


Reactions of Trifluorotriacetic Acid Lactone and Hexafluorodehydroacetic Acid with Amines: Synthesis of Trifluoromethylated 4-Pyridones and Aminoenones

Abstract
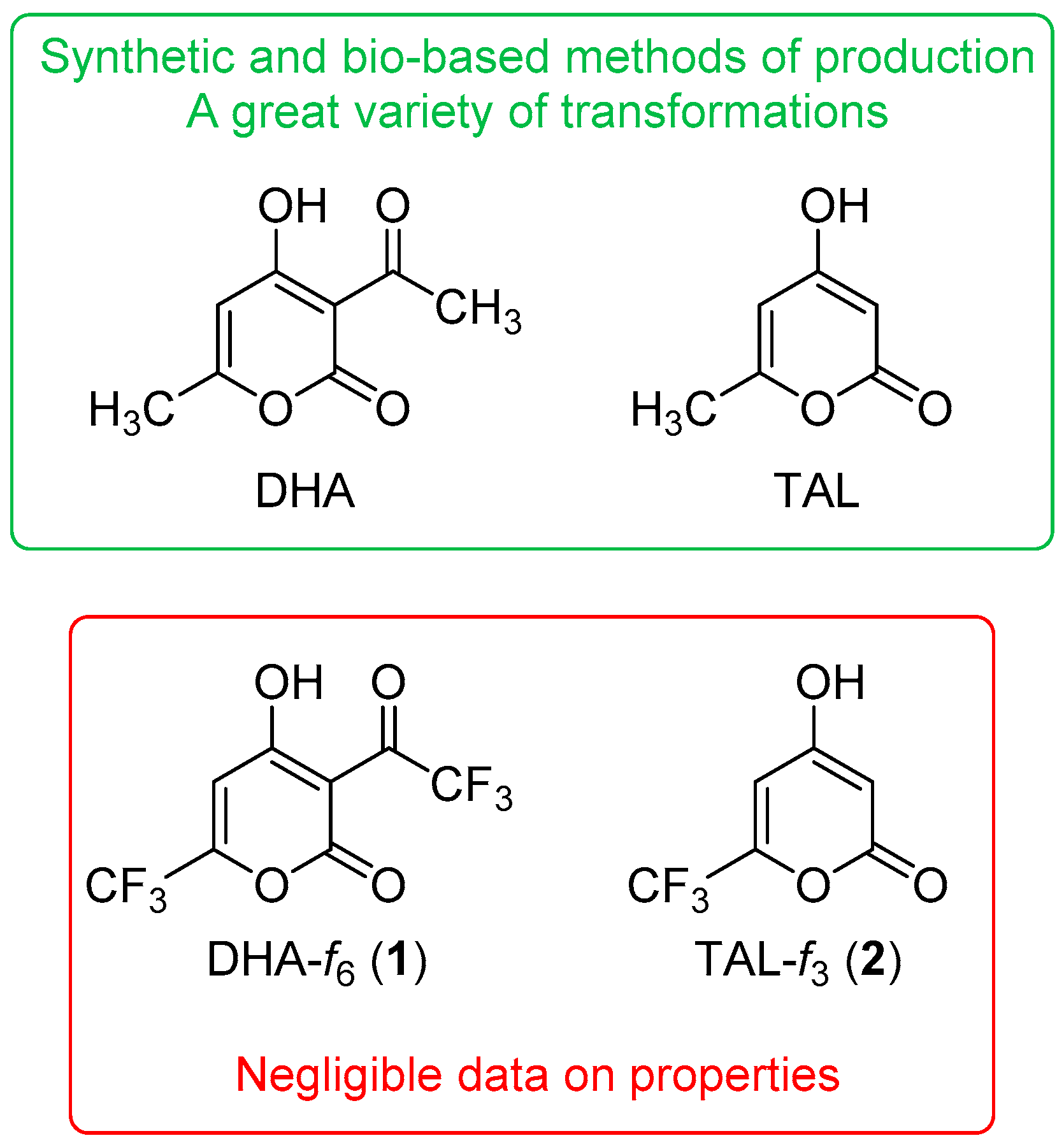
:1. Introduction
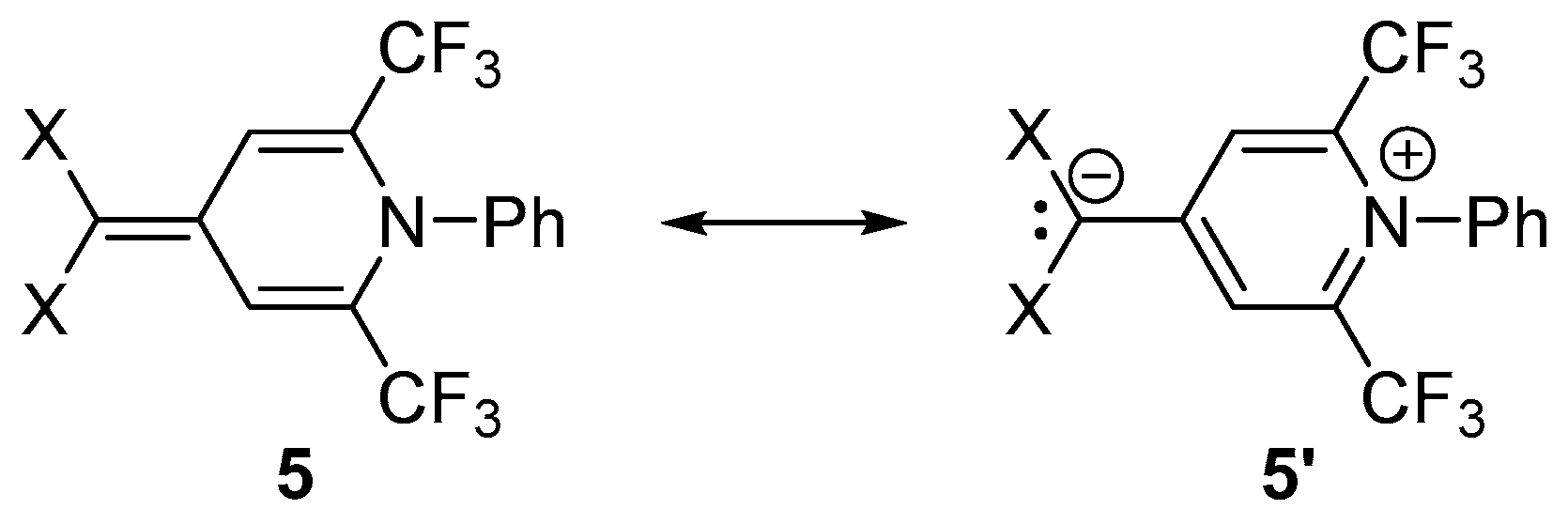
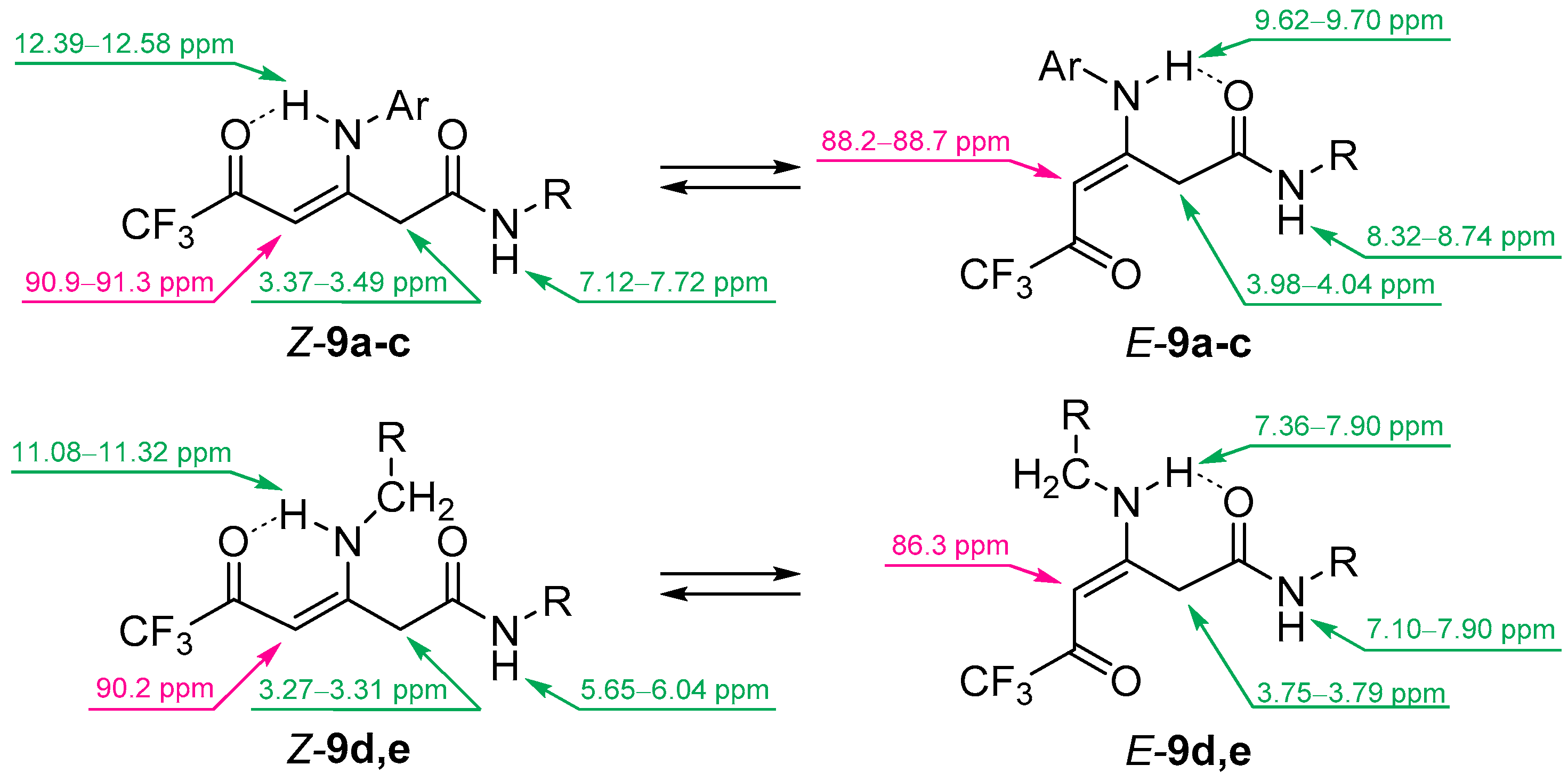
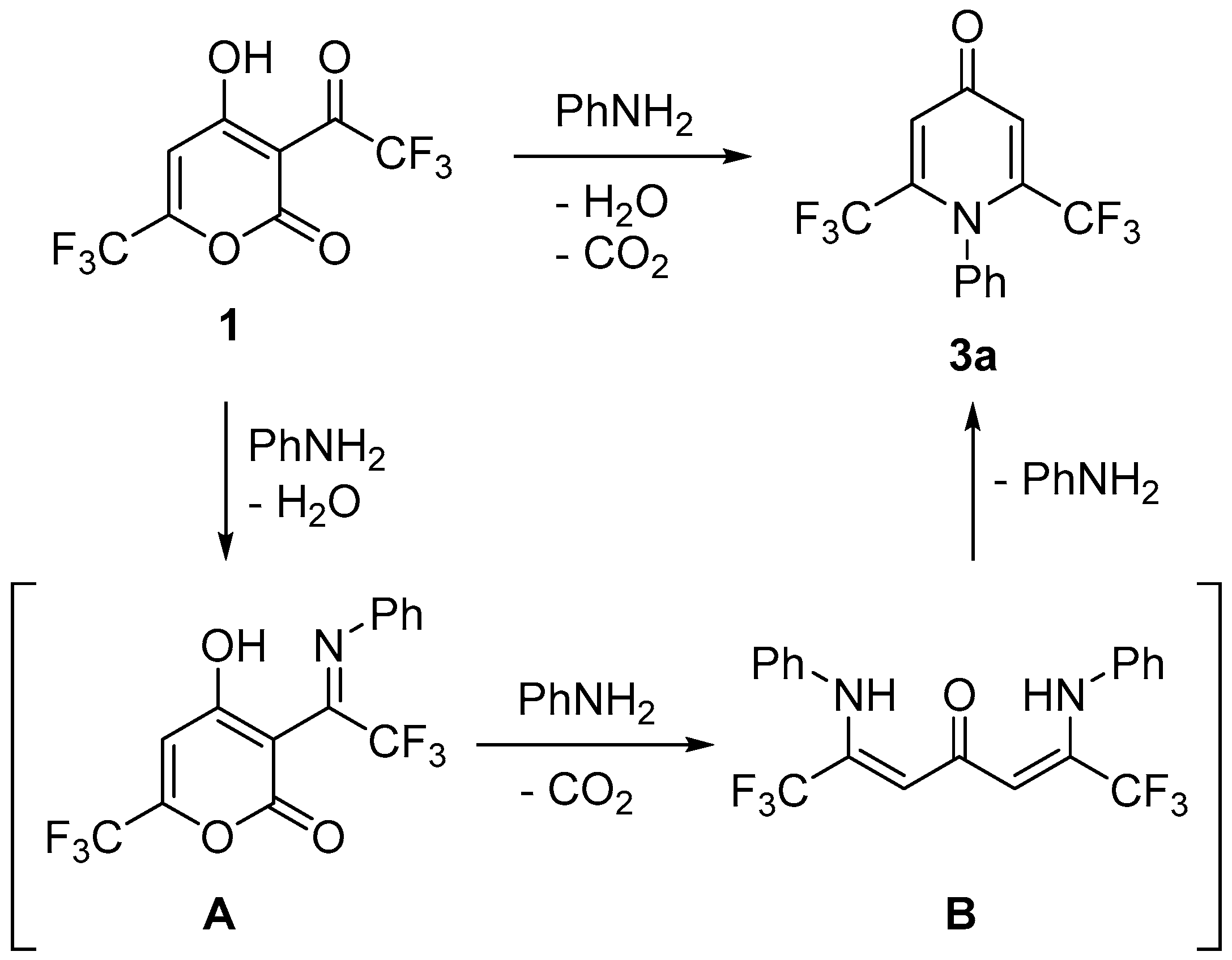
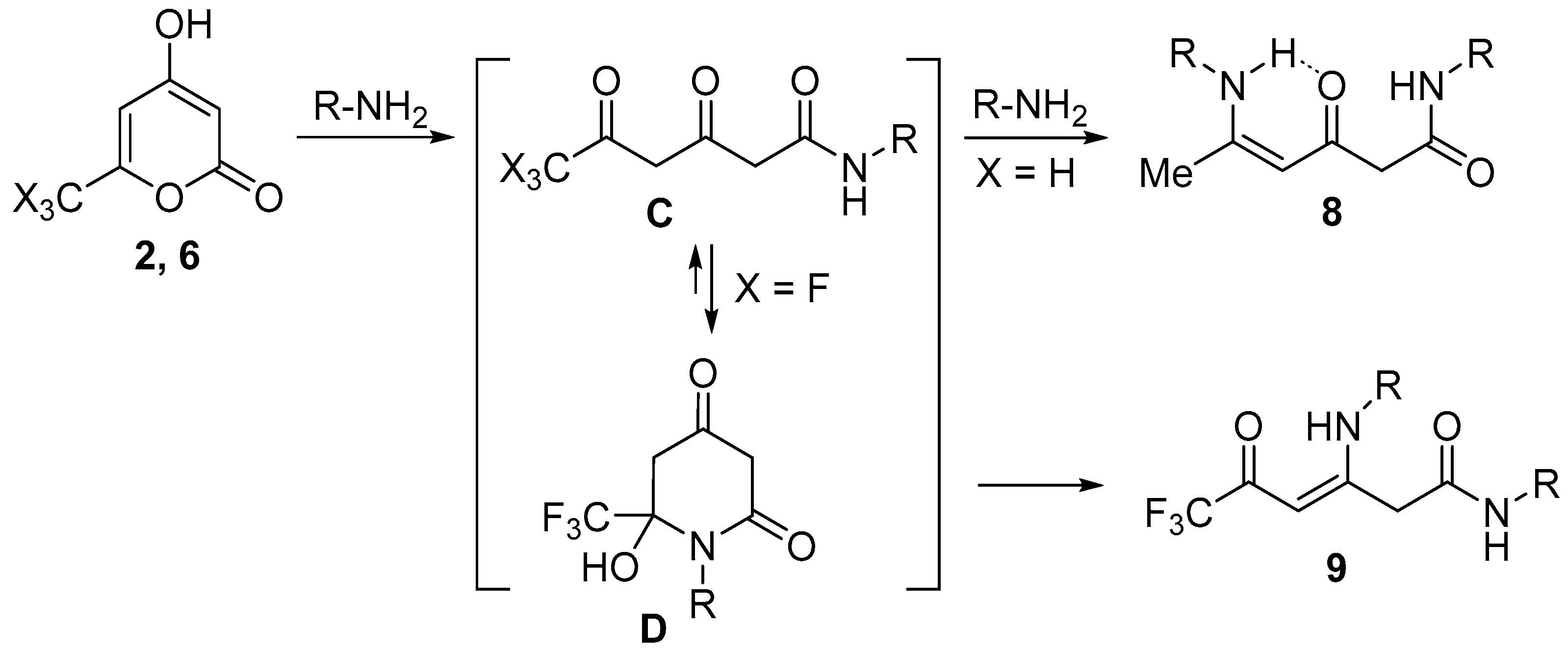
2. Results and Discussion
3. Materials and Methods
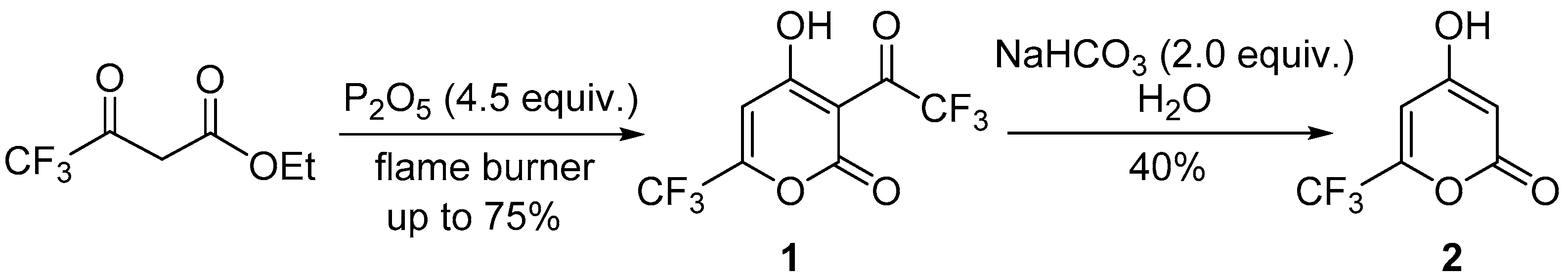
3.1. Synthesis of Pyrones 1 and 2
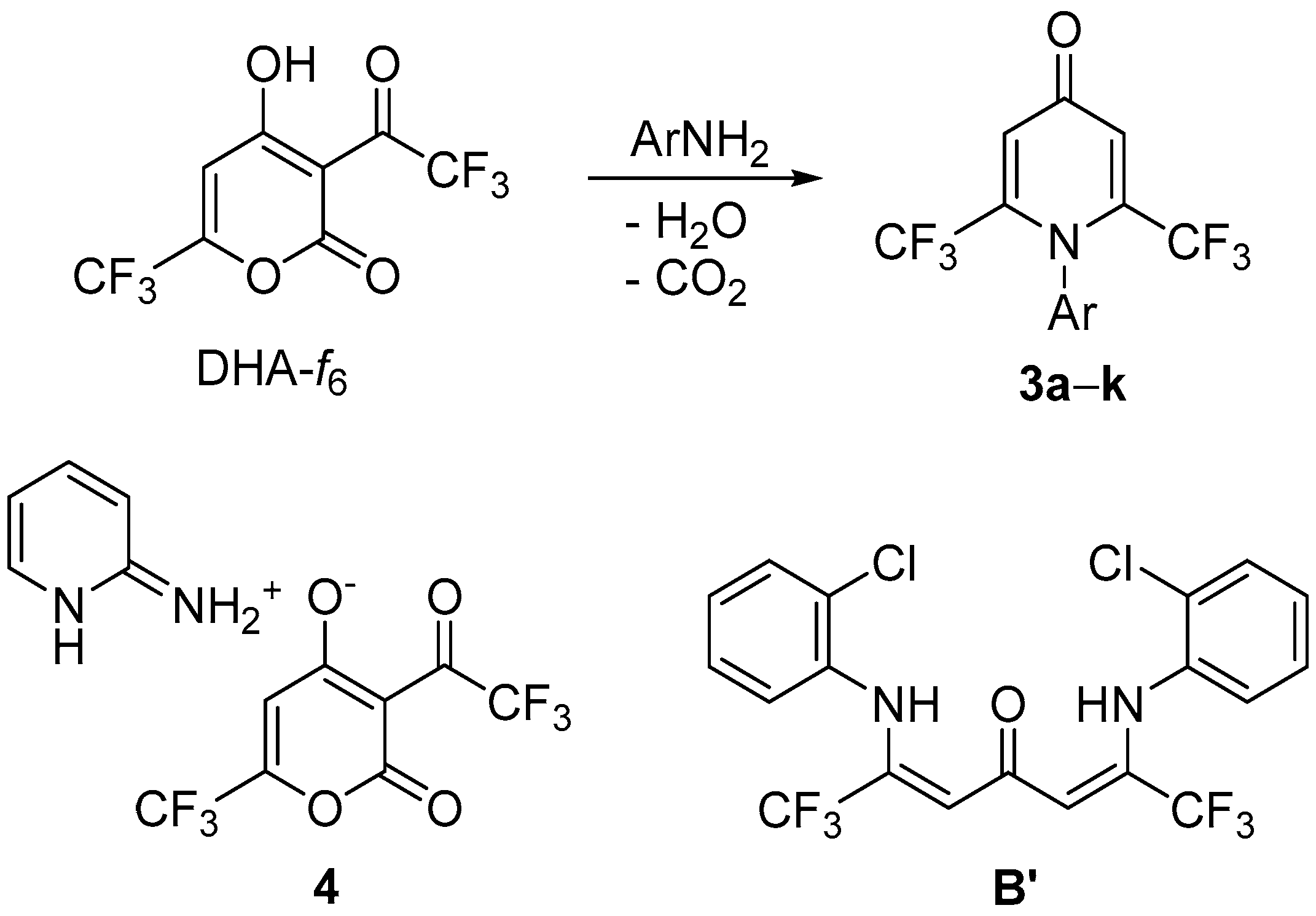
3.2. Synthesis of Compounds 3a–k
General Procedure
3.3. Synthesis of Compound B′
3.4. Synthesis of Compound 4
3.5. Synthesis of Compounds 5
General Procedure
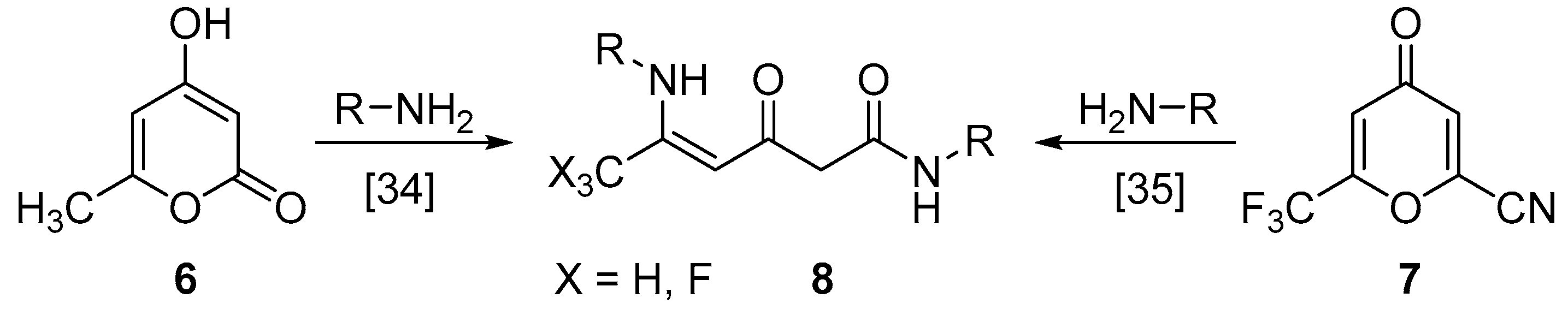
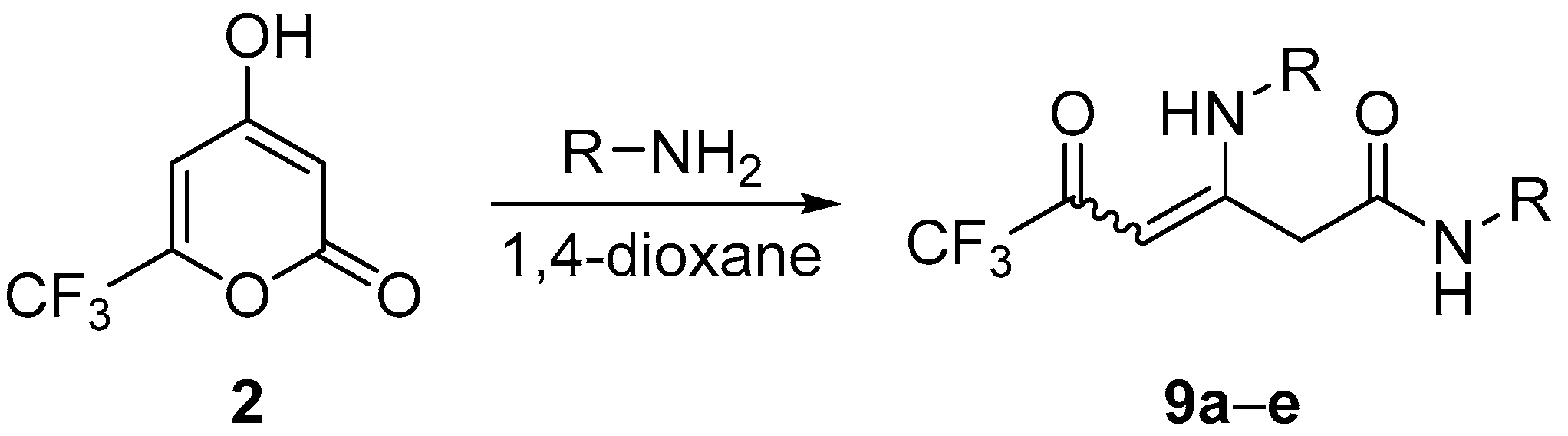
3.6. Synthesis of Compounds 9
General Procedure
3.7. Synthesis of Compound 10
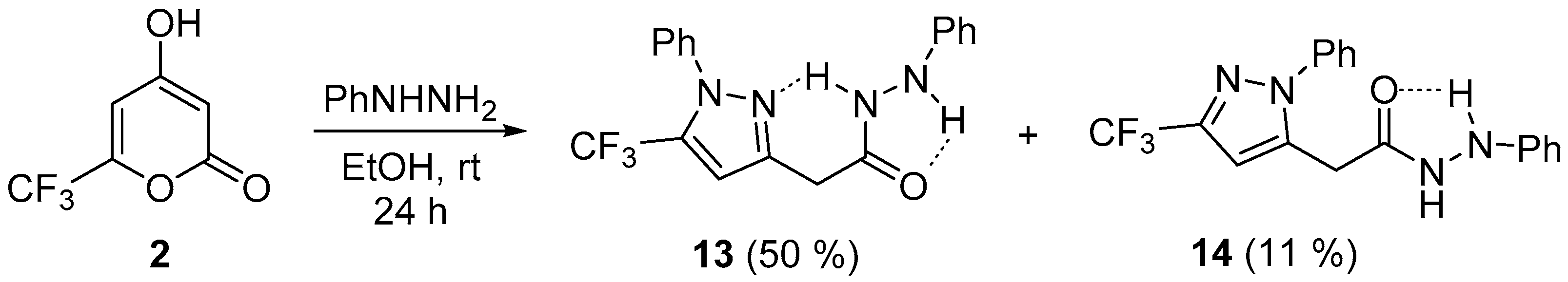
3.8. Synthesis of Compounds 13 and 14
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- McGlacken, G.P.; Fairlamb, I.J.S. 2-Pyrone natural products and mimetics: Isolation, characterisation and biological activity. Nat. Prod. Rep. 2005, 22, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Ram, V.J. Natural and synthetic 2H-pyran-2-ones and their versatility in organic synthesis. Tetrahedron 2009, 65, 7865–7913. [Google Scholar] [CrossRef]
- Pratap, R.; Ram, V.J. 2H-Pyran-2-ones and their annelated analogs as multifaceted building blocks for the fabrication of diverse heterocycles. Tetrahedron 2017, 73, 2529–2590. [Google Scholar] [CrossRef]
- Dobler, D.; Leitner, M.; Moor, N.; Reiser, O. 2-Pyrone—A privileged heterocycle and widespread motif in nature. Eur. J. Org. Chem. 2021, 2021, 6180–6205. [Google Scholar] [CrossRef]
- Hickey, A.; Pardo, L.M.; Reen, F.J.; McGlacken, G.P. Pyrones identified as LuxR signal molecules in photorhabdus and their synthetic analogues can alter multicellular phenotypic behavior of Bacillus atropheaus. ACS Omega 2021, 6, 33141–33148. [Google Scholar] [CrossRef]
- Yadav, J.S.; Ganganna, B.; Dutta, P.; Singarapu, K.K. Synthesis and determination of absolute configuration of α-pyrones isolated from Penicillium corylophilum. J. Org. Chem. 2014, 79, 10762–10771. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.K.; Yun, B.S. Styrylpyrone-class compounds from medicinal fungi Phellinus and Inonotus spp., and their medicinal importance. J. Antibiot. 2011, 64, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Bhat, Z.S.; Rather, M.A.; Maqbool, M.; Lah, H.U.; Yousuf, S.K.; Ahmad, Z. α-pyrones: Small molecules with versatile structural diversity reflected in multiple pharmacological activities-an update. Biomed. Pharmacother. 2017, 91, 265–277. [Google Scholar] [CrossRef]
- Aggarwal, R.; Walia, S.; Rani, C. Dehydroacetic acid and its derivatives as starting synthons for synthesis of heterocyclic compounds. Heterocycles 2017, 94, 1197–1244. [Google Scholar] [CrossRef]
- Obydennov, D.L.; El-Tantawy, A.I.; Sosnovskikh, V.Y. Triacetic acid lactone as a bioprivileged molecule in organic synthesis. Mendeleev Commun. 2019, 29, 1–10. [Google Scholar] [CrossRef]
- Prakash, O.; Kumar, A.; Singh, S.P. Synthesis of heterocyclic compounds from the reactions of dehydroacetic acid (DHA) and its derivatives. Heterocycles 2004, 63, 1193–1220. [Google Scholar] [CrossRef]
- Moreno-Mañas, M.; Pleixats, R. Dehydroacetic acid, triacetic acid lactone, and related pyrones. Adv. Heterocycl. Chem. 1992, 53, 1–84. [Google Scholar] [CrossRef]
- Politanskaya, L.V.; Selivanova, G.A.; Panteleeva, E.V.; Tretyakov, E.V.; Platonov, V.E.; Nikul’shin, P.V.; Vinogradov, A.S.; Zonov, Y.V.; Karpov, V.M.; Mezhenkova, T.V.; et al. Organofluorine chemistry: Promising growth areas and challenges. Russ. Chem. Rev. 2019, 88, 425–569. [Google Scholar] [CrossRef]
- Han, J.; Remete, A.M.; Dobson, L.S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V.A.; O’Hagan, D. Next generation organofluorine containing blockbuster drugs. J. Fluor. Chem. 2020, 239, 109639. [Google Scholar] [CrossRef]
- Isanbor, C.; O’Hagan, D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; O’Hagan, D. Successful fluorine-containing herbicide agrochemicals. J. Fluor. Chem. 2014, 167, 16–29. [Google Scholar] [CrossRef]
- Jeschke, P. The unique role of fluorine in the design of active ingredients for modern crop protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Organic fluorine compounds: A great opportunity for enhanced materials properties. Chem. Soc. Rev. 2011, 40, 3496–3508. [Google Scholar] [CrossRef] [PubMed]
- Granqvist, L.; Virta, P. Characterization of G-quadruplex/hairpin transitions of RNAs by 19F NMR spectroscopy. Chem. Eur. J. 2016, 22, 15360–15372. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.N.G.; Suzuki, Y. Using 19F NMR to probe biological interactions of proteins and peptides. ACS Chem. Biol. 2014, 9, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Resnick, P.R. Practical uses of fluorinated heterocycles. In Fluorinated Heterocyclic Compounds; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 493–506. [Google Scholar]
- German, L.S.; Sterlin, S.R.; Cherstkov, V.F. Hexafluorodehydroacetic acid. Bull. Acad. Sci. USSR Div. Chem. Sci. 1982, 31, 1476–1477. [Google Scholar] [CrossRef]
- Li, C.-S.; Lau, C.K.; Therien, M.; Prasit, P. Pyrones as Inhibitors of Cyclooxygenase-2. U.S. Patent 0058690, 16 May 2002. [Google Scholar]
- Hanselmann, P.; Wenger, W. Method for the Production of 6,6,6-Trihalo-3,5-dioxohexanoic Acid Esters. WO Patent 040087, 6 May 2005. [Google Scholar]
- Garratt, S. The Mechanism of the reaction between dehydroacetic acid and alkylamines. J. Org. Chem. 1963, 28, 1886–1888. [Google Scholar] [CrossRef]
- Cook, D. The preparation, properties, and structure of 2,6-bis-(alkylamino)-2,5-heptadien-4-ones. Can. J. Chem. 1963, 41, 1435–1440. [Google Scholar] [CrossRef] [Green Version]
- Pierce, J.B.; Ariyan, Z.S.; Ovenden, G.S. Preparation and antiinflammatory activity of 2- and 4-pyridones. J. Med. Chem. 1982, 25, 131–136. [Google Scholar] [CrossRef]
- Schmidt, R.; Stolte, M.; Grüne, M.; Würthner, F. Hydrogen-bond-directed formation of supramolecular polymers incorporating head-to-tail oriented dipolar merocyanine dyes. Macromolecules 2011, 44, 3766–3776. [Google Scholar] [CrossRef]
- Würthner, F.; Schmidt, J.; Stolte, M.; Wortmann, R. Hydrogen-bond-directed head-to-tail orientation of dipolar merocyanine dyes: A strategy for the design of electrooptical materials. Angew. Chem. Int. Ed. 2006, 45, 3842–3846. [Google Scholar] [CrossRef] [PubMed]
- Eiden, F. Untersuchungen an γ-Pyronen. 4. Mitteilung: Die Kondensation von γ-Pyronen mit cyclischen Malonsäureamiden. Arch. Pharm. 1960, 293, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Obydennov, D.L.; El-Tantawy, A.I.; Sosnovskikh, V.Y. Bio-based triacetic acid lactone in the synthesis of azaheterocycles via a ring-opening transformation. New J. Chem. 2018, 42, 8943–8952. [Google Scholar] [CrossRef]
- Usachev, B.I.; Obydennov, D.L.; Röschenthaler, G.-V.; Sosnovskikh, V.Y. 2-Cyano-6-(trifluoromethyl)-4H-pyran-4-one: A novel versatile CF3-containing building block. J. Fluor. Chem. 2012, 137, 22–26. [Google Scholar] [CrossRef]
- Zheglova, D.K.; Genov, D.G.; Gribanov, A.V.; Kol’tsov, A.I.; Smirnov, S.N. NMR spectroscopic study of the (Z)/(E)-isomerism of 1-aryl-3-arylamino-2-propen-1-ones in solution and in the crystalline state. Monatsh. Chem. 1994, 125, 1443–1446. [Google Scholar] [CrossRef]
- Gerus, I.I.; Balabon, O.A.; Pazenok, S.V.; Lui, N.; Kondratov, I.S.; Tarasenko, K.V.; Shaitanova, E.N.; Ivasyshyn, V.E.; Kukhar, V.P. Synthesis and properties of polyfunctional cyclic β-alkoxy-α,β-unsaturated ketones based on 4-methylene-1,3-dioxolanes. Eur. J. Org. Chem. 2018, 2018, 3853–3861. [Google Scholar] [CrossRef]
- Wöjcik, J.; Domalewski, W.; Kamieńska-Trela, K.; Stefaniak, L.; Vdovienko, S.I.; Gerus, I.I.; Gorbunova, M.G. Conformational behaviour of some trifluoroenaminones by multinuclear magnetic resonance. Magn. Reson. Chem. 1993, 31, 808–814. [Google Scholar] [CrossRef]
- Kiang, A.K.; Tan, S.F.; Wong, W.S. Reactions of acetonedicarboxylic anhydride (tetrahydropyrantrione) and its mono- and di-acetyl derivatives with amines. J. Chem. Soc. C 1971, 2721–2726. [Google Scholar] [CrossRef]
- Usachev, S.A.; Usachev, B.I.; Sosnovskikh, V.Y. Synthesis of 6-hydroxy-5,6-dihydro-2-pyrones and -pyridones by reaction of 4-aryl-6-trifluoromethyl-2-pyrones with water, hydrazine, and hydroxylamine. Chem. Heterocycl. Compd. 2017, 53, 1294–1301. [Google Scholar] [CrossRef]
- Gerus, I.I.; Tolmachova, N.A.; Vdovenko, S.I.; Fröhlich, R.; Haufe, G. A convenient synthesis and chemical properties of 3-acylamino-6-poly-fluoroalkyl-2H-pyran-2-ones. Synthesis 2005, 2005, 1269–1278. [Google Scholar] [CrossRef]
- Ye, W.-P.; Mu, H.-L.; Shi, X.-C.; Cheng, Y.-X.; Li, Y.-S. Synthesis and characterization of the titanium complexes bearing two regioisomeric trifluoromethyl-containing enaminoketonato ligands and their behavior in ethylene polymerization. Dalt. Trans. 2009, 2009, 9452–9465. [Google Scholar] [CrossRef] [PubMed]
- Desens, W.; Winterberg, M.; Michalik, D.; Langer, P. Synthesis and solution structure of 1H-benzo-1,5-diazepine derivatives with a perfluoroalkyl side chain. Helv. Chim. Acta 2016, 99, 361–372. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
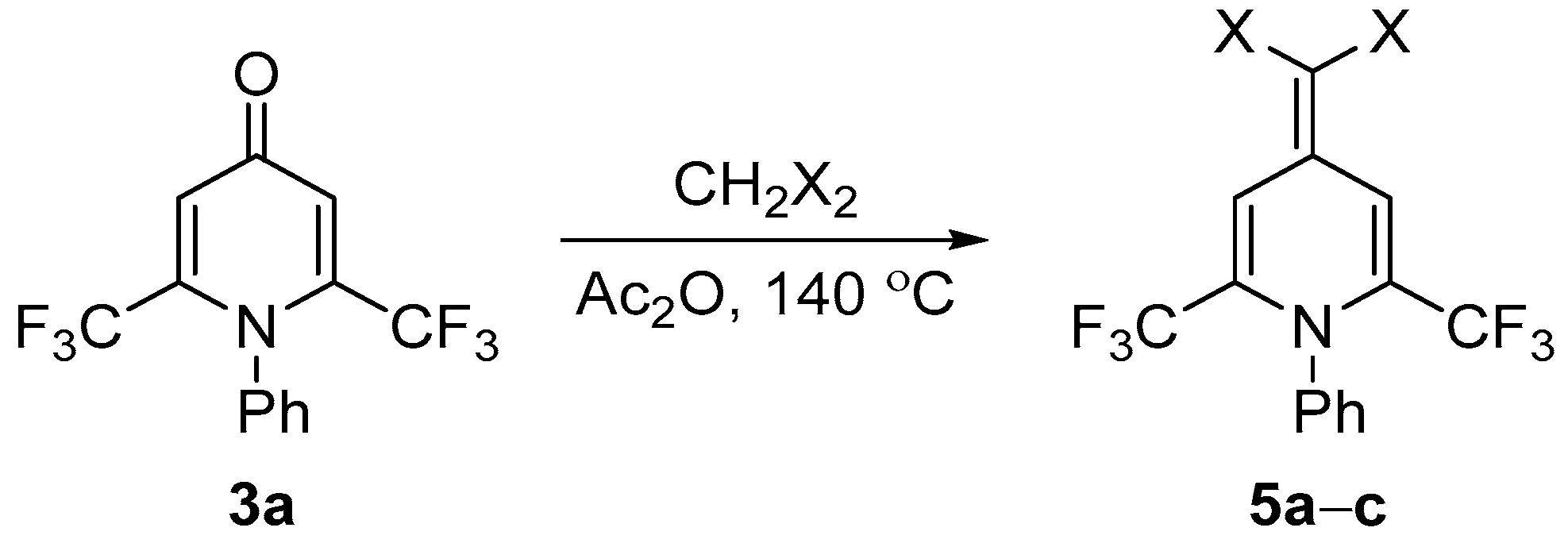{kind=link}
{kind=link}
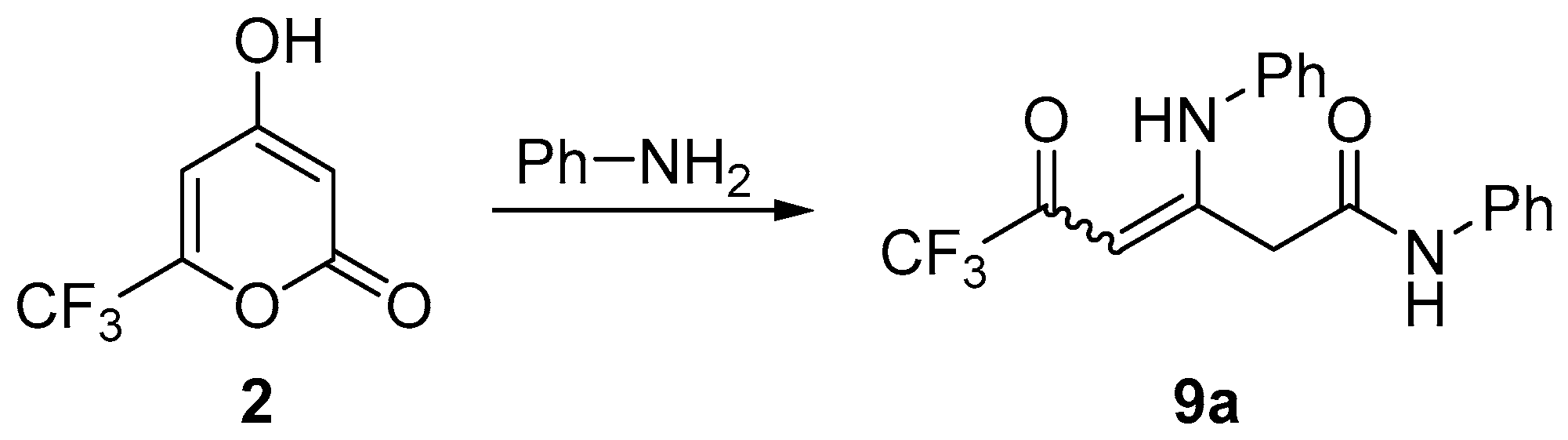{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | PhNH2, Equiv. | Solvent | Yield 3a, % |
|---|---|---|---|
| 1 | 1.5 | HCl (0.5 M) b | n.r. c |
| 2 | 2.3 | ethanol | 60 |
| 3 | 3.5 | ethanol | 40 |
| 4 | 4.0 | ethanol | 45 |
| 5 | 2.3 | ethanol | 37 d |
| 6 | 4.0 | 1,4-dioxane | 36 |
| 7 | 3.5 | 1,4-dioxane | 27 e |
| 8 | 4.0 | toluene | 17 |
| 9 | 2.3 | toluene | 19 |
| 10 | 2.3 | neat | 25 |
| Pyridone | Ar | Time, h | Yield, % | Mp, °C |
|---|---|---|---|---|
| 3a | Ph | 24 | 60 | 135–136 |
| 3b | 4-MeOC6H4 | 8 | 56 | 125–126 |
| 3c | 3,4-F2C6H3 | 6 | 57 | 142–143 |
| 3d | 4-BrC6H4 | 24 | 47 | 129–131 |
| 3e | 3-AcC6H4 | 24 | 47 | 118–119 |
| 3f | 3-F3CC6H4 | 24 | 47 | 91–92 |
| 3g | 2,5-F2C6H3 | 48 | 46 | 116–119 |
| 3h | 2-ClC6H4 | 40 | 49 | 132–136 |
| 3i | 2-MeC6H4 | 24 | 65 | Liq. |
| 3j | 2,5-Me2C6H3 | 24 | 48 | 105–107 |
| 3k | 4-O2NC6H4 | 144 | 8 b | – c |
| Compound | Structure | Time, h | Yield, % | Mp, °C |
|---|---|---|---|---|
| 5a |  | 10 | 65 | 172–173 |
| 5b |  | 4 | 58 | 270–271 |
| 5c |  | 6 | 43 | 208–209 |
| Entry | PhNH2, Equiv. | Solvent | Yield 9a, % |
|---|---|---|---|
| 1 | 5 | ethanol | 43 |
| 2 | 3.5 | ethanol | 63 |
| 3 | 2.1 | ethanol | 64 |
| 4 | 2.1 | 1,4-dioxane | 72 |
| 5 | 2.1 | toluene | 30 |
| 6 | 2.1 | neat | 12 |
| Enaminone | R | Time, h | Yield, % | Z/E ratio b |
|---|---|---|---|---|
| 9a | Ph | 24 | 72 | 59:41 (44:56 c) |
| 9b | 4-MeOC6H4 | 17 | 69 | 65:35 |
| 9c | 4-BrC6H4 | 15 | 41 | 59:41 |
| 9d | Bn | 17 | 24 | 58:42 (67:33 c) |
| 9e | Bu | 72 | 7 d | 65:35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedin, V.V.; Usachev, S.A.; Obydennov, D.L.; Sosnovskikh, V.Y. Reactions of Trifluorotriacetic Acid Lactone and Hexafluorodehydroacetic Acid with Amines: Synthesis of Trifluoromethylated 4-Pyridones and Aminoenones. Molecules 2022, 27, 7098. https://doi.org/10.3390/molecules27207098
Fedin VV, Usachev SA, Obydennov DL, Sosnovskikh VY. Reactions of Trifluorotriacetic Acid Lactone and Hexafluorodehydroacetic Acid with Amines: Synthesis of Trifluoromethylated 4-Pyridones and Aminoenones. Molecules. 2022; 27(20):7098. https://doi.org/10.3390/molecules27207098
Chicago/Turabian StyleFedin, Vladislav V., Sergey A. Usachev, Dmitrii L. Obydennov, and Vyacheslav Y. Sosnovskikh. 2022. "Reactions of Trifluorotriacetic Acid Lactone and Hexafluorodehydroacetic Acid with Amines: Synthesis of Trifluoromethylated 4-Pyridones and Aminoenones" Molecules 27, no. 20: 7098. https://doi.org/10.3390/molecules27207098
APA StyleFedin, V. V., Usachev, S. A., Obydennov, D. L., & Sosnovskikh, V. Y. (2022). Reactions of Trifluorotriacetic Acid Lactone and Hexafluorodehydroacetic Acid with Amines: Synthesis of Trifluoromethylated 4-Pyridones and Aminoenones. Molecules, 27(20), 7098. https://doi.org/10.3390/molecules27207098