
Selective Oxidation of Glycerol via Acceptorless Dehydrogenation Driven by Ir(I)-NHC Catalysts


 and
and 
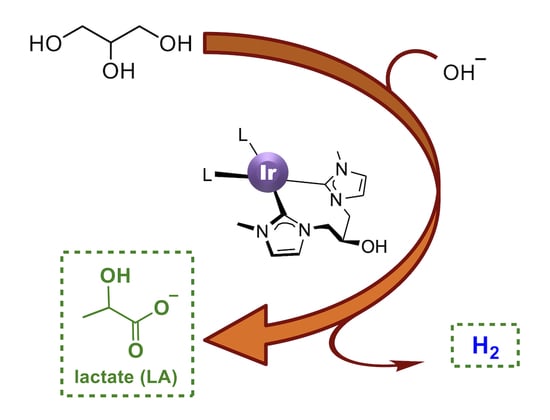
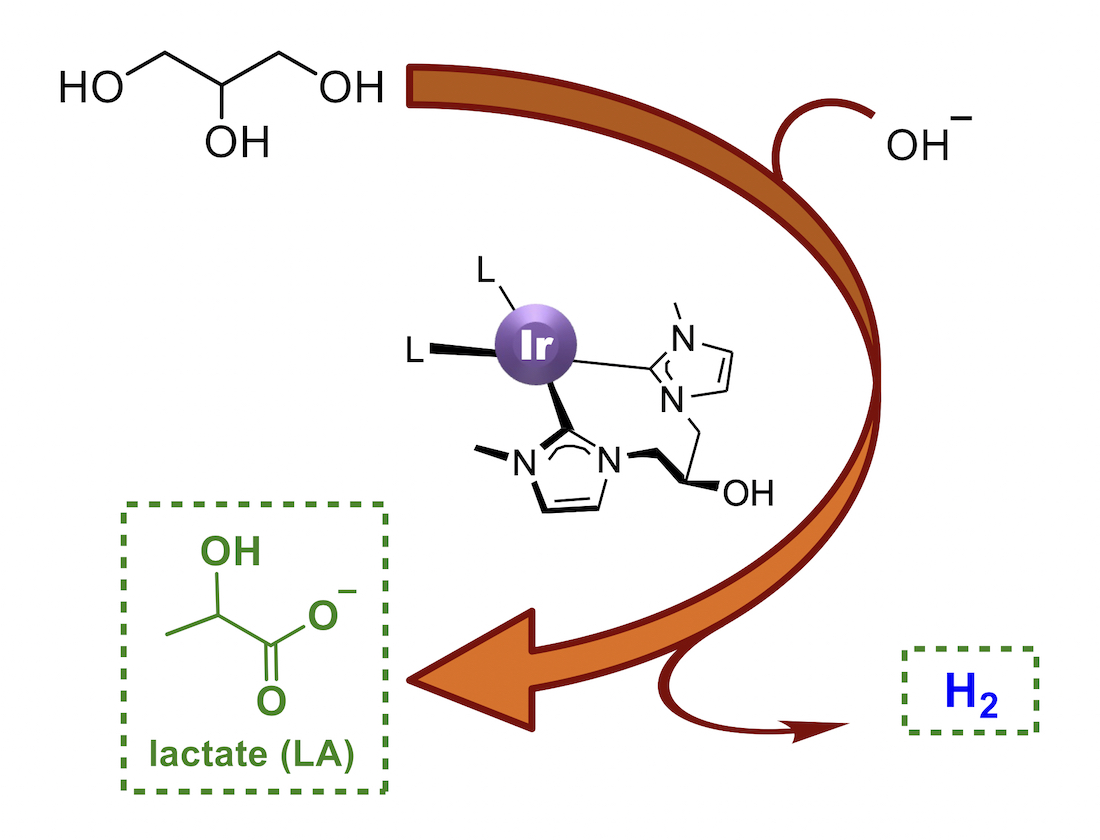
Abstract
:
1. Introduction
2. Results and Discussion
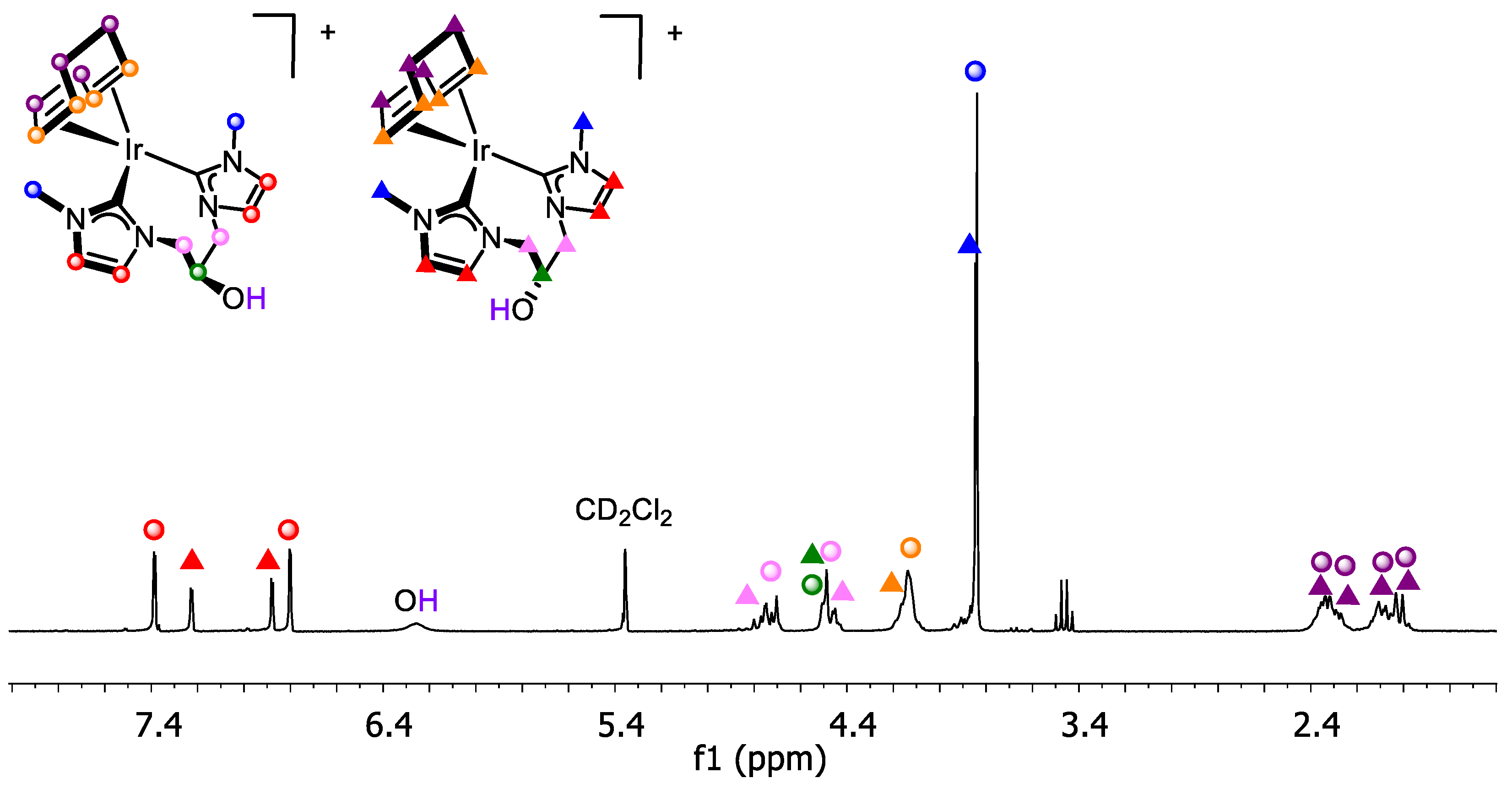
2.1. Synthesis and Characterization of bis-NHC Iridium(I) Complexes
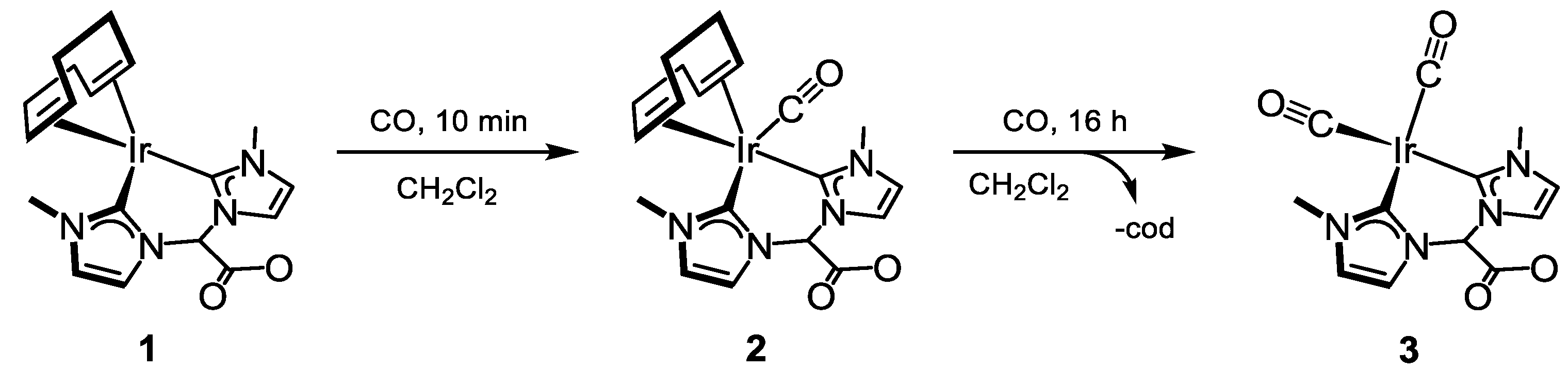
2.1.1. Carbonylation of [Ir(cod){(MeIm)2CHCOO}]
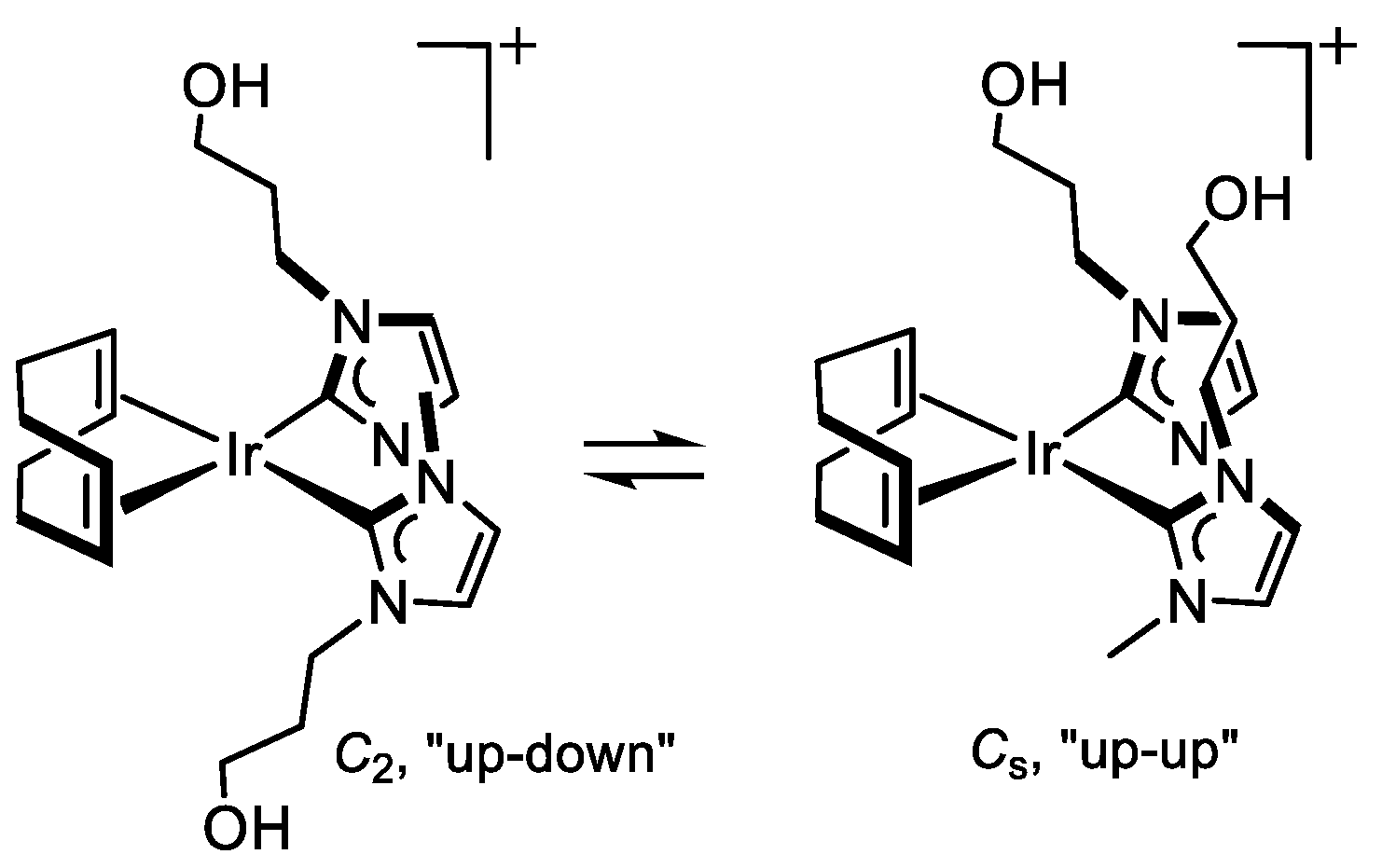
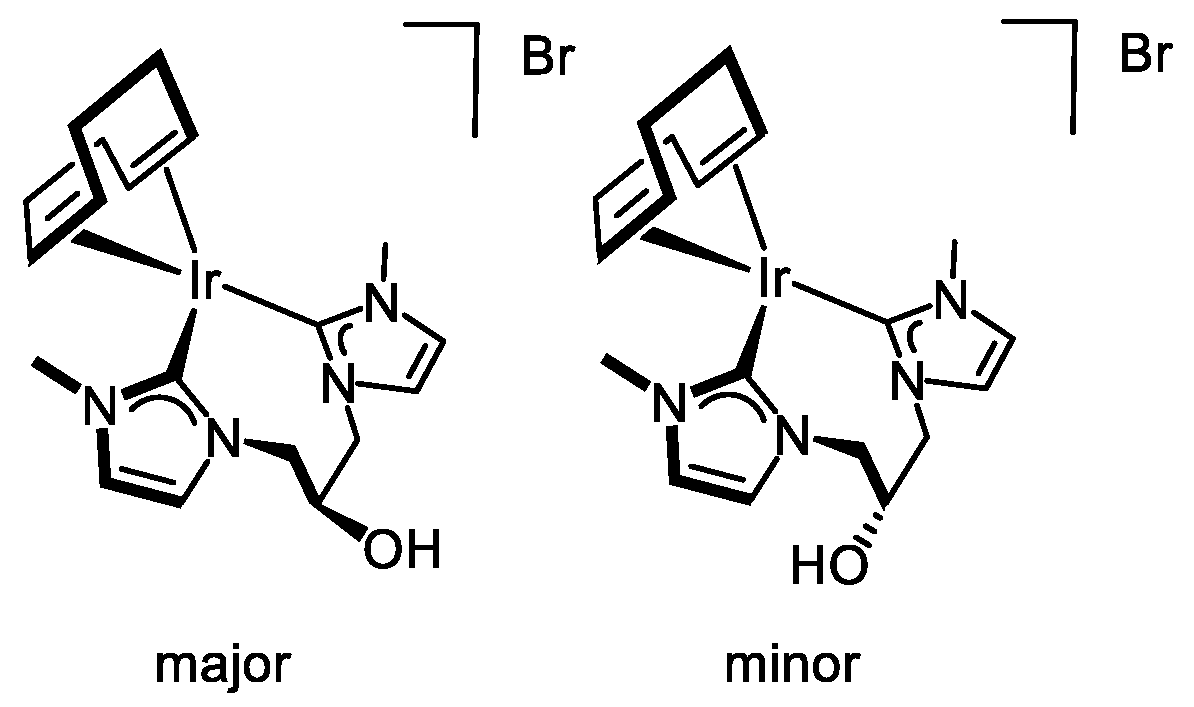
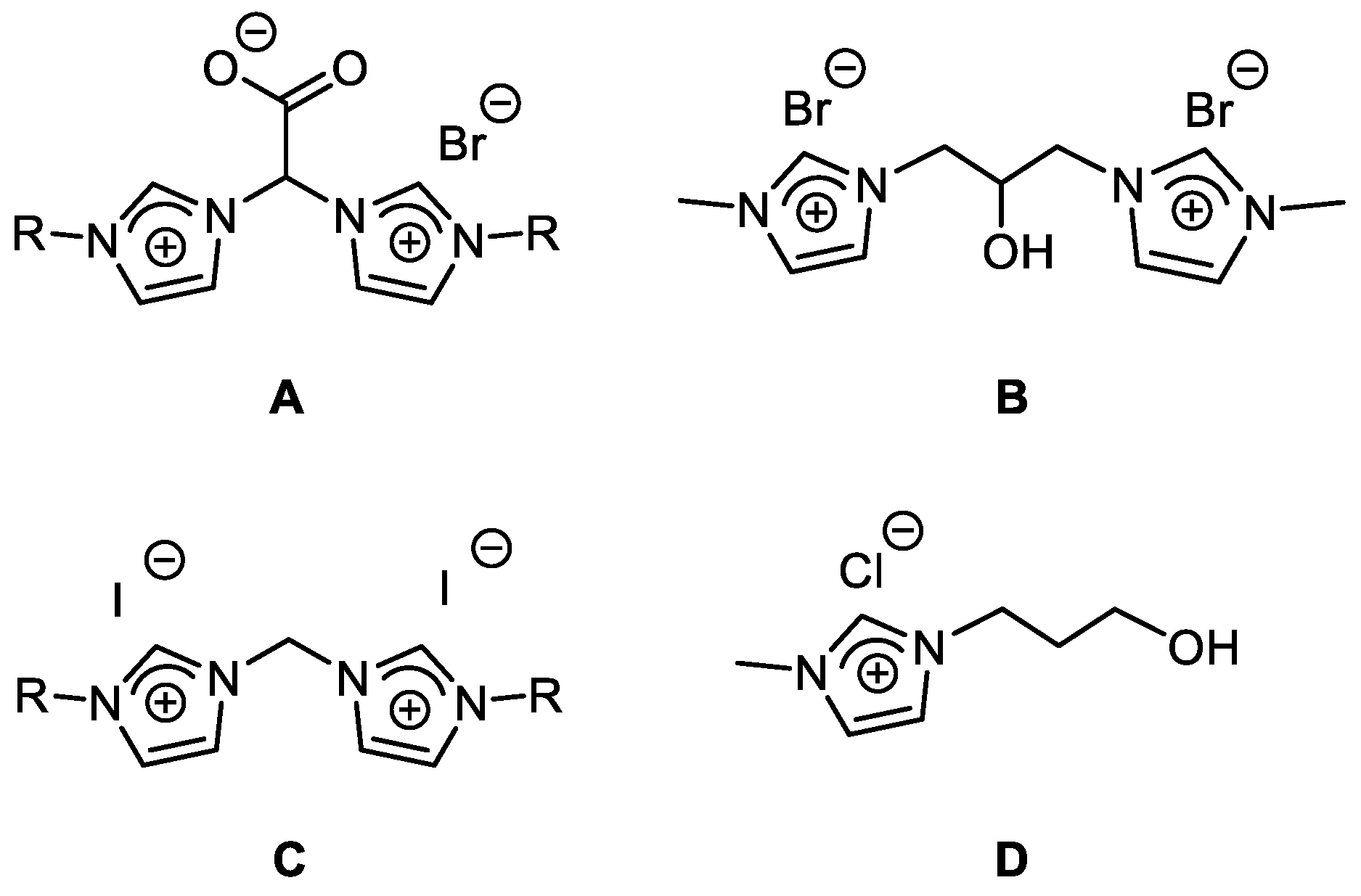
2.1.2. Synthesis and Reactivity of [Ir(cod){(MeImCH2)2CHOH}]Br
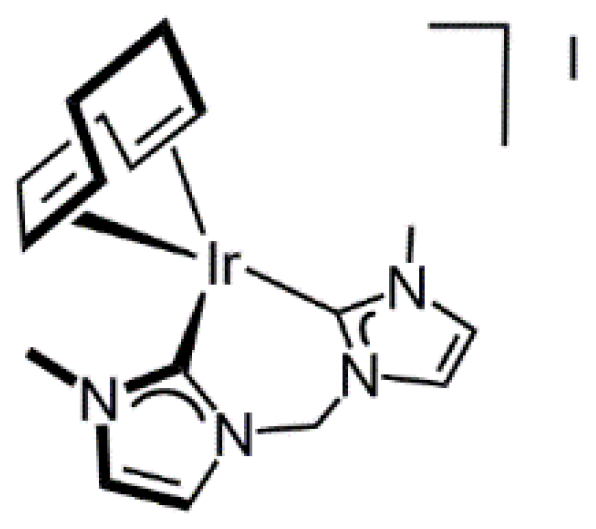
2.1.3. Synthesis of [Ir(cod){(MeIm)2CH2}]I
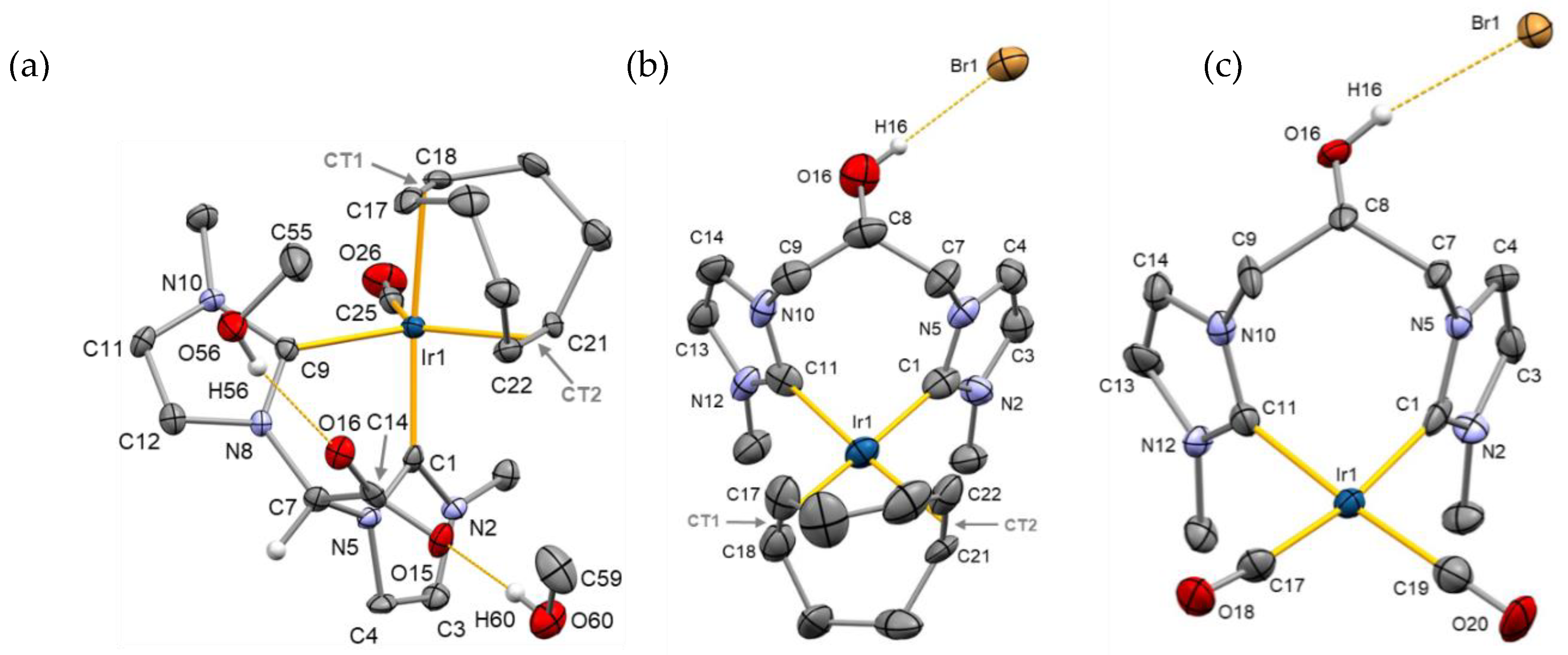
2.1.4. Crystal and Molecular Structure of bis-NHC Iridium(I) Compounds
2.2. Synthesis and Characterization of NHC Iridium(I) Complexes
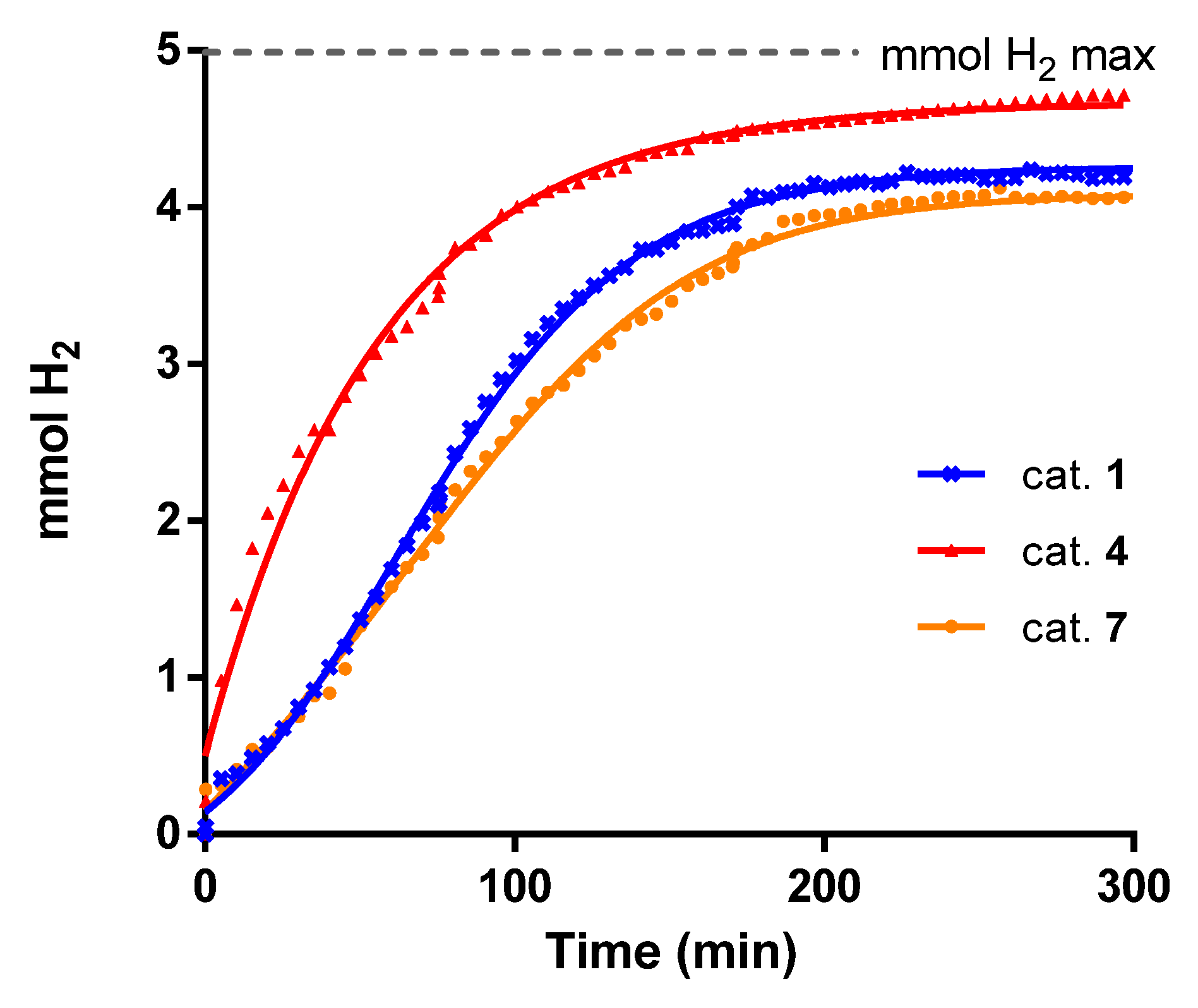
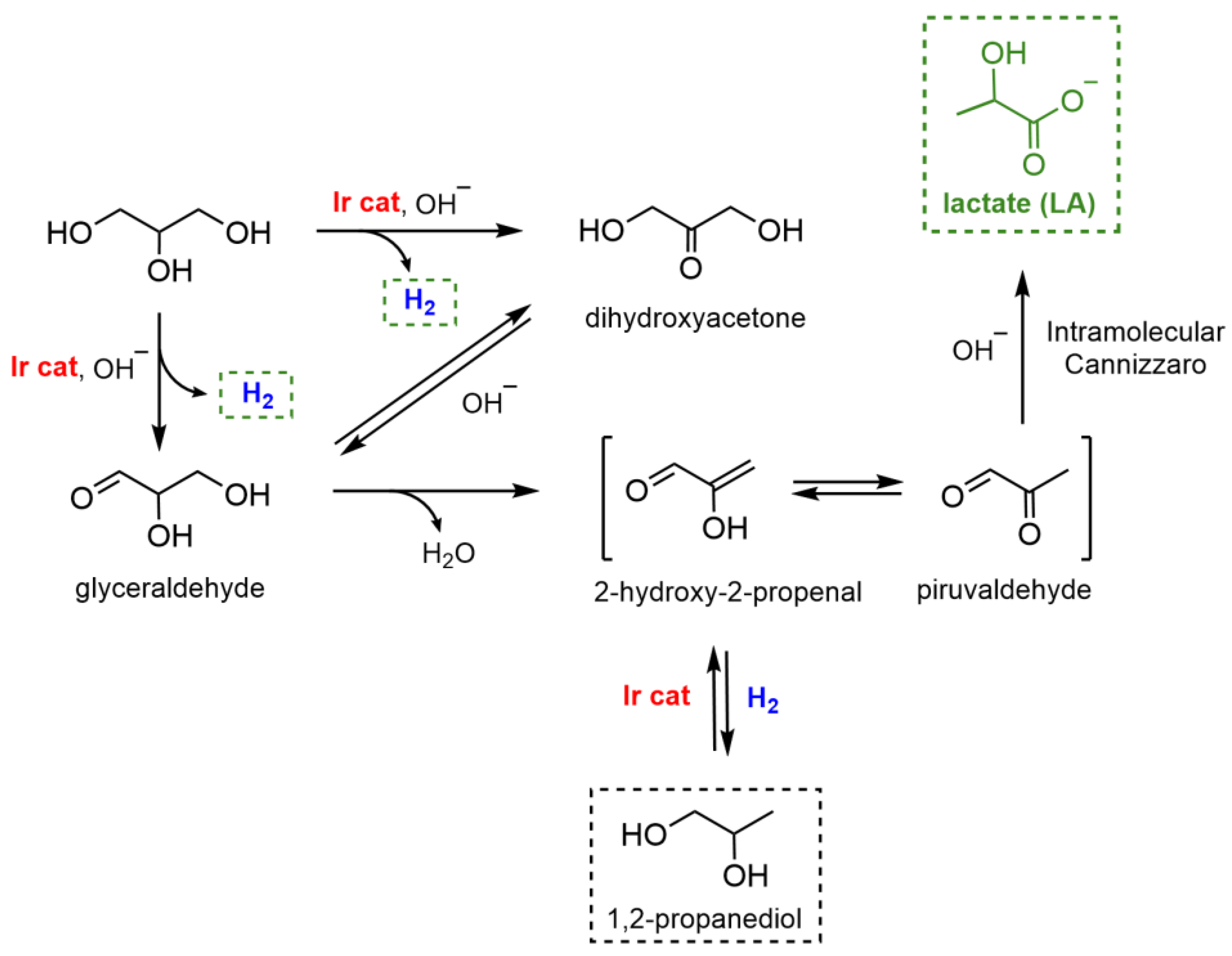
2.3. Dehydrogenation of Glycerol to Lactic Acid
3. Experimental Section
3.1. Materials and Methods
3.2. Scientific Equipment
3.3. Compound Synthesis
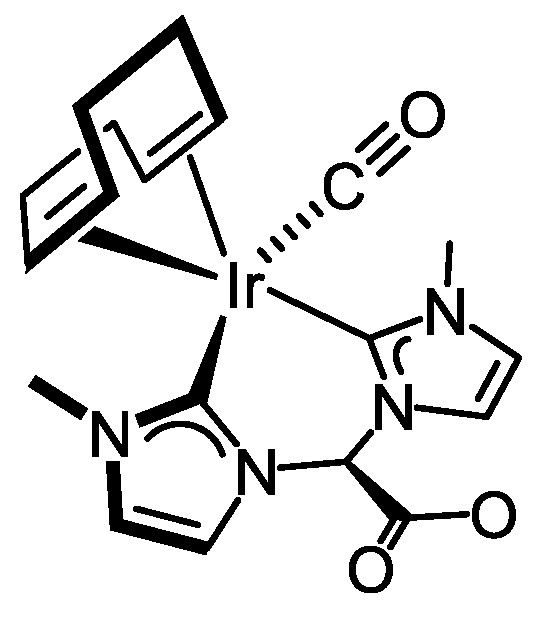
3.3.1. Synthesis of [Ir(CO)(cod){(MeIm)2CHCOO}] (2), Figure 5

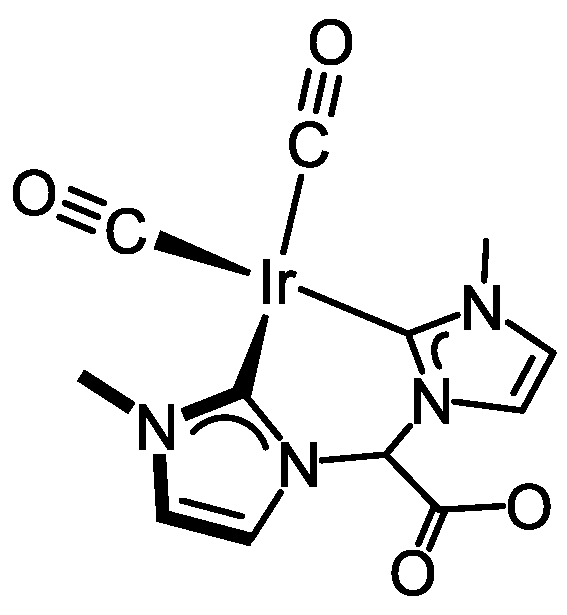
3.3.2. Synthesis of [Ir(CO)2{(MeIm)2CHCOO}] (3), Figure 6

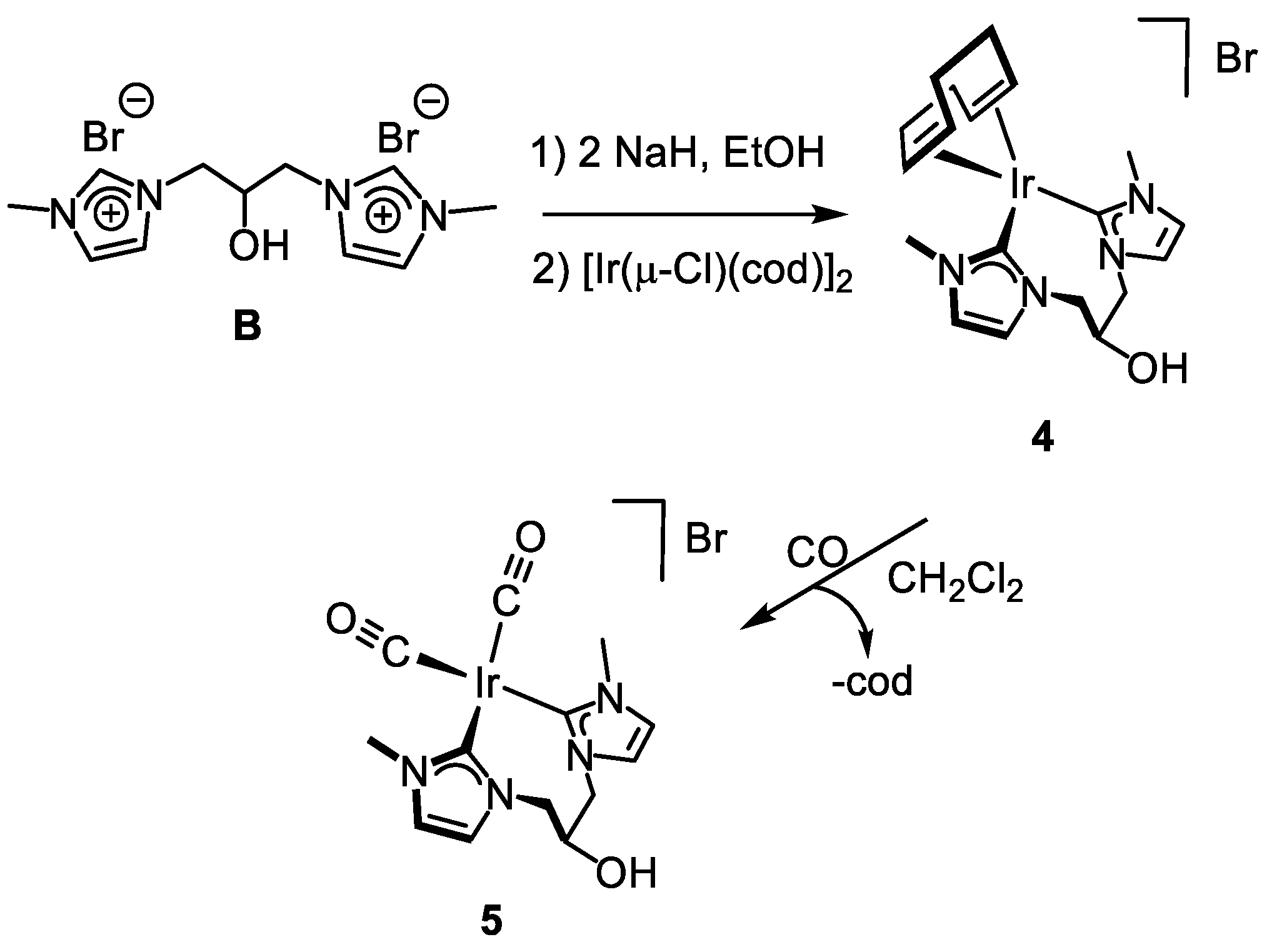
3.3.3. Synthesis of [Ir(cod){(MeImCH2)2CHOH}]Br (4), Figure 7

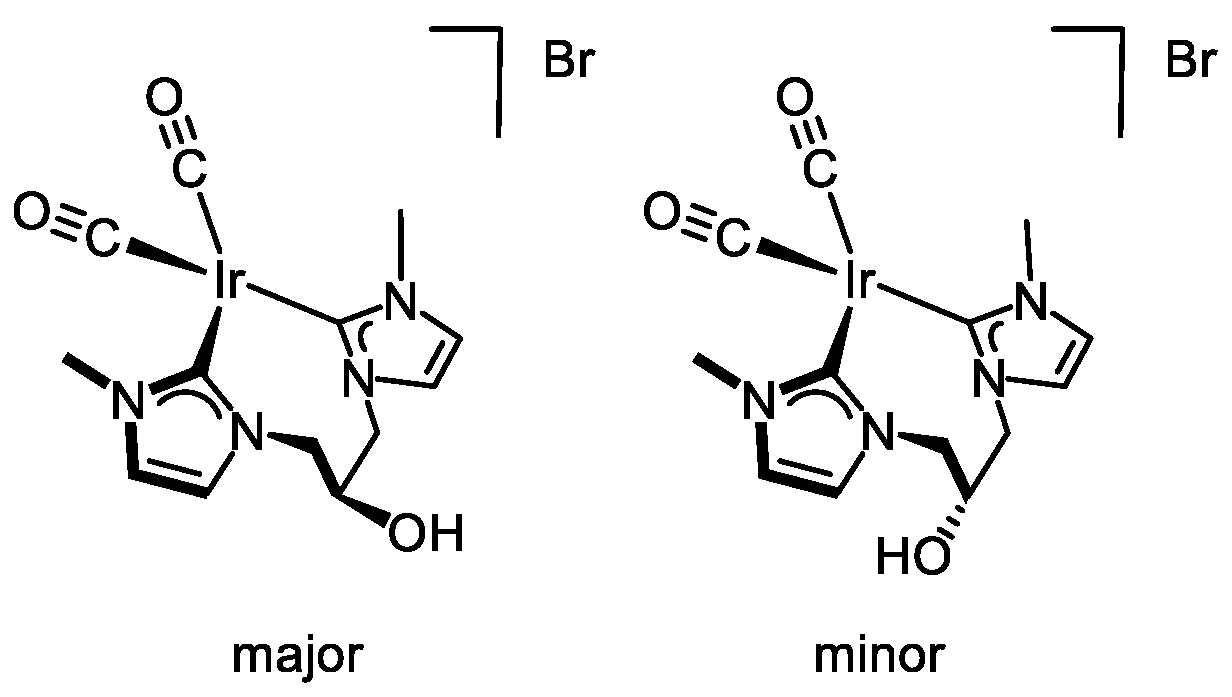
3.3.4. Synthesis of [Ir(CO)2{(MeImCH2)2CHOH}]Br (5), Figure 8

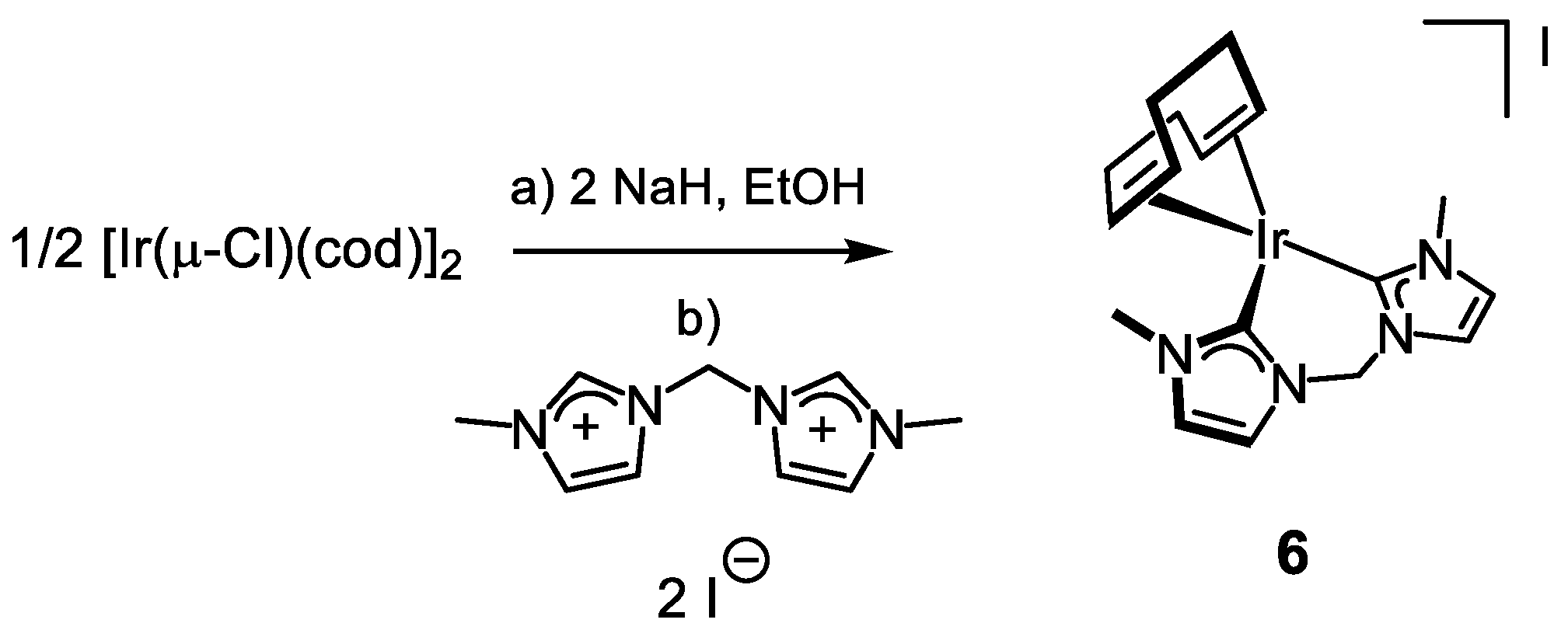
3.3.5. Synthesis of [Ir(cod){(MeIm)2CH2}]I (6), Figure 9

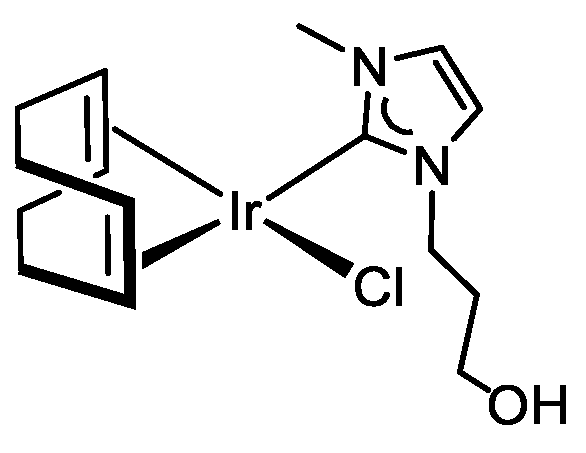
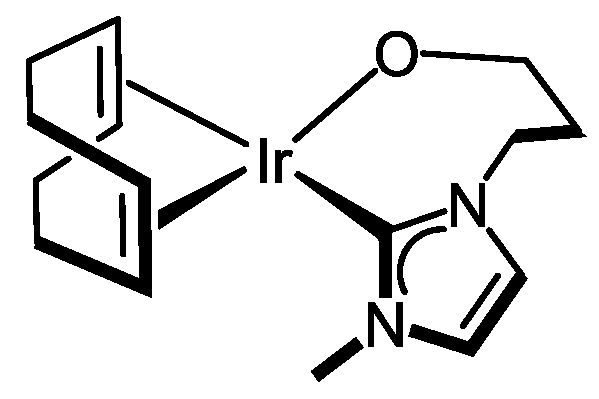
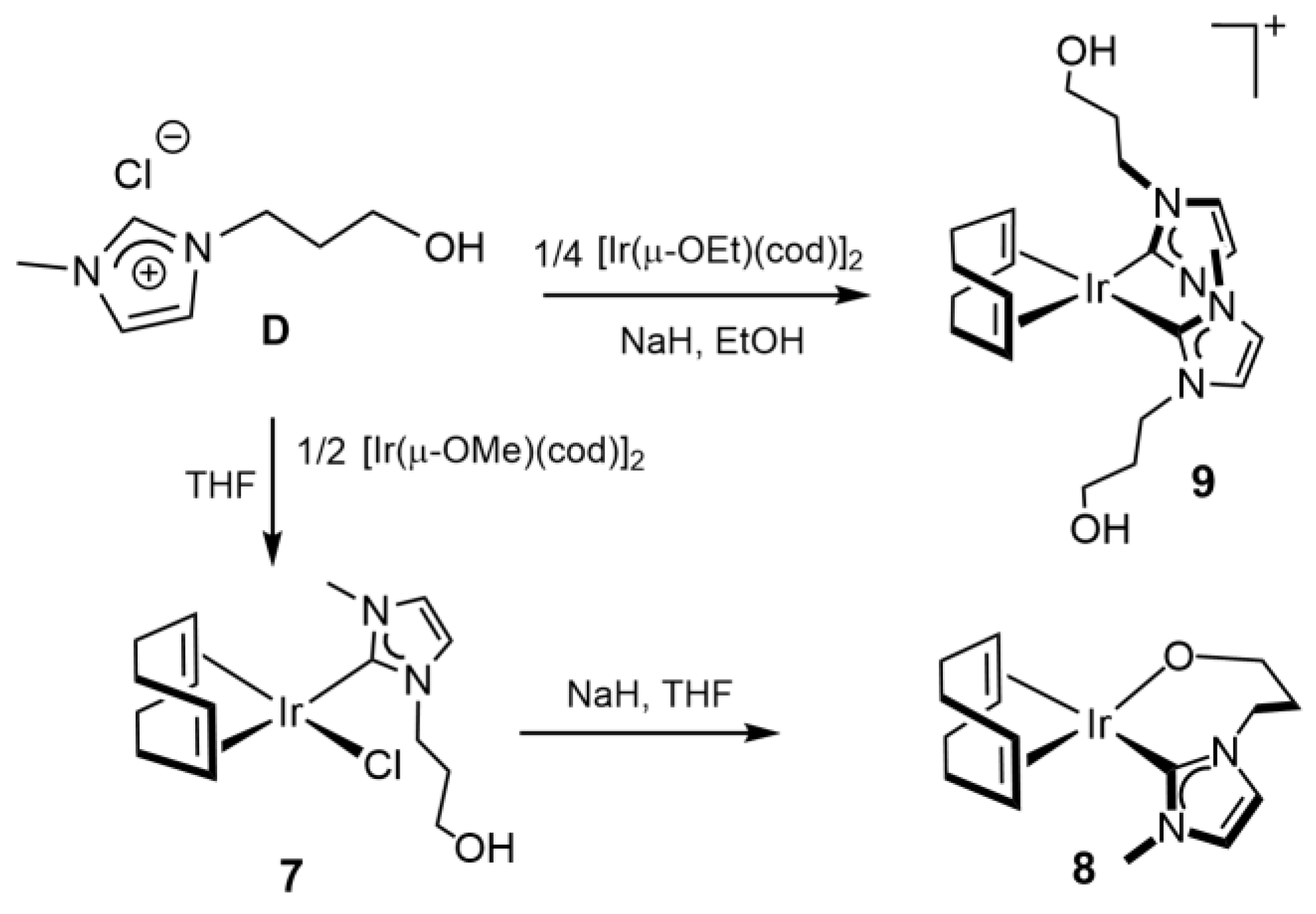
3.3.6. Synthesis of [IrCl(cod){MeIm(CH2)3OH}] (7), Figure 10

3.3.7. Synthesis of [Ir(cod){κ2C,O-{MeIm(CH2)3O}] (8), Figure 11

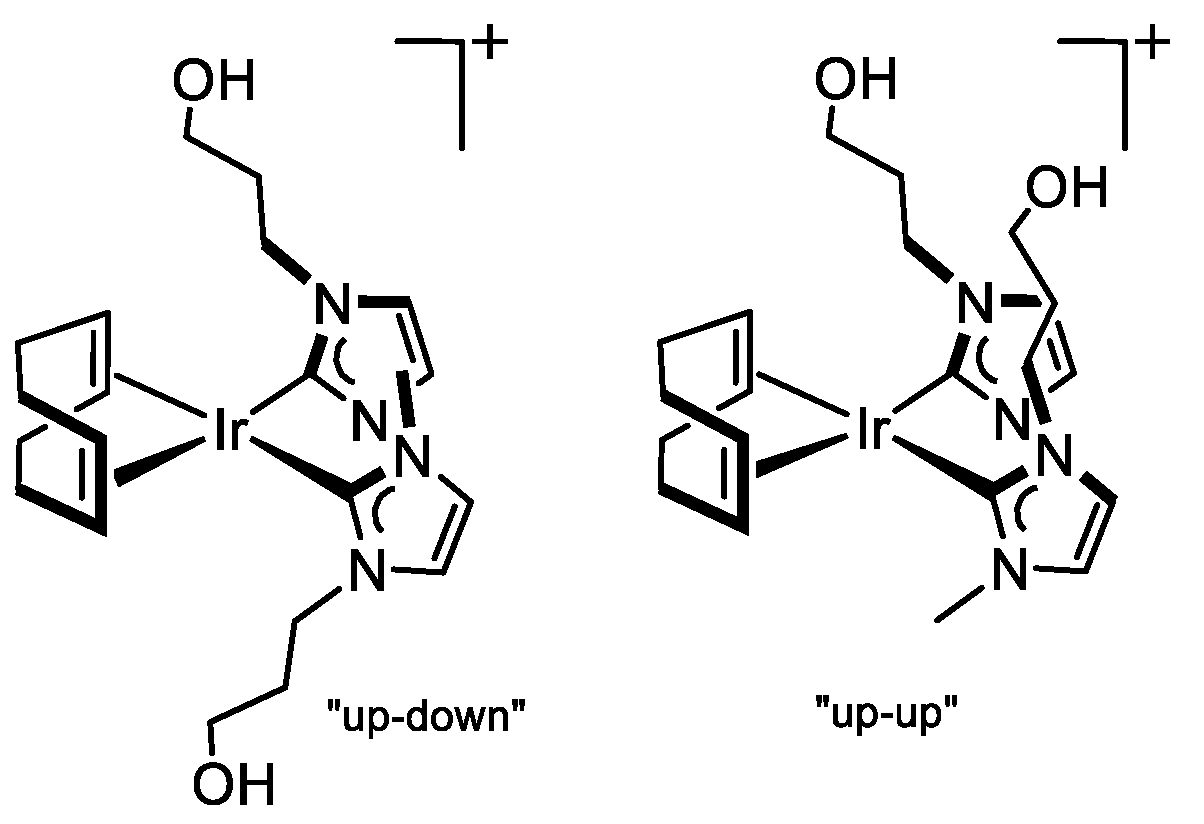
3.3.8. Synthesis of [Ir(cod){MeIm(CH2)3OH}2]Cl (9), Figure 12

3.4. General Procedure for the Acceptorless Dehydrogenation of Glycerol
3.5. Crystal Structure Determination
3.5.1. Crystal Data and Structure Refinement for 2
3.5.2. Crystal Data and Structure Refinement for 4
3.5.3. Crystal Data and Structure Refinement for 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takkellapati, S.; Li, T.; González, M.A. An Overview of Biorefinery Derived Platform Chemicals from a Cellulose and Hemicellulose Biorefinery. Clean Technol Env. Policy 2018, 20, 1615–1630. [Google Scholar] [CrossRef]
- Chilakamarry, C.R.; Sakinah, A.M.M.; Zularisam, A.W.; Pandey, A. Glycerol waste to value added products and its potential applications. Syst. Microbiol. Biomanuf. 2021, 1, 378–396. [Google Scholar] [CrossRef]
- Dusselier, M.; Van Wouwe, P.; Dewaele, A.; Makshina, E.; Sels, B.F. Lactic acid as a platform chemical in the biobased economy: The role of chemocatalysis. Energy Environ. Sci. 2014, 6, 1415–1442. [Google Scholar] [CrossRef]
- Morales, M.; Dapsens, P.Y.; Giovinazzo, I.; Witte, J.; Mondelli, C.; Papadokonstantakis, S.; Hungerbuhler, K.; Perez-Ramirez, J. Environmental and economic assessment of lactic acid production from glycerol using cascade bio- and chemocatalysis. Energy Environ. Sci. 2015, 8, 558–567. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Yang, Z.; Ma, Y.; Zhao, W.; Sun, J.; Lu, T.; He, H. Recent advances in the utilization of glycerol for the production of lactic acid by catalysis. Biofuels Bioprod. Bioref. 2022, 16, 1428–1454. [Google Scholar] [CrossRef]
- Montassier, C.; Menezo, J.C.; Hoang, L.C.; Renaud, C.; Barbier, J. Aqueous polyol conversions on ruthenium and on sulfur-modified ruthenium. J. Mol. Catal. 1991, 70, 99–110. [Google Scholar] [CrossRef]
- Maris, E.P.; Davis, R.J. Hydrogenolysis of glycerol over carbon-supported Ru and Pt catalysts. J. Catal. 2007, 249, 328–337. [Google Scholar] [CrossRef]
- Roy, D.; Subramaniam, B.; Chaudhari, R.V. Kinetic Modeling of Aqueous-Phase Glycerol Hydrogenolysis in a Batch Slurry Reactor. ACS Catal. 2011, 1, 548–551. [Google Scholar] [CrossRef]
- Akbulut, D.; Ozkar, S. A review of the catalytic conversion of glycerol to lactic acid in the presence of aqueous base. RSC Adv. 2022, 12, 18864–18883. [Google Scholar] [CrossRef]
- Kumar, A.; Gao, G. Homogeneous (De)hydrogenative Catalysis for Circular Chemistry—Using Waste as a Resource. ChemCatChem 2021, 13, 1105–1134. [Google Scholar] [CrossRef]
- Lu, Z.; Cherepakhin, V.; Demianets, I.; Lauridsen, P.J.; Williams, T.J. Iridium-based hydride transfer catalysts: From hydrogen storage to fine chemicals. Chem. Commun. 2018, 54, 7711–7724. [Google Scholar] [CrossRef] [PubMed]
- Bielinski, E.A.; Lagaditis, P.O.; Zhang, Y.; Mercado, B.Q.; Würtele, C.; Bernskoetter, W.H.; Hazari, N.; Schneider, S. Lewis Acid-Assisted Formic Acid Dehydrogenation Using a Pincer-Supported Iron Catalyst. J. Am. Chem. Soc. 2014, 136, 10234–10237. [Google Scholar] [CrossRef]
- Chakraborty, S.; Lagaditis, P.O.; Förster, M.; Bielinski, E.A.; Hazari, N.; Holthausen, M.C.; Jones, W.D.; Schneider, S. Well-Defined Iron Catalysts for the Acceptorless Reversible Dehydrogenation-Hydrogenation of Alcohols and Ketones. ACS Catal. 2014, 4, 3994–4003. [Google Scholar] [CrossRef]
- Bielinski, E.A.; Förster, M.; Zhang, Y.; Bernskoetter, W.H.; Hazari, N.; Holthausen, M.C. Base-Free Methanol Dehydrogenation Using a Pincer-Supported Iron Compound and Lewis Acid Co-catalyst. ACS Catal. 2015, 5, 2404–2415. [Google Scholar] [CrossRef]
- Alberico, E.; Sponholz, P.; Cordes, C.; Nielsen, M.; Drexler, H.J.; Baumann, W.; Junge, H.; Beller, M. Selective Hydrogen Production from Methanol with a Defined Iron Pincer Catalyst under Mild Conditions. Angew. Chem. Int. Ed. 2013, 52, 14162–14166. [Google Scholar] [CrossRef]
- Chakraborty, S.; Brennessel, W.W.; Jones, W.D.A. Molecular Iron Catalyst for the Acceptorless Dehydrogenation and Hydrogenation of N-Heterocycles. J. Am. Chem. Soc. 2014, 136, 8564–8567. [Google Scholar] [CrossRef]
- Sharninghausen, L.S.; Mercado, B.Q.; Crabtree, R.H.; Hazari, N. Selective conversion of glycerol to lactic acid with iron pincer precatalysts. Chem. Commun. 2015, 51, 16201–16204. [Google Scholar] [CrossRef]
- Li, Y.; Nielsen, M.; Li, B.; Dixneuf, P.H.; Junge, H.; Beller, M. Ruthenium-catalyzed hydrogen generation from glycerol and selective synthesis of lactic acid. Green Chem. 2015, 17, 193198. [Google Scholar] [CrossRef]
- Kirchhecker, S.; Spiegelberg, B.; de Vries, J.G. Homogenous Iridium Catalysts for Biomass Conversion. In Iridium Catalysts for Organic Reactions; Oro, L.A., Claver, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2020; Volume 69, pp. 341–395. [Google Scholar]
- Sharninghausen, L.S.; Campos, J.; Manas, M.G.; Crabtree, R.H. Efficient selective and atom economic catalytic conversion of glycerol to lactic acid. Nat. Commun. 2014, 5, 5084. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Demianets, I.; Hamze, R.; Terrile, N.J.; Williams, T.J. A Prolific Catalyst for Selective Conversion of Neat Glycerol to Lactic Acid. ACS Catal. 2016, 6, 2014–2017. [Google Scholar] [CrossRef]
- Finn, M.; Ridenour, J.A.; Heltzel, J.; Cahill, C.; Voutchkova-Kostal, A. Next-Generation Water-Soluble Homogeneous Catalysts for Conversion of Glycerol to Lactic Acid. Organometallics 2018, 37, 1400–1409. [Google Scholar] [CrossRef]
- Puerta-Oteo, R.; Ojeda-Amador, A.I.; Jiménez, M.V.; Pérez-Torrente, J.J. Catalytic applications of zwitterionic transition metal compounds. Dalton Trans. 2022, 51, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Poyatos, M.; Mata, J.A.; Peris, E. Complexes with Poly(N-heterocyclic carbene) Ligands: Structural Features and Catalytic Applications. Chem. Rev. 2009, 109, 3677–3707. [Google Scholar] [CrossRef]
- Cheong, Y.-J.; Sung, K.; Kim, J.-A.; Kim, Y.K.; Jang, H.-Y. Highly Efficient Iridium-Catalyzed Production of Hydrogen and Lactate from Glycerol: Rapid Hydrogen Evolution by Bimetallic Iridium Catalysts. Eur. J. Inorg. Chem. 2020, 4064–4068. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, Y.; Chen, J.; Huang, C.; Tu, T. Robust Iridium Coordination Polymers: Highly Selective, Efficient, and Recyclable Catalysts for Oxidative Conversion of Glycerol to Potassium Lactate with Dihydrogen Liberation. ACS Catal. 2015, 5, 6573–6578. [Google Scholar] [CrossRef]
- Puerta-Oteo, R.; Jiménez, M.V.; Lahoz, F.J.; Modrego, F.J.; Passarelli, V.; Pérez-Torrente, J.J. Zwitterionic rhodium and iridium complexes based on a carboxylate bridge-functionalized bis-N-heterocyclic carbene ligand: Synthesis, structure, dynamic behavior, and reactivity. Inorg. Chem. 2018, 57, 5526–5543. [Google Scholar] [CrossRef] [Green Version]
- Puerta-Oteo, R.; Munarriz, J.; Polo, V.; Jiménez, M.V.; Pérez-Torrente, J.J. Carboxylate-Assisted β-(Z) Stereoselective Hydrosilylation of Terminal Alkynes Catalyzed by a Zwitterionic Bis-NHC Rhodium(III) Complex. ACS Catal. 2020, 10, 7367–7380. [Google Scholar] [CrossRef]
- Ojeda-Amador, A.I.; Munarriz, J.; Alamán-Valtierra, P.; Polo, V.; Puerta-Oteo, R.; Jiménez, M.V.; Fernández-Alvarez, F.J.; Pérez-Torrente, J.J. Mechanistic Insights on the Functionalization of CO2 with Amines and Hydrosilanes Catalyzed by a Zwitterionic Iridium Carboxylate-Functionalized Bis-NHC Catalyst. ChemCatChem 2019, 11, 5524–5535. [Google Scholar] [CrossRef]
- Puerta-Oteo, R.; Hölscher, M.; Jiménez, M.V.; Leitner, W.; Passarelli, V.; Pérez-Torrente, J.J. Experimental and Theoretical Mechanistic Investigation on the Catalytic CO2 Hydrogenation to Formate by a Carboxylate-Functionalized Bis(N-heterocyclic carbene) Zwitterionic Iridium(I) Compound. Organometallics 2018, 37, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Puerta-Oteo, R.; Jiménez, M.V.; Pérez-Torrente, J.J. Molecular water oxidation catalysis by zwitterionic carboxylate bridge-functionalized bis-NHC iridium complexes. Catal. Sci. Technol. 2018, 9, 1437–1450. [Google Scholar] [CrossRef]
- Desai, S.P.; Mondal, M.; Choudhury, J. Chelating Bis-N-heterocyclic Carbene–Palladium(II) Complexes for Oxidative Arene C–H Functionalization. Organometallics 2015, 34, 2731–2736. [Google Scholar] [CrossRef]
- Zhong, R.; Pöthig, A.; Mayer, D.C.; Jandl, C.; Altmann, P.J.; Herrmann, W.A.; Kühn, F.E. Spectroscopic and Structural Properties of Bridge-Functionalized Dinuclear Coinage-Metal (Cu, Ag, and Au) NHC Complexes: A Comparative Study. Organometallics 2015, 34, 2573–2579. [Google Scholar] [CrossRef]
- Zhong, R.; Pöthig, A.; Haslinger, S.; Hofmann, B.; Raudaschl-Sieber, G.; Herdtweck, E.; Herrmann, W.A.; Kühn, F.E. Toward Tunable Immobilized Molecular Catalysts: Functionalizing the Methylene Bridge of Bis(N-heterocyclic carbene) Ligands. ChemPlusChem 2014, 79, 1294–1303. [Google Scholar] [CrossRef]
- Quezada, C.; Garrison, J.C.; Tessier, C.A.; Youngs, W.J. Synthesis and structural characterization of two bis(imidazol-2-ylidene) complexes of Pt(II). J. Organomet. Chem. 2003, 671, 183–186. [Google Scholar] [CrossRef]
- Bekhouche, M.; Blum, L.J.; Doumèche, B. Ionic liquid-inspired cations covalently bound to FDH improve its stability and activity in IL. ChemCatChem 2011, 3, 875–882. [Google Scholar] [CrossRef]
- Blanco, M.; Alvarez, P.; Blanco, C.; Jiménez, M.V.; Fernández-Tornos, J.; Pérez-Torrente, J.J.; Oro, L.A.; Menendez, R. Enhanced hydrogen-transfer catalytic activity of iridium N-heterocyclic carbenes by covalent attachment on carbon nanotubes. ACS Catalysis 2013, 3, 1307–1317. [Google Scholar] [CrossRef] [Green Version]
- Couzijn, E.P.A.; Slootweg, J.C.; Ehlers, A.W.; Lammertsma, K. Stereomutation of Pentavalent Compounds: Validating the Berry Pseudorotation, Redressing Ugi’s Turnstile Rotation, and Revealing the Two- and Three-Arm Turnstiles. J. Am. Chem. Soc. 2010, 132, 18127–18140. [Google Scholar] [CrossRef]
- Straubinger, C.S.; Jokić, N.B.; Högerl, M.P.; Herdtweck, E.; Herrmann, W.A.; Kühn, F.E. Bridge functionalized bis-N-heterocyclic carbene rhodium(I) complexes and their application in catalytic hydrosilylation. J. Organomet. Chem. 2011, 696, 687–692. [Google Scholar] [CrossRef]
- Jiménez, M.V.; Fernández-Tornos, J.; Pérez-Torrente, J.J.; Modrego, F.J.; García-Orduña, P.; Oro, L.A. Mechanistic Insights into Transfer Hydrogenation Catalysis by [Ir(cod)(NHC)2]+ Complexes with Functionalized N-Heterocyclic Carbene Ligands. Organometallics 2015, 34, 926–940. [Google Scholar] [CrossRef] [Green Version]
- Brookhart, M.; Green, M.L.H.; Parkin, G. Agostic interactions in transition metal compounds. Proc. Natl. Acad. Sci. USA 2007, 104, 6908–6914. [Google Scholar] [CrossRef] [PubMed]
- Frey, G.D.; Rentzsch, C.F.; von Preysing, D.; Scherg, T.; Mühlhofer, M.; Herdtweck, E.; Herrmann, W.A. Rhodium and iridium complexes of N-heterocyclic carbenes: Structural investigations and their catalytic properties in the borylation reaction. J. Organomet. Chem. 2006, 691, 5725–5738. [Google Scholar] [CrossRef]
- Rentzsch, C.F.; Tosh, E.; Herrmann, W.A.; Kühn, F.E. Iridium complexes of N-heterocyclic carbenes in C–H borylation using energy efficient microwave technology: Influence of structure, ligand donor strength and counter ion on catalytic activity. Green Chem. 2009, 11, 1610–1617. [Google Scholar] [CrossRef]
- Jiménez, M.V.; Pérez-Torrente, J.J.; Bartolomé, M.I.; Gierz, V.; Lahoz, F.J.; Oro, L.A. Rhodium (I) complexes with hemilabile N-heterocyclic carbenes: Efficient alkyne hydrosilylation catalysts. Organometallics 2008, 27, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Herde, J.L.; Lambert, J.C.; Senoff, C.V.; Cushing, M.A. Cyclooctene and 1,5-Cyclooctadiene Complexes of Iridium(I). Inorg. Synth. 2007, 15, 18–20. [Google Scholar]
- Usón, R.; Oro, L.A.; Cabeza, J.A. Dinuclear methoxy, cyclooctadiene, and barrelene complexes of rhodium(I) and iridium(I). Inorg. Synth. 1985, 23, 126–127. [Google Scholar]
- Ulmer, L.; Mattay, J.; Torres-García, H.G.; Luftmann, H. The Use of 2-[(2E)-3-(4-Tert-Butylphenyl)-2-Methylprop-2-Enylidene]Malononitrile as a Matrix for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Eur. J. Mass Spectrom. 2000, 6, 49–52. [Google Scholar] [CrossRef]
- Blanco, M.; Álvarez, P.; Blanco, C.; Jiménez, M.V.; Fernández-Tornos, J.; Pérez-Torrente, J.J.; Blasco, J.; Subías, G.; Cuartero, V.; Oro, L.A.; et al. Effect of structural differences of carbon nanotubes and graphene based iridium-NHC materials on the hydrogen transfer catalytic activity. Carbon 2016, 96, 66–74. [Google Scholar] [CrossRef] [Green Version]
- SAINT+: Area-Detector Integration Software, version 6.01; Bruker AXS: Madison, WI, USA, 2001.
- Sheldrick, G.M. SADABS Program; University of Göttingen: Göttingen, Germany, 1999. [Google Scholar]
- Sheldrick, G.M. SHELXS 97, Program for the Solution of Crystal Structure; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Azpíroz, R.; Rubio-Pérez, L.; Di Giuseppe, A.; Passarelli, V.; Lahoz, F.J.; Castarlenas, R.; Pérez-Torrente, J.J.; Oro, L.A. Rhodium(I)-N-Heterocyclic Carbene Catalyst for Selective Coupling of N-Vinylpyrazoles with Alkynes via C−H Activation. ACS Catal. 2014, 4, 4244–4253. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2 | 4 | 5 | |
|---|---|---|---|
| Ir–C(1) | 2.023(6) | 2.025(13) | 2.056(14) |
| C(9)–Ir(1) | 2.089(6) | - | - |
| C(25)–Ir(1) | 1.929(6) | - | - |
| C(25)–O(26) | 1.139(7) | - | - |
| Ir–C(11) | - | 2.011(12) | 2.083(13) |
| Ir–C(17) | - | - | 1.876(14) |
| Ir–C(19) | - | - | 1.883(15) |
| Ir1–CT01 | 2.1679(2) | 2.0378(5) | - |
| Ir1–CT02 | 2.0183(2) | 2.0708(5) | - |
| C(17)–C(18) | 1.373(8) | 1.352(19) | - |
| C(21)–C(22) | 1.446(8) | 1.391(18) | - |
| C(14)–O(16) | 1.239(7) | - | - |
| C(14)–O(15) | 1.245(7) | - | - |
| C(17)–O(18) | - | - | 1.131(16) |
| C(19)–O(20) | - | - | 1.151(17) |
| C(1)–Ir(1)–C(11) | - | 84.1(5) | 80.4(5) |
| C(1)–Ir(1)–C(9) | 83.6(2) | - | - |
| CT01–Ir1–CT02 | 83.559(8) | 86.476(18) | - |
| C1–Ir1–CT01 | 171.44(15) | - | - |
| C25–Ir1–CT02 | 128.21(17) | - | - |
| CT02–Ir1–C9 | 129.68(15) | - | - |
| C(25)–Ir(1)–C(9) | 102.0(2) | - | - |
| C(17)–Ir(1)–C(19) | - | - | 93.6(6) |
| C(17)–Ir(1)–C(1) | - | - | 171.1(5) |
| C(19)–Ir(1)–C(11) | - | - | 174.0(6) |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Base (mmol) | t [h] | T [°C] | Conv. [%] b | Select. LA(%) b | TONLA c |
| 1 | 1 (1) | - | 66 | 115 | 0 | 0 | 0 |
| 2 | 4 (1) | - | 66 | 115 | 0 | 0 | 0 |
| 3 | - | KOH (5) | 66 | 115 | 0 | 0 | 0 |
| 4 | 3 (1) d | KOH (5) | 36 | 115 | 67 | 59 | 198 |
| 5 | 5 (1) d | KOH (5) | 36 | 115 | 69 | 76 | 262 |
| 6 | 1 (0.2) | KOH (5) | 2 | 130 | 24 | 93 | 112 |
| 7 | 3 (0.2) | KOH (5) | 2 | 130 | 30 | 98 | 147 |
| 8 | 4 (0.2) | KOH (5) | 2 | 130 | 61 | 93 | 284 |
| 9 | 5 (0.2) | KOH (5) | 2 | 130 | 91 | 92 | 419 |
| 10 | 6 (0.2) | KOH (5) | 2 | 130 | 15 | 100 | 75 |
| 11 | 7 (0.2) | KOH (5) | 2 | 130 | 80 | 86 | 344 |
| 12 | 8 (0.2) | KOH (5) | 2 | 130 | 35 | 98 | 172 |
| 13 | 9 (0.2) | KOH (5) | 2 | 130 | 56 | 92 | 258 |
| 14 | 7 e | NaOH (5) | 6 | 130 | 78 | 80 | 312 |
| 15 | 7 e | Cs2CO3 (5) | 25 | 130 | 23 | 84 | 97 |
| 16 | 1 (0.2) | KOH (5) | 0,75 | 150 | 31 | 93 | 144 |
| 17 | 3 (0.2) | KOH (5) | 0,75 | 150 | 40 | 94 | 188 |
| 18 | 4 (0.2) | KOH (5) | 0,75 | 150 | 71 | 92 | 327 |
| 19 | 5 (0.2) | KOH (5) | 0,75 | 150 | 100 | 93 | 465 |
| 20 | 7 (0.2) | KOH (5) | 0,75 | 150 | 70 | 92 | 322 |
| 21 | 5 (0.2) | KOH (0.5) | 0,75 | 150 | 15 | 59 | 44 |
| 22 | 5 (0.2) | KOH (2.5) | 0,75 | 150 | 80 | 57 | 228 |
| 23 | 5 (0.2) | KOH (6) | 0,75 | 150 | 91 | 91 | 414 |
| 24 | 5 (0.07) | KOH (5) | 24 | 150 | 91 | 71 | 923 |
| 25 | 5 (0.07) d | KOH (5) | 24 | 150 | 68 | 100 | 971 |
| 26 | 5 (0.07) | KOH (5) | 72 | 150 | 100 | 70 | 1000 |
| 27 | 5 (0.007) | KOH (5) | 72 | 150 | 74 | 100 | 10,571 |
| 28 | 5 (0.0014) | KOH (5) | 72 | 150 | 21 | 100 | 15,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez, M.V.; Ojeda-Amador, A.I.; Puerta-Oteo, R.; Martínez-Sal, J.; Passarelli, V.; Pérez-Torrente, J.J. Selective Oxidation of Glycerol via Acceptorless Dehydrogenation Driven by Ir(I)-NHC Catalysts. Molecules 2022, 27, 7666. https://doi.org/10.3390/molecules27227666
Jiménez MV, Ojeda-Amador AI, Puerta-Oteo R, Martínez-Sal J, Passarelli V, Pérez-Torrente JJ. Selective Oxidation of Glycerol via Acceptorless Dehydrogenation Driven by Ir(I)-NHC Catalysts. Molecules. 2022; 27(22):7666. https://doi.org/10.3390/molecules27227666
Chicago/Turabian StyleJiménez, M. Victoria, Ana I. Ojeda-Amador, Raquel Puerta-Oteo, Joaquín Martínez-Sal, Vincenzo Passarelli, and Jesús J. Pérez-Torrente. 2022. "Selective Oxidation of Glycerol via Acceptorless Dehydrogenation Driven by Ir(I)-NHC Catalysts" Molecules 27, no. 22: 7666. https://doi.org/10.3390/molecules27227666
APA StyleJiménez, M. V., Ojeda-Amador, A. I., Puerta-Oteo, R., Martínez-Sal, J., Passarelli, V., & Pérez-Torrente, J. J. (2022). Selective Oxidation of Glycerol via Acceptorless Dehydrogenation Driven by Ir(I)-NHC Catalysts. Molecules, 27(22), 7666. https://doi.org/10.3390/molecules27227666