

B3Al4+: A Three-Dimensional Molecular Reuleaux Triangle

 , ,
, , 


Abstract
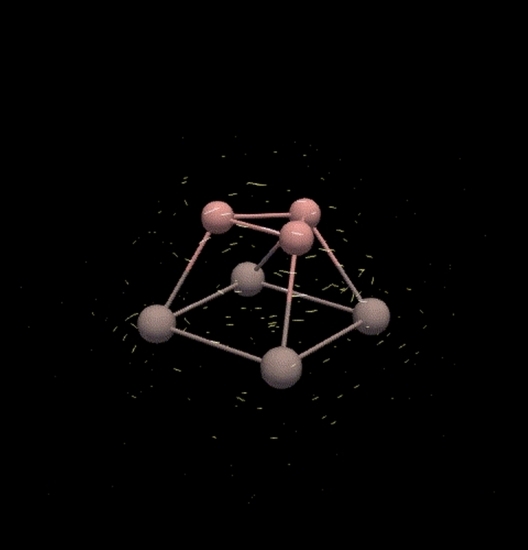
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Details
3. Discussion
4. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Huang, W.; Sergeeva, A.P.; Zhai, H.-J.; Averkiev, B.B.; Wang, L.-S.; Boldyrev, A.I. A concentric planar doubly π-aromatic B19− cluster. Nat. Chem. 2010, 2, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Halla, J.O.C.; Islas, R.; Heine, T.; Merino, G. B19−: An aromatic Wankel motor. Angew. Chem. Int. Ed. Engl. 2010, 49, 5668–5671. [Google Scholar] [CrossRef]
- Moreno, D.; Pan, S.; Zeonjuk, L.L.; Islas, R.; Osorio, E.; Martínez-Guajardo, G.; Chattaraj, P.K.; Heine, T.; Merino, G. B182−: A quasi-planar bowl member of the Wankel motor family. Chem. Commun. 2014, 50, 8140–8143. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Zhao, X.-Y.; Chen, Q.; Zhai, H.-J.; Li, S.-D. B11−: A moving subnanoscale tank tread. Nanoscale 2015, 7, 16054–16060. [Google Scholar] [CrossRef]
- Wang, Y.-J.; You, X.-R.; Chen, Q.; Feng, L.-Y.; Wang, K.; Ou, T.; Zhao, X.-Y.; Zhai, H.-J.; Li, S.-D. Chemical bonding and dynamic fluxionality of a B15+ cluster: A nanoscale double-axle tank tread. Phys. Chem. Chem. Phys. 2016, 18, 15774–15782. [Google Scholar] [CrossRef]
- Yang, Y.; Jia, D.; Wang, Y.-J.; Zhai, H.-J.; Man, Y.; Li, S.-D. A universal mechanism of the planar boron rotors B11−, B13+, B15+, and B19−: Inner wheels rotating in pseudo-rotating outer bearings. Nanoscale 2017, 9, 1443–1448. [Google Scholar] [CrossRef]
- Liu, L.; Moreno, D.; Osorio, E.; Castro, A.C.; Pan, S.; Chattaraj, P.K.; Heine, T.; Merino, G. Structure and bonding of IrB12−: Converting a rigid boron B12 platelet to a Wankel motor. RSC Adv. 2016, 6, 27177–27182. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Feng, L.-Y.; Zhai, H.-J. Starting a subnanoscale tank tread: Dynamic fluxionality of boron-based B10Ca alloy cluster. Nanoscale Adv. 2019, 1, 735–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Guajardo, G.; Sergeeva, A.P.; Boldyrev, A.I.; Heine, T.; Ugalde, J.M.; Merino, G. Unravelling phenomenon of internal rotation in B13+ through chemical bonding analysis. Chem. Commun. 2011, 47, 6242–6244. [Google Scholar] [CrossRef] [PubMed]
- Merino, G.; Heine, T. And yet it rotates: Thestarter for a molecular Wankel motor. Angew. Chem. Int. Ed. Engl. 2012, 51, 10226–10227. [Google Scholar] [CrossRef]
- Fagiani, M.R.; Song, X.; Petkov, P.; Debnath, S.; Gewinner, S.; Schöllkopf, W.; Heine, T.; Fielicke, A.; Asmis, K.R. Structure and fluxionality of B13+ probed by infrared photodissociation spectroscopy. Chem. Int. Ed. Engl. 2017, 56, 501–504. [Google Scholar] [CrossRef]
- Pan, S.; Barroso, J.; Jalife, S.; Heine, T.; Asmis, K.R.; Merino, G. Fluxional boron clusters: From theory to reality. Acc. Chem. Res. 2019, 52, 2732–2744. [Google Scholar] [CrossRef]
- Jalife, S.; Liu, L.; Pan, S.; Cabellos, J.L.; Osorio, E.; Lu, C.; Heine, T.; Donald, K.J.; Merino, G. Dynamical behavior of boron clusters. Nanoscale 2016, 8, 17639–17644. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Feng, L.-Y.; Guo, J.-C.; Zhai, H.-J. Dynamic Mg2B8 cluster: A nanoscale compass. Chem. Asian J. 2017, 12, 2899–2903. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Guo, J.-C. Dynamic fluxionality of ternary Mg2BeB8 cluster: A nanocompass. J. Mol. Model. 2020, 26, 30. [Google Scholar] [CrossRef]
- Li, W.-L.; Jian, T.; Chen, X.; Li, H.-R.; Chen, T.-T.; Luo, X.-M.; Li, S.-D.; Li, J.; Wang, L.-S. Observation of a metal-centered B2-Ta@B18− tubular molecular rotor and a perfect Ta@B20− boron drum with the record coordination number of twenty. Chem. Commun. 2017, 53, 1587–1590. [Google Scholar] [CrossRef]
- Martínez-Guajardo, G.; Cabellos, J.L.; Díaz-Celaya, A.; Pan, S.; Islas, R.; Chattaraj, P.K.; Heine, T.; Merino, G. Dynamical behavior of borospherene: A nanobubble. Sci. Rep. 2015, 5, 11287. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.-C.; Feng, L.-Y.; Wang, Y.-J.; Jalife, S.; Vásquez-Espinal, A.; Cabellos, J.L.; Pan, S.; Merino, G.; Zhai, H.-J. Coaxial triple-layered versus helical Be6B11− clusters: Dual structural fluxionality and multifold aromaticity. Angew. Chem. Int. Ed. Engl. 2017, 56, 10174–10177. [Google Scholar] [CrossRef]
- Feng, L.-Y.; Guo, J.-C.; Li, P.-F.; Zhai, H.-J. Boron-based binary Be6B102− cluster: Three-layered aromatic sandwich, electronic transmutation, and dynamic structural fluxionality. Phys. Chem. Chem. Phys. 2018, 20, 22719–22729. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Feng, L.-Y.; Zhai, H.-J. Sandwich-type Na6B7− and Na8B7+ clusters: Charge-transfer complexes, four-fold π/σ aromaticity, and dynamic fluxionality. Phys. Chem. Chem. Phys. 2019, 21, 18338–18345. [Google Scholar] [CrossRef]
- Barroso, J.; Pan, S.; Merino, G. Structural transformations in boron clusters induced by metal doping. Chem. Soc. Rev. 2022, 51, 1098–1123. [Google Scholar] [CrossRef]
- Yu, R.; Barroso, J.; Wang, M.-H.; Liang, W.-Y.; Chen, C.; Zarate, X.; Orozco-Ic, M.; Cui, Z.-H.; Merino, G. Structure and bonding of molecular stirrers with formula B7M2− and B8M2 (M = Zn, Cd, Hg). Phys. Chem. Chem. Phys. 2020, 22, 12312–12320. [Google Scholar]
- Wen, L.; Zhou, D.; Yang, L.-M.; Li, G.; Ganz, E. Atomistic structures, stabilities, electronic properties, and chemical bonding of boron–aluminum mixed clusters B3Aln0/−/+ (n = 2–6). J. Cluster Sci. 2021, 32, 1261–1276. [Google Scholar]
- Ortiz-Chi, F.; Merino, G. A Hierarchical Algorithm for Molecular Similarity (H-FORMS); GLOMOS: Mérida, Mexico, 2020. [Google Scholar]
- Grande-Aztatzi, R.; Martínez-Alanis, P.R.; Cabellos, J.L.; Osorio, E.; Martínez, A.; Merino, G. Structural evolution of small gold clusters doped by one and two boron atoms. J. Comput. Chem. 2014, 35, 2288–2296. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar]
- Dunning, T.H., Jr.; Hay, P.J. Modern Theoretical Chemistry; Schaefer, H.F., III, Ed.; Plenum: New York, NY, USA, 1977; Volume 3, pp. 1–28. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar]
- Purvis, G.D., III; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar]
- Millam, J.M.; Bakken, V.; Chen, W.; Hase, W.L.; Schlegel, H.B. Ab initio classical trajectories on the Born-Oppenheimer surface: Hessian-based integrators using fifth-order polynomial and rational function fits. J. Chem. Phys. 1999, 111, 3800–3805. [Google Scholar] [CrossRef]
- Jusélius, J.; Sundholm, D.; Gauss, J. Calculation of current densities using gauge-including atomic orbitals. J. Chem. Phys. 2004, 121, 3952–3963. [Google Scholar] [CrossRef] [PubMed]
- Fliegl, H.; Taubert, S.; Lehtonen, O.; Sundholm, D. The gauge including magnetically induced current method. Phys. Chem. Chem. Phys. 2011, 13, 20500–20518. [Google Scholar] [CrossRef] [PubMed]
- Sundholm, D.; Fliegl, H.; Berger, R.J.F. Calculations of magnetically induced current densities: Theory and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 639–678. [Google Scholar] [CrossRef]
- Sundholm, D.; Dimitrova, M.; Berger, R.J.F. Current density and molecular magnetic properties. Chem. Commun. 2021, 57, 12362–12378. [Google Scholar] [CrossRef] [PubMed]
- Merino, G.; Heine, T.; Seifert, G. The induced magnetic field in cyclic molecules. Chem.-Eur. J. 2004, 10, 4367–4371. [Google Scholar] [CrossRef] [PubMed]
- Heine, T.; Islas, R.; Merino, G. σ and π contributions to the induced magnetic field: Indicators for the mobility of electrons in molecules. J. Comput. Chem. 2007, 28, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Islas, R.; Heine, T.; Merino, G. The induced magnetic field. Acc. Chem. Res. 2012, 45, 215–228. [Google Scholar] [CrossRef]
- Orozco-Ic, M.; Cabellos, J.L.; Merino, G. Aromagnetic; Cinvestav-Mérida: Mérida, Mexico, 2016. [Google Scholar]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lehtola, S.; Dimitrova, M.; Fliegl, H.; Sundholm, D. Benchmarking magnetizabilities with recent density functionals. J. Chem. Theory Comput. 2021, 17, 1457–1468. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Navarro, F.; Martínez-Guajardo, G.; Osorio, E.; Moreno, D.; Tiznado, W.; Islas, R.; Donald, K.J.; Merino, G. Stop rotating! One substitution halts the B19− motor. Chem. Commun. 2014, 50, 10680–10682. [Google Scholar] [CrossRef]
- Berger, R.J.F.; Dimitrova, M.; Nasibullin, R.T.; Valiev, R.R.; Sundholm, D. Integration of global ring currents using the Ampère-Maxwell law. Phys. Chem. Chem. Phys. 2022, 24, 624–628. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, L.-X.; Orozco-Ic, M.; Zarate, X.; Sundholm, D.; Pan, S.; Guo, J.-C.; Merino, G. B3Al4+: A Three-Dimensional Molecular Reuleaux Triangle. Molecules 2022, 27, 7407. https://doi.org/10.3390/molecules27217407
Bai L-X, Orozco-Ic M, Zarate X, Sundholm D, Pan S, Guo J-C, Merino G. B3Al4+: A Three-Dimensional Molecular Reuleaux Triangle. Molecules. 2022; 27(21):7407. https://doi.org/10.3390/molecules27217407
Chicago/Turabian StyleBai, Li-Xia, Mesías Orozco-Ic, Ximena Zarate, Dage Sundholm, Sudip Pan, Jin-Chang Guo, and Gabriel Merino. 2022. "B3Al4+: A Three-Dimensional Molecular Reuleaux Triangle" Molecules 27, no. 21: 7407. https://doi.org/10.3390/molecules27217407
APA StyleBai, L. -X., Orozco-Ic, M., Zarate, X., Sundholm, D., Pan, S., Guo, J. -C., & Merino, G. (2022). B3Al4+: A Three-Dimensional Molecular Reuleaux Triangle. Molecules, 27(21), 7407. https://doi.org/10.3390/molecules27217407