


Environmentally Sustainable Achiral and Chiral Chromatographic Analysis of Amino Acids in Food Supplements
 , ,
, ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
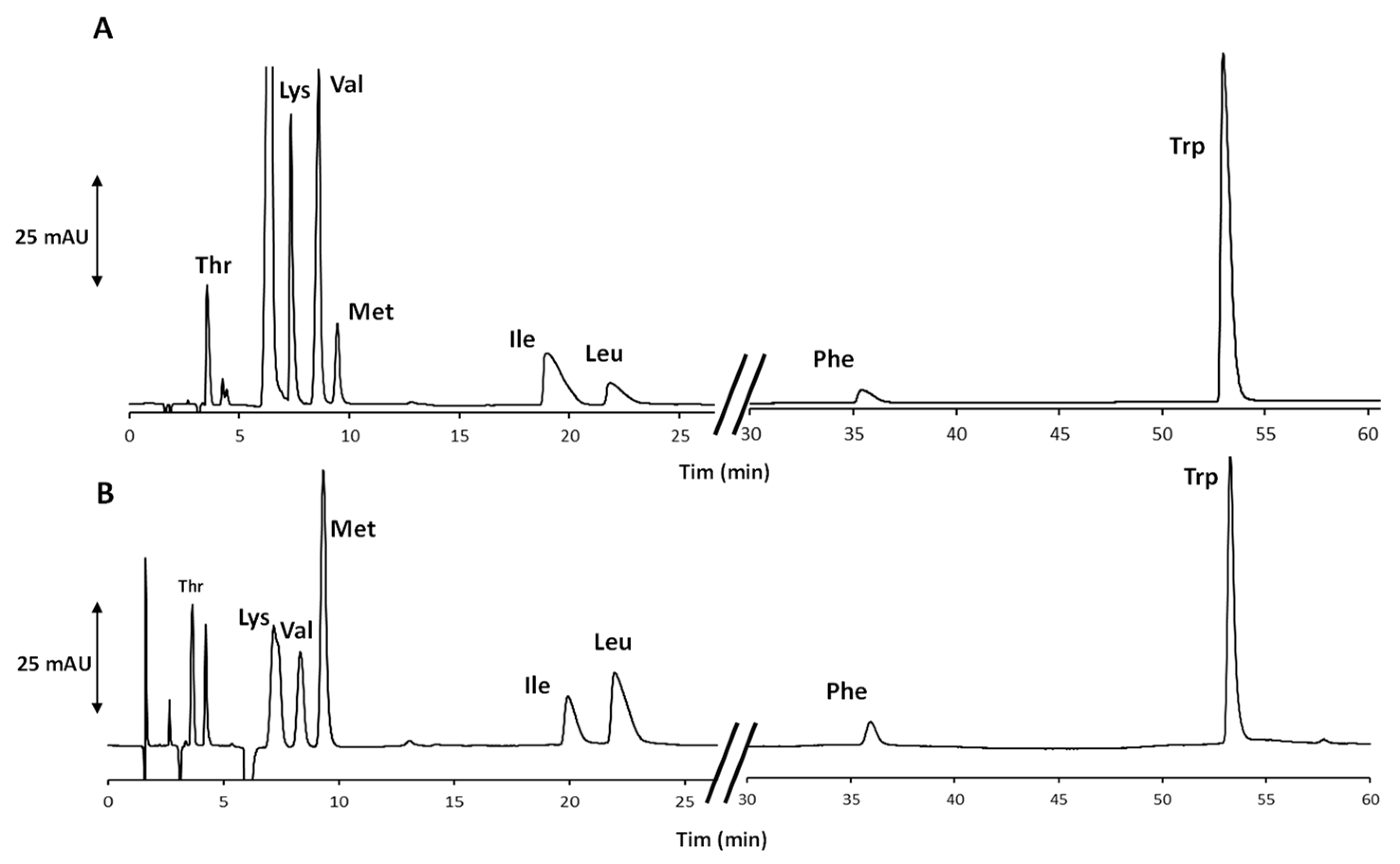
2.1. Development of the Achiral IP-RP-HPLC Method
2.2. Validation of the Optimized Achiral IP-RP-HPLC Method and Quantitative Analysis of the AAs in the Food Supplement
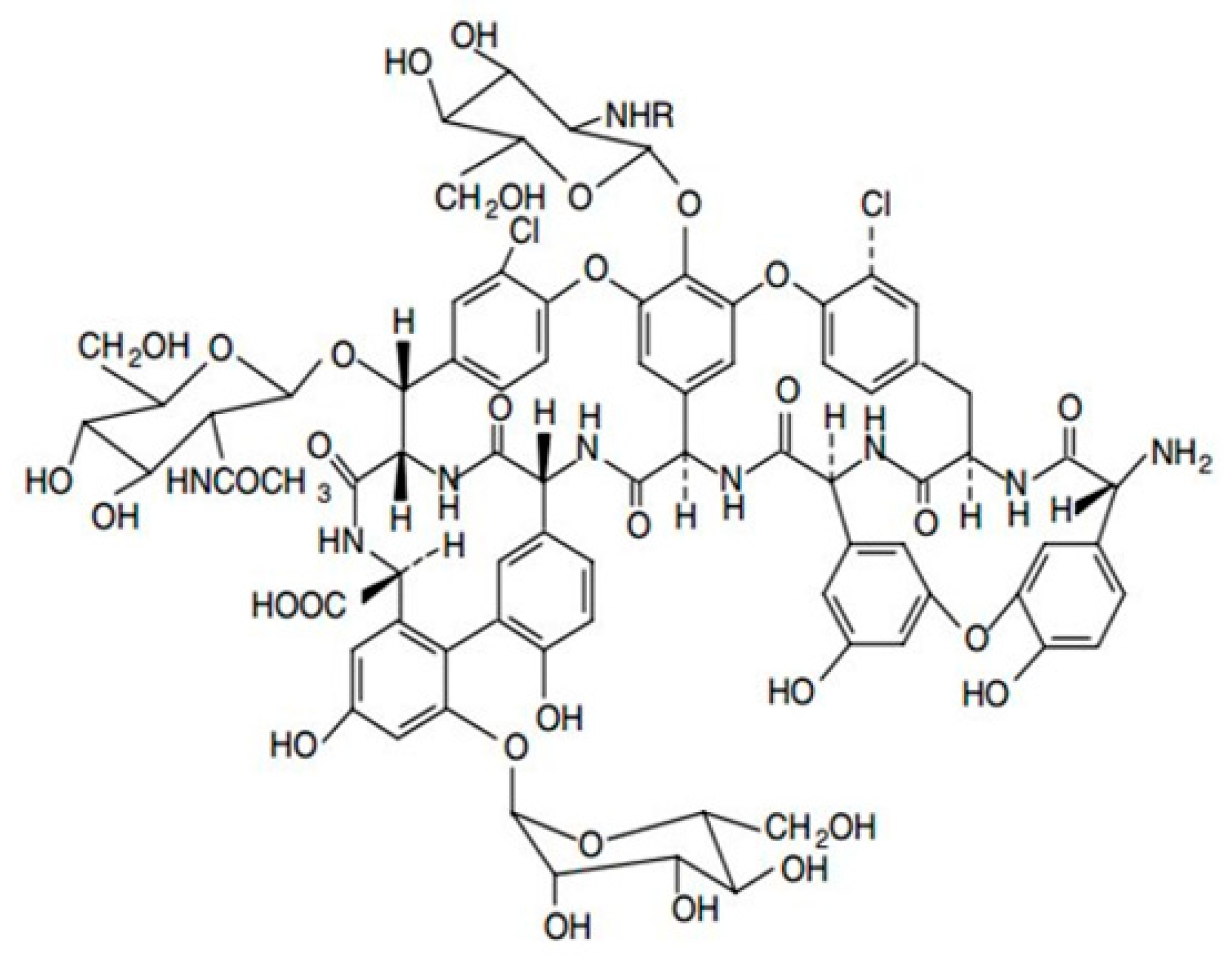
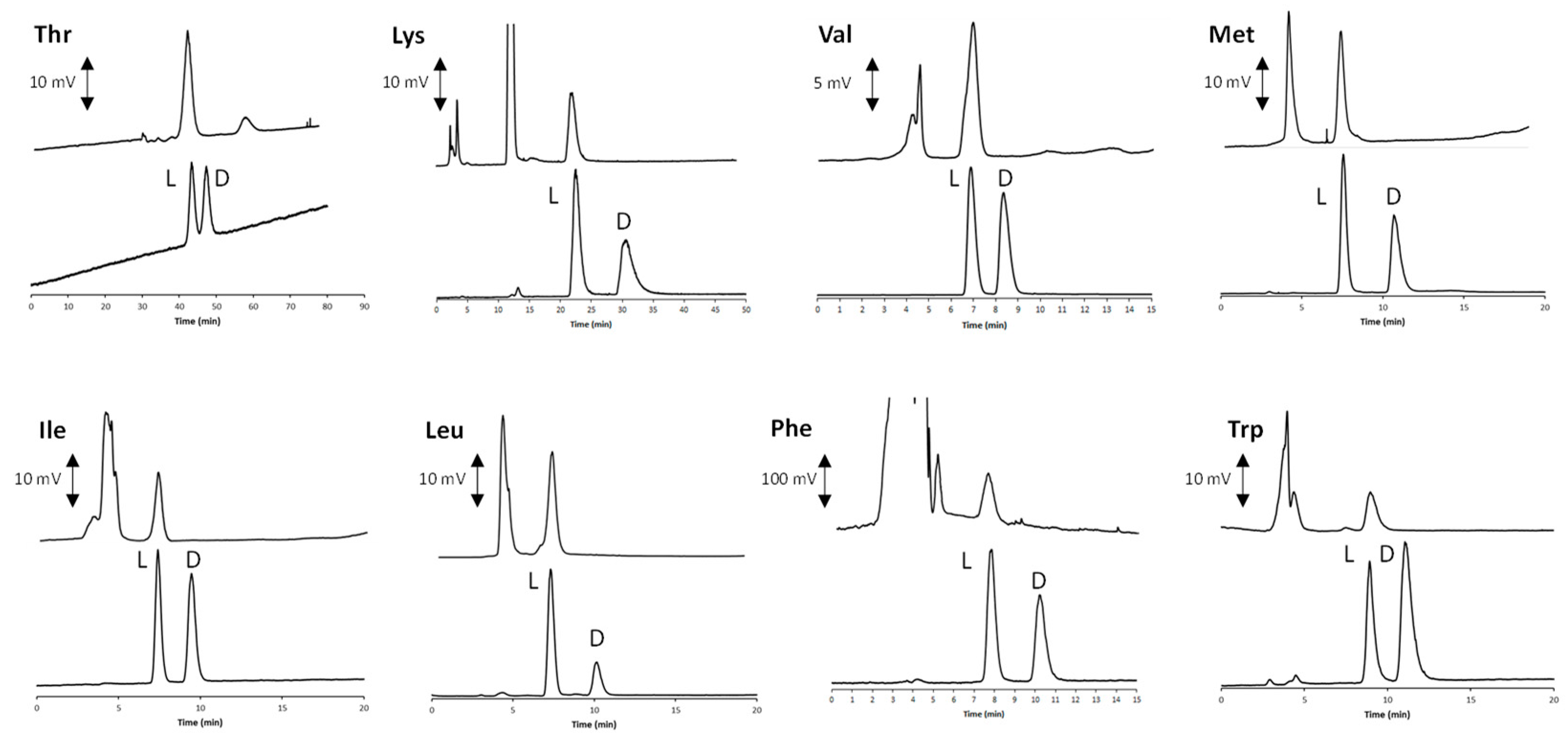
2.3. Development of the Enantioselective Liquid-Chromatographic Method and Application to the Real Sample
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Extraction of the AA Pool from the Food Supplement
3.3. Calibration Curves for Quantitative Analysis
3.4. Achiral and Chiral HPLC Analysis
3.5. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wu, G. Functional amino acids in nutrition and health. Amino Acids 2013, 45, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerksick, C.M.; Arent, S.; Schoenfeld, B.J.; Stout, J.R.; Campbell, B.; Wilborn, C.D.; Taylor, L.; Kalman, D.; Smith-Ryan, A.E.; Kreider, R.B.; et al. International society of sports nutrition position stand: Nutrient timing. J. Int. Soc. Sports Nutr. 2017, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Beaudart, C.; Dawson, A.; Shaw, S.C.; Harvey, N.C.; Kanis, J.A.; Binkley, N.J.; Reginster, Y.; Chapurlat, R.; Chan, D.C.; Bruyère, O.; et al. Nutrition and physical activity in the prevention and treatment of sarcopenia: Systematic review. Osteoporos. Int. 2017, 28, 1817–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronis, M.J.J.; Pedersen, K.B.; Watt, J. Adverse Effects of Nutraceuticals and Dietary Supplements. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 583–601. [Google Scholar] [CrossRef]
- Wierzejska, R.E. Dietary Supplements-For Whom? The Current State of Knowledge about the Health Effects of Selected Supplement Use. Int. J. Environ. Res. Public Health 2021, 18, 8897. [Google Scholar] [CrossRef]
- Xue, A.S.; Dayan, E.; Rohrich, R.J. Dietary Supplements and Current Available Evidence. Plast. Reconstr. Surg. 2020, 146, 474e–481e. [Google Scholar] [CrossRef]
- Fibigr, J.; Šatínský, D.; Solich, P. Current trends in the analysis and quality control of food supplements based on plant extracts. Anal. Chim. Acta 2018, 1036, 1–15. [Google Scholar] [CrossRef]
- Bastings, J.J.A.J.; van Eijk, H.M.; Olde Damink, S.W.; Rensen, S.S. 1D-Amino Acids in Health and Disease: A Focus on Cancer. Nutrients 2019, 11, 2205. [Google Scholar] [CrossRef] [Green Version]
- Friedman, M.; Levin, C.E. Nutritional and medicinal aspects of D-amino acids. Amino Acids 2012, 42, 1553–1582. [Google Scholar] [CrossRef]
- Grishin, D.V.; Zhdanov, D.D.; Pokrovskaya, M.V.; Sokolov, N.N. D-amino acids in nature, agriculture and biomedicine. All Life 2020, 13, 11–22. [Google Scholar] [CrossRef]
- Genchi, G. An overview on D-amino acids. Amino Acids 2017, 49, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Olives, A.I.; González-Ruiz, V.; Martín, M.A. Sustainable and Eco-Friendly Alternatives for Liquid Chromatographic Analysis. ACS Sustain. Chem. Eng. 2017, 5, 5618–5634. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Kloskowskic, A.; Namieśnik, J. Perspectives on the replacement of harmful organic solvents in analytical methodologies: A framework toward the implementation of a generation of eco-friendly alternatives. Green Chem. 2015, 17, 3687–3705. [Google Scholar] [CrossRef]
- Tobiszewski, M.; Marć, M.; Gałuszka, A.; Namieśnik, J. Green Chemistry Metrics with Special Reference to Green Analytical Chemistry. Molecules 2015, 20, 10928–10946. [Google Scholar] [CrossRef]
- Capello, C.; Fischer, U.; Hungerbuhler, K. What is a green solvent? A comprehensive framework for the environmental assessment of solvents. Green Chem. 2007, 9, 927–934. [Google Scholar] [CrossRef]
- Sajida, M.; Płotka-Wasylka, J. Green analytical chemistry metrics: A review. Talanta 2022, 238, 123046. [Google Scholar] [CrossRef]
- Płotka, J.; Tobiszewski, M.; Sulej, A.M.; Kupska, M.; Górecki, T.; Namieśnika, J. Green chromatography. J. Chromatogr. A 2013, 1307, 1–20. [Google Scholar] [CrossRef]
- Petritis, K.; Chaimbault, P.; Elfakir, C.; Dreux, M. Ion-pair reversed-phase liquid chromatography for determination of polar underivatized amino acids using perfluorinated carboxylic acids as ion pairing agent. J. Chromatogr. A 1999, 833, 147–155. [Google Scholar] [CrossRef]
- Petritis, K.; de Person, M.; Elfakir, C.; Dreux, M. Validation of an Ion-Interaction Chromatography Analysis of Underivatized Amino Acids in Commercial Preparation Using Evaporative Light Scattering Detection. Chromatographia 2004, 60, 293–298. [Google Scholar] [CrossRef]
- Ianni, F.; Lisanti, A.; Marinozzi, M.; Camaioni, E.; Pucciarini, L.; Massoli, A.; Sardella, R.; Concezzi, L.; Natalini, B. Hydrophobic Amino Acid Content in Onions as Potential Fingerprints of Geographical Origin: The Case of Rossa da Inverno sel. Rojo Duro. Molecules 2018, 23, 1259. [Google Scholar] [CrossRef]
- Ianni, F.; Sechi, P.; La Mantia, A.; Pucciarini, L.; Camaioni, E.; Cenci Goga, B.T.; Sardella, R.; Natalini, B. The Relationships between Somatic Cells and Isoleucine, Leucine and Tyrosine Content in Cow Milk. Appl. Sci. 2019, 9, 349. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.D. Research and Evaluation of Organic Hazardous Air Pollutant Source Emission Test Methods. J. Air Waste Manag. Ass. 1996, 46, 1135–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilisz, I.; Berkecz, R.; Péter, A. HPLC separation of amino acid enantiomers and small peptides on macrocyclic antibiotic-based chiral stationary phases: A review. J. Sep. Sci 2006, 29, 1305–1321. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, F.; Jankowski, V.; Günthner, T.; Salem, S.; Nierhaus, M.; Schulz, A.; Zidek, W.; Jankowski, J. Replacement of acetonitrile by ethanol as solvent in reversed phase chromatography of biomolecules. J. Chromatogr. B 2010, 878, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Sardella, R.; Lisanti, A.; Marinozzi, M.; Ianni, F.; Natalini, B.; Blanch, G.P.; Ruiz del Castillo, M.L. Combined monodimensional chromatographic approaches to monitor the presence of d-amino acids in cheese. Food Control 2013, 34, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Shibue, M.; Mant, C.T.; Hodges, R.S. Effect of anionic ion-pairing reagent hydrophobicity on selectivity of peptide separations by reversed-phase liquid chromatography. J. Chromatogr. A 2005, 1080, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Barra, R.; González, P. Sustainable chemistry challenges from a developing country perspective: Education, plastic pollution, and beyond. Curr. Opin. Green Sustain. Chem. 2018, 9, 40–44. [Google Scholar] [CrossRef]
- Matlin, S.A.; Mehta, G.; Hopf, H.; Krief, A.; Keßler, L.; Kümmerer, K. Material circularity and the role of the chemical sciences as a key enabler of a sustainable post-trash age. Sustain. Chem. Pharm. 2020, 17, 100312. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, A.; Wang, W.Y.; Zhao, Y.-L.; Chiang, P.-C. Implementing Green Chemistry Principles for Circular Economy Towards Sustainable Development Goals. Chem. Eng. Trans. 2021, 88, 955–960. [Google Scholar]
- Davies, R.L. D-Lysine, Alloisoleucine and Lysinoalanine in Supplementary Proteins with Different Lysine Avdlabilities. J. Sci. Food Agric. 1993, 61, 151–154. [Google Scholar] [CrossRef]
- Bellmaine, S.; Schnellbaecher, A.; Zimmer, A. Reactivity and degradation products of tryptophan in solution and proteins. Free Radic. Biol. Med. 2020, 160, 696–718. [Google Scholar] [CrossRef] [PubMed]
- Chumsae, C.; Gaza-Bulseco, G.; Sun, J.; Liu, H. Comparison of methionine oxidation in thermal stability and chemically stressed samples of a fully human monoclonal antibody. J. Chromatogr. B 2007, 850, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Ilisz, I.; Aranyi, A.; Pataj, Z.; Péter, A. Recent advances in the direct and indirect liquid chromatographic enantioseparation of amino acids and related compounds: A review. J. Pharma. Biol. Anal. 2012, 69, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Natalini, B.; Sardella, R.; Pellicciari, R. Cysteine-based chiral selectors for the ligand-exchange separation of amino acids. J. Chromatogr. B 2008, 875, 108–117. [Google Scholar] [CrossRef]
- Hoffman, C.V.; Pell, R.; Lämmerhofer, M.; Lindner, W. Synergistic Effects on Enantioselectivity of Zwitterionic Chiral Stationary Phases for Separations of Chiral Acids, Bases, and Amino Acids by HPLC. Anal. Chem. 2008, 80, 8780–8789. [Google Scholar] [CrossRef] [PubMed]
- Hyun, M.H.; Jin, J.S.; Lee, W. Liquid chromatographic resolution of racemic amino acids and their derivatives on a new chiral stationary phase based on crown ether. J. Chromatogr. A 1998, 822, 155–161. [Google Scholar] [CrossRef]
- Berthod, A.; Liu, Y.; Bagwill, C.; Armstrong, D.W. Facile liquid chromatographic enantioresolution of native amino acids and peptides using a teicoplanin chiral stationary phase. J. Chromatogr. A 1996, 731, 123–137. [Google Scholar] [CrossRef]
- Fumagalli, L.; Pucciarini, L.; Regazzoni, L.; Gilardoni, E.; Carini, M.; Vistoli, G.; Aldini, G.; Sardella, R. Direct HPLC separation of carnosine enantiomers with two chiral stationary phases based on penicillamine and teicoplanin derivatives. J. Sep. Sci. 2018, 41, 1240–1246. [Google Scholar] [CrossRef]
- Ali, I.; Suhail, M.; Asnin, L. Chiral separation and modeling of quinolones on teicoplanin macrocyclic glycopeptide antibiotics CSP. Chirality 2018, 30, 1304–13111. [Google Scholar] [CrossRef]
- Lämmerhofer, M. Liquid chromatographic enantiomer separation with special focus on zwitterionic chiral ion-exchangers. Anal. Bioanal. Chem. 2014, 406, 6095–6103. [Google Scholar] [CrossRef]
- Varfaj, I.; Protti, M.; Di Michele, A.; Macchioni, A.; Lindner, W.; Carotti, A.; Sardella, R.; Mercolini, L. Efficient enantioresolution of aromatic a-hydroxy acids with Cinchona alkaloid-based zwitterionic stationary phases and volatile polar-ionic eluents. Anal. Chim. Acta 2021, 1180, 338928. [Google Scholar] [CrossRef] [PubMed]
- Dugo, P.; Cacciola, F.; Kumm, T.; Dugo, G.; Mondello, L. Comprehensive multidimensional liquid chromatography: Theory and applications. J. Pharm. Biol. Anal. 2015, 116, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Koga, R.; Oyama, T.; Han, H.; Ueno, K.; Masuyama, K.; Itoh, Y.; Hamase, K. HPLC analysis of naturally occurring free d-amino acids in mammals. J. Pharm. Biol. Anal. 2012, 69, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Le, B.V.; Hajare, H.S.; Baxter, R.H.G.; Miller, S.J. X-ray Crystal Structure of Teicoplanin A2-2 Bound to a Catalytic Peptide Sequence via the Carrier Protein Strategy. J. Org. Chem. 2014, 79, 8550–8556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milletti, F.; Vulpetti, A. Tautomer Preference in PDB Complexes and its Impact on Structure-Based Drug Discovery. J. Chem. Inf. Model. 2010, 50, 1062–1074. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.; Russell, E.; Von Bargen, C.; Abel, R.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mobile Phase Composition | ||||||||
| a | b | c | |||||||
| k | α | Rs | k | α | Rs | k | α | Rs | |
| Thr | 0.62 | 0.51 | 0.60 | ||||||
| Lys | 1.71 | 2.75 | 6.39 | 1.21 | 2.35 | 4.61 | 2.07 | 3.46 | 8.59 |
| Val | 1.71 | 1.00 | N.R. | 1.46 | 1.21 | 1.22 | 2.38 | 1.15 | 1.50 |
| Met | 2.73 | 1.60 | 3.63 | 2.20 | 1.51 | 3.31 | 2.68 | 1.13 | 1.81 |
| Ile | 4.19 | 1.53 | 3.94 | 3.10 | 1.41 | 3.76 | 5.36 | 2.00 | 9.85 |
| Leu | 5.01 | 1.20 | 1.71 | 3.64 | 1.18 | 1.99 | 6.35 | 1.18 | 2.52 |
| Phe | 6.07 | 1.21 | 1.68 | 4.32 | 1.19 | 2.20 | 8.71 | 1.37 | 5.45 |
| Trp | 10.98 | 1.81 | 4.86 | 11.21 | 2.59 | 9.43 | 25.06 | 2.88 | 17.71 |
| Compound | Mobile Phase Composition | ||||||||
| d | e | f | |||||||
| k | α | Rs | k | α | Rs | k | α | Rs | |
| Thr | 0.48 | 0.48 | 0.44 | ||||||
| Lys | 2.07 | 4.29 | 14.66 | 2.07 | 4.29 | 14.66 | 2.01 | 4.60 | 14.95 |
| Val | 2.54 | 1.23 | 4.22 | 2.54 | 1.23 | 4.22 | 2.51 | 1.25 | 4.10 |
| Met | 2.99 | 1.18 | 3.60 | 2.99 | 1.18 | 3.60 | 2.86 | 1.14 | 2.45 |
| Ile | 7.91 | 2.64 | 22.52 | 7.91 | 2.64 | 22.52 | 6.80 | 2.38 | 9.56 |
| Leu | 8.97 | 1.13 | 3.32 | 8.97 | 1.13 | 3.32 | 8.02 | 1.18 | 2.01 |
| Phe | 15.56 | 1.77 | 18.27 | 15.56 | 1.13 | 18.27 | 13.27 | 1.65 | 10.01 |
| Trp | N.E. | 18.92 | 1.35 | 26.98 | 20.13 | 1.53 | 15.04 | ||
| Compound | Theoretical Conc. (mg/mL) | Intra-Day Mean Conc. (mg/mL) | Intra-Day Precision (RSD%) | Intra-Day Accuracy (Recovery%) | Inter-Day Mean Conc. (mg/mL) | Inter-Day Precision (RSD%) | Inter-Day Accuracy (Recovery%) |
|---|---|---|---|---|---|---|---|
| Leu | 0.11 | 0.11 | 1.29 | 99.39 | 0.11 | 1.87 | 100.11 |
| 0.11 | 1.32 | 99.44 | |||||
| 0.11 | 2.33 | 101.50 | |||||
| 0.45 | 0.45 | 0.96 | 99.69 | 0.45 | 1.53 | 99.61 | |
| 0.44 | 2.45 | 98.75 | |||||
| 0.45 | 0.66 | 100.39 | |||||
| Phe | 0.02 | 0.02 | 1.82 | 99.08 | 0.02 | 1.77 | 98.31 |
| 0.02 | 1.97 | 96.77 | |||||
| 0.02 | 1.04 | 99.07 | |||||
| 0.16 | 0.16 | 1.78 | 99.51 | 0.16 | 1.48 | 100.62 | |
| 0.16 | 0.72 | 101.09 | |||||
| 0.16 | 1.58 | 101.26 |
| Mobile Phase | Compound | k2d | α | N2d | RS |
|---|---|---|---|---|---|
| a | Lys | 7.66 | 1.31 | 912 | 1.58 |
| Phe | 1.55 | 1.29 | 2285 | 1.79 | |
| Thr | 0.34 | 1.39 | 2681 | 0.99 | |
| Val | 0.64 | 1.48 | 1316 | 1.44 | |
| b | Lys | 12.9 | 1.32 | 1437 | 1.80 |
| Phe | 1.57 | 1.77 | 3565 | 4.81 | |
| Thr | 0.52 | 1.44 | 4765 | 1.95 | |
| Val | 0.95 | 1.59 | 2302 | 2.78 | |
| Ile | 1.28 | 1.63 | 2849 | 3.31 | |
| Leu | 1.5 | 1.94 | 3603 | 5.28 | |
| Met | 1.45 | 2.16 | 1681 | 4.24 | |
| Trp | 1.68 | 1.74 | 3193 | 4.66 | |
| c | Lys | 4.32 | 1.39 | 722 | 1.54 |
| Phe | 1.49 | 1.31 | 2118 | 1.84 | |
| Thr | 0.4 | 1.4 | N.C. e | N.C. e | |
| Val | 0.79 | 1.53 | 699 | 1.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varfaj, I.; Carotti, A.; Mangiapelo, L.; Cossignani, L.; Taticchi, A.; Macchiarulo, A.; Ianni, F.; Sardella, R. Environmentally Sustainable Achiral and Chiral Chromatographic Analysis of Amino Acids in Food Supplements. Molecules 2022, 27, 7724. https://doi.org/10.3390/molecules27227724
Varfaj I, Carotti A, Mangiapelo L, Cossignani L, Taticchi A, Macchiarulo A, Ianni F, Sardella R. Environmentally Sustainable Achiral and Chiral Chromatographic Analysis of Amino Acids in Food Supplements. Molecules. 2022; 27(22):7724. https://doi.org/10.3390/molecules27227724
Chicago/Turabian StyleVarfaj, Ina, Andrea Carotti, Luciano Mangiapelo, Lina Cossignani, Agnese Taticchi, Antonio Macchiarulo, Federica Ianni, and Roccaldo Sardella. 2022. "Environmentally Sustainable Achiral and Chiral Chromatographic Analysis of Amino Acids in Food Supplements" Molecules 27, no. 22: 7724. https://doi.org/10.3390/molecules27227724
APA StyleVarfaj, I., Carotti, A., Mangiapelo, L., Cossignani, L., Taticchi, A., Macchiarulo, A., Ianni, F., & Sardella, R. (2022). Environmentally Sustainable Achiral and Chiral Chromatographic Analysis of Amino Acids in Food Supplements. Molecules, 27(22), 7724. https://doi.org/10.3390/molecules27227724