer–water mixtures and its relationship to the hydrophilic and hydrophobic properties of polymers. J. Chem. Phys. 2002, 116, 8610. [Google Scholar] [CrossRef]
- Malik, M.I.; Mays, J.; Shah, M.R. (Eds.) Molecular Characterization of Polymers; Elsevier: Amsterdam, The Netherlands, 2021; ISBN 9780128197684. [Google Scholar]
- Mastai, Y.; Rudloff, J.; Cölfen, H.; Antoniette, M. Control over the structure of ice and water by block copolymer additives. ChemPhysChem 2002, 3, 119–123. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, X.; Duan, B.; Yu, Z.; Cheng, T.; Yu, L.; Liu, L.; Liu, K. Polymer-Water Interaction Enabled Intelligent Moisture Regulation in Hydrogels. J. Phys. Chem. Lett. 2021, 12, 2587–2592. [Google Scholar] [CrossRef]
- Dong, W.; Yan, M.; Zhang, M.; Liu, Z.; Li, Y. A computational and experimental investigation of the interaction between the template molecule and the functional monomer used in the molecularly imprinted polymer. Anal. Chim. Acta 2005, 542, 186–192. [Google Scholar] [CrossRef]
- Taylor, L.S.; Langkilde, F.W.; Zografi, G. Fourier transform Raman spectroscopic study of the interaction of water vapor with amorphous polymers. J. Pharm. Sci. 2001, 90, 888–901. [Google Scholar] [CrossRef]
- Schmidt, P.; Dybal, J.; Trchová, M. Investigations of the hydrophobic and hydrophilic interactions in polymer–water systems by ATR FTIR and Raman spectroscopy. Vib. Spectrosc. 2006, 42, 278–283. [Google Scholar] [CrossRef]
- Maeda, Y.; Kitano, H. The structure of water in polymer systems as revealed by Raman spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1995, 51, 2433–2446. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Huck, C.W. Near-Infrared Spectroscopy in Bio-Applications. Molecules 2020, 25, 2948. [Google Scholar] [CrossRef]
- Bokobza, L. Some Applications of Vibrational Spectroscopy for the Analysis of Polymers and Polymer Composites. Polymers 2019, 11, 1159. [Google Scholar] [CrossRef]
- Du Yi, P.; Liang, Y.Z.; Kasemsumran, S.; Maruo, K.; Ozaki, Y. Removal of interference signals due to water from in vivo near-infrared (NIR) spectra of blood glucose by region orthogonal signal correction (ROSC). Anal. Sci. 2004, 20, 1339–1345. [Google Scholar] [CrossRef] [Green Version]
- Schwanninger, M.; Rodrigues, J.C.; Fackler, K. A Review of Band Assignments in near Infrared Spectra of Wood and Wood Components. J. Near Infrared Spectrosc. 2011, 19, 287–308. [Google Scholar] [CrossRef]
- Czarnecki, M.A.; Beć, K.B.; Grabska, J.; Hofer, T.S.; Ozaki, Y. Overview of Application of NIR Spectroscopy to Physical Chemistry. In Near-Infrared Spectroscopy; Ozaki, Y., Huck, C., Tsuchikawa, S., Engelsen, S.B., Eds.; Springer: Singapore, 2021; pp. 297–330. ISBN 978-981-15-8647-7. [Google Scholar]
- Schuler, M.J.; Hofer, T.S.; Morisawa, Y.; Futami, Y.; Huck, C.W.; Ozaki, Y. Solvation effects on wavenumbers and absorption intensities of the OH-stretch vibration in phenolic compounds—Electrical- and mechanical anharmonicity via a combined DFT/Numerov approach. Phys. Chem. Chem. Phys. 2020, 22, 13017–13029. [Google Scholar] [CrossRef] [PubMed]
- Futami, Y.; Ozaki, Y.; Hamada, Y.; Wojcik, M.J.; Ozaki, Y. Frequencies and absorption intensities of fundamentals and overtones of NH stretching vibrations of pyrrole and pyrrole–pyridine complex studied by near-infrared/infrared spectroscopy and density-functional-theory calculations. Chem. Phys. Lett. 2009, 482, 320–324. [Google Scholar] [CrossRef]
- Lachenal, G.; Ozaki, Y. Advantages of near infrared spectroscopy for the analysis of polymers and composites. Macromol. Symp. 1999, 141, 283–292. [Google Scholar] [CrossRef]
- Zhang, X.; He, A.; Guo, R.; Zhao, Y.; Yang, L.; Morita, S.; Xu, Y.; Noda, I.; Ozaki, Y. A new approach to removing interference of moisture from FTIR spectrum. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 265, 120373. [Google Scholar] [CrossRef]
- Margenot, A.J.; Calderón, F.J.; Parikh, S.J. Limitations and Potential of Spectral Subtractions in Fourier-Transform Infrared Spectroscopy of Soil Samples. Soil Sci. Soc. Am. J. 2016, 80, 10–26. [Google Scholar] [CrossRef]
- Chen, D.; Hu, B.; Shao, X.; Su, Q. Removal of major interference sources in aqueous near-infrared spectroscopy techniques. Anal. Bioanal. Chem. 2004, 379, 143–148. [Google Scholar] [CrossRef]
- Ramoelo, A.; Skidmore, A.K.; Schlerf, M.; Mathieu, R.; Heitkönig, I.M. Water-removed spectra increase the retrieval accuracy when estimating savanna grass nitrogen and phosphorus concentrations. ISPRS J. Photogramm. Remote Sens. 2011, 66, 408–417. [Google Scholar] [CrossRef]
- Yoon, G.; Amerov, A.K.; Jeon, K.J.; Kim, Y.-J. Determination of glucose concentration in a scattering medium based on selected wavelengths by use of an overtone absorption band. Appl. Opt. 2002, 41, 1469–1475. [Google Scholar] [CrossRef]
- Wold, S.; Antti, H.; Lindgren, F.; Öhman, J. Orthogonal signal correction of near-infrared spectra. Chemom. Intell. Lab. Syst. 1998, 44, 175–185. [Google Scholar] [CrossRef]
- Gao, B.-C.; Goetzt, A.F. Retrieval of equivalent water thickness and information related to biochemical components of vegetation canopies from AVIRIS data. Remote Sens. Environ. 1995, 52, 155–162. [Google Scholar] [CrossRef]
- Tsenkova, R.; Munćan, J.; Pollner, B.; Kovacs, Z. Essentials of Aquaphotomics and Its Chemometrics Approaches. Front. Chem. 2018, 6, 363. [Google Scholar] [CrossRef]
- Tan, J.; Sun, Y.; Ma, L.; Feng, H.; Guo, Y.; Cai, W.; Shao, X. Knowledge-based genetic algorithm for resolving the near-infrared spectrum and understanding the water structures in aqueous solution. Chemom. Intell. Lab. Syst. 2020, 206, 104150. [Google Scholar] [CrossRef]
- Czarnik-Matusewicz, B.; Pilorz, S. Study of the temperature-dependent near-infrared spectra of water by two-dimensional correlation spectroscopy and principal components analysis. Vib. Spectrosc. 2006, 40, 235–245. [Google Scholar] [CrossRef]
- Sandorfy, C.; Buchet, R.; Lachenal, G. Principles of Molecular Vibrations for Near-Infrared Spectroscopy. In Near-Infrared Spectroscopy in Food Science and Technology; Ozaki, Y., McClure, W.F., Christy, A.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 11–46. ISBN 9780470047705. [Google Scholar]
- Beganović, A.; Moll, V.; Huck, C.W. Comparison of Multivariate Regression Models Based on Water- and Carbohydrate-Related Spectral Regions in the Near-Infrared for Aqueous Solutions of Glucose. Molecules 2019, 24, 2696. [Google Scholar] [CrossRef]
- Pazderka, T.; Kopecky, V., Jr. 2D Correlation Spectroscopy and Its Application in Vibrational Spectroscopy Using Matlab. Institute of Physics, Faculty of Mathematics and Physics, Charles University: Prague, Czech Republic, 2008; pp. 978–998. [Google Scholar]
- Dharmaratne, N.U.; Jouaneh, T.M.M.; Kiesewetter, M.K.; Mathers, R.T. Quantitative Measurements of Polymer Hydrophobicity Based on Functional Group Identity and Oligomer Length. Macromolecules 2018, 51, 8461–8468. [Google Scholar] [CrossRef]
- Figg, C.A.; Carmean, R.N.; Bentz, K.C.; Mukherjee, S.; Savin, D.A.; Sumerlin, B.S. Tuning Hydrophobicity To Program Block Copolymer Assemblies from the Inside Out. Macromolecules 2017, 50, 935–943. [Google Scholar] [CrossRef]
- Piao, C.; Winandy, J.E.; Shupe, T.F. From Hydrophilicity to Hydrophobicity: A Critical Review: Part I. Wettability and Surface Behavior. Wood Fiber Sci. 2010, 42, 490–510. [Google Scholar]
- Hou, X.; Deem, P.T.; Choy, K.-L. Hydrophobicity study of polytetrafluoroethylene nanocomposite films. Thin Solid Films 2012, 520, 4916–4920. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavenumber/cm−1 | Assignment | Polymer/Water |
|---|---|---|
| 10,000–9000 | 3 ν (OH); hydrogen-bonded | |
| 8600–8200 [21,39] 8250 [21] | 3 ν (CH3 [21,39], CH2 [39]) 2 ν + 2 δ (CH3, CH2) | all |
| 7200–7000 | 2 ν (free OH) 2 ν + δ (CH3, CH2) | Lig, Chi, Cell all |
| 7200–6800 [25] | νs + νas (OH) | water |
| 7000–6200 | 2 ν (OH); hydrogen-bonded ν (OH) + ν (CH) | all |
| 6900 [40] | 2 ν CH + δ CH | all |
| 6700–6500 | 2 ν (NH); free | PA, Chi |
| 6600–6300 | 2 ν (NH); hydrogen-bonded | |
| 6500 | 2 ν (OH); carbohydrates, polyphenols, …; hydrogen-bonded | Lig, Chi, Cell |
| 6200 | ν (CH3, CH2) | all |
| 6000–5600 | 2 ν (CH3, CH2) νs + 2 δ (CH3, CH2) | |
| 5300–5000 [25] 5200 [11,39,40] | νas + δ (OH) | water |
| 5280 [11] | Hydrogen-bonded water | water |
| 5190 [11,39] | νas + δ (OH) water molecule trapped in Polymer | Cell + water |
| 5150 [28] | νas + δ (OH); water molecule trapped in Polymer | PA + water |
| 4900–4600 | ν + δ (NH) | PA, Chi |
| 4500 | ν (CH3, CH2) | all |
| 4400–4200 | ν + δ (CH3, CH2) | all |
| Polymer | Shifts for Overtone Water Band/cm−1 | Shifts for Combination Water Band/cm−1 |
|---|---|---|
| Polypropylene | 6796–6836 | 5180/not shifted |
| Polystyrene | 6812–6852 | 5248–5176 |
| Polyvinylchloride | 6808–6842 | 5256–5180 |
| Polyoxymethylene | 6822–6826 | 5228–5204 |
| Polyamide 6 | 6828–6840 | 5140–5164 |
| (Lignin) | (7084–7064) | (5224–5208) |
| Chitin | 7048–7024 | 5176–5164 |
| Cellulose | 7100–7120 | 5180–5172 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moll, V.; Beć, K.B.; Grabska, J.; Huck, C.W. Investigation of Water Interaction with Polymer Matrices by Near-Infrared (NIR) Spectroscopy. Molecules 2022, 27, 5882. https://doi.org/10.3390/molecules27185882
Moll V, Beć KB, Grabska J, Huck CW. Investigation of Water Interaction with Polymer Matrices by Near-Infrared (NIR) Spectroscopy. Molecules. 2022; 27(18):5882. https://doi.org/10.3390/molecules27185882
Chicago/Turabian StyleMoll, Vanessa, Krzysztof B. Beć, Justyna Grabska, and Christian W. Huck. 2022. "Investigation of Water Interaction with Polymer Matrices by Near-Infrared (NIR) Spectroscopy" Molecules 27, no. 18: 5882. https://doi.org/10.3390/molecules27185882
APA StyleMoll, V., Beć, K. B., Grabska, J., & Huck, C. W. (2022). Investigation of Water Interaction with Polymer Matrices by Near-Infrared (NIR) Spectroscopy. Molecules, 27(18), 5882. https://doi.org/10.3390/molecules27185882