


Resin Glycosides from Convolvulaceae Family: An Update


Abstract
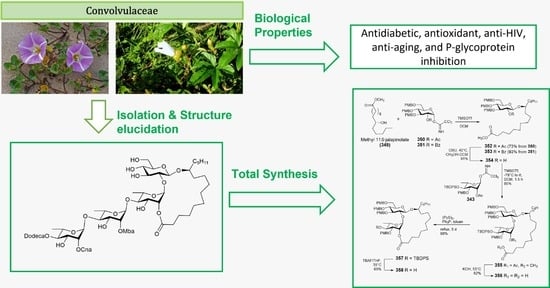
:
1. Introduction
2. Classification
2.1. Monosaccharides
2.2. Trisaccharides
2.3. Tetrasaccharides
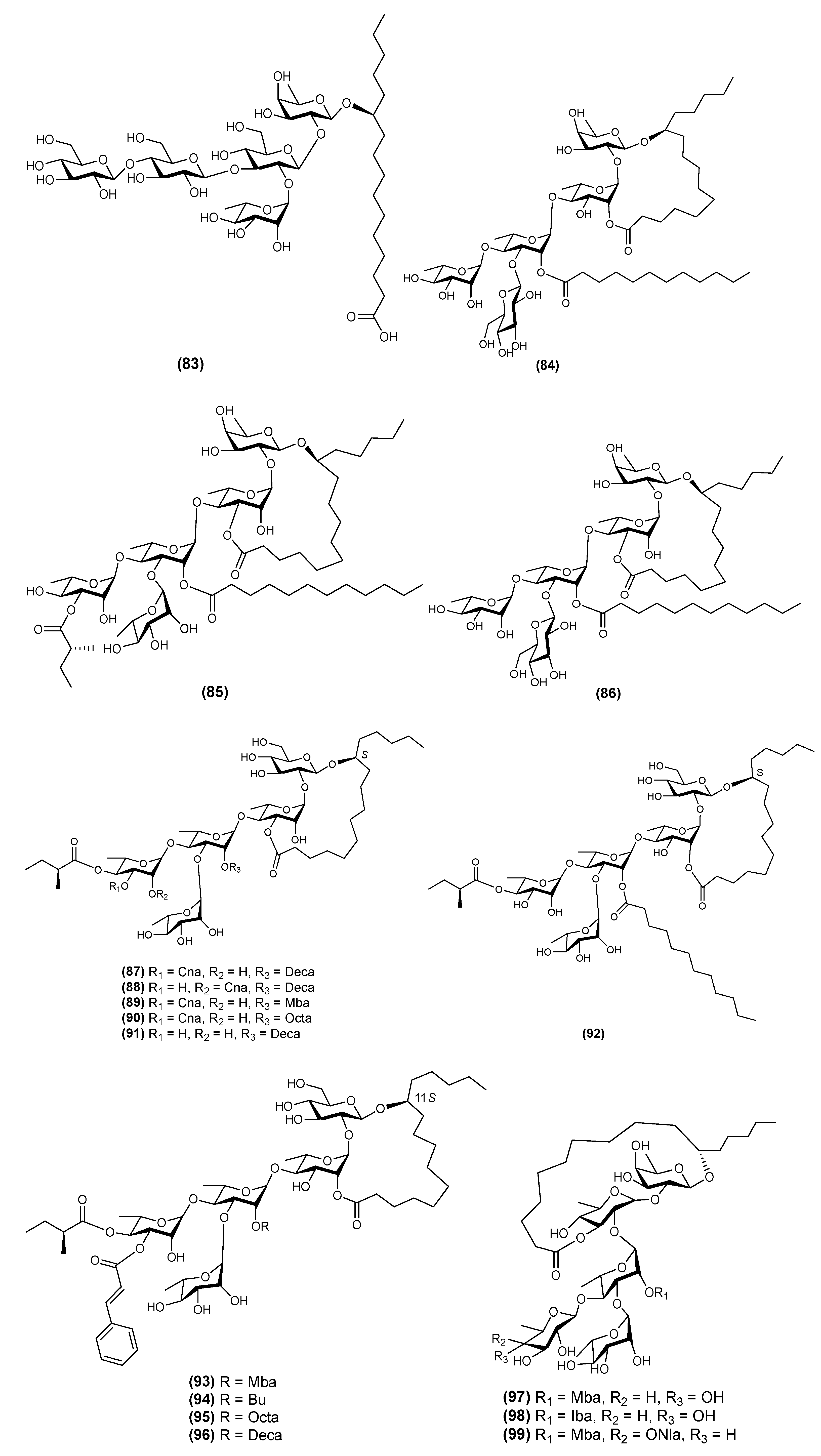
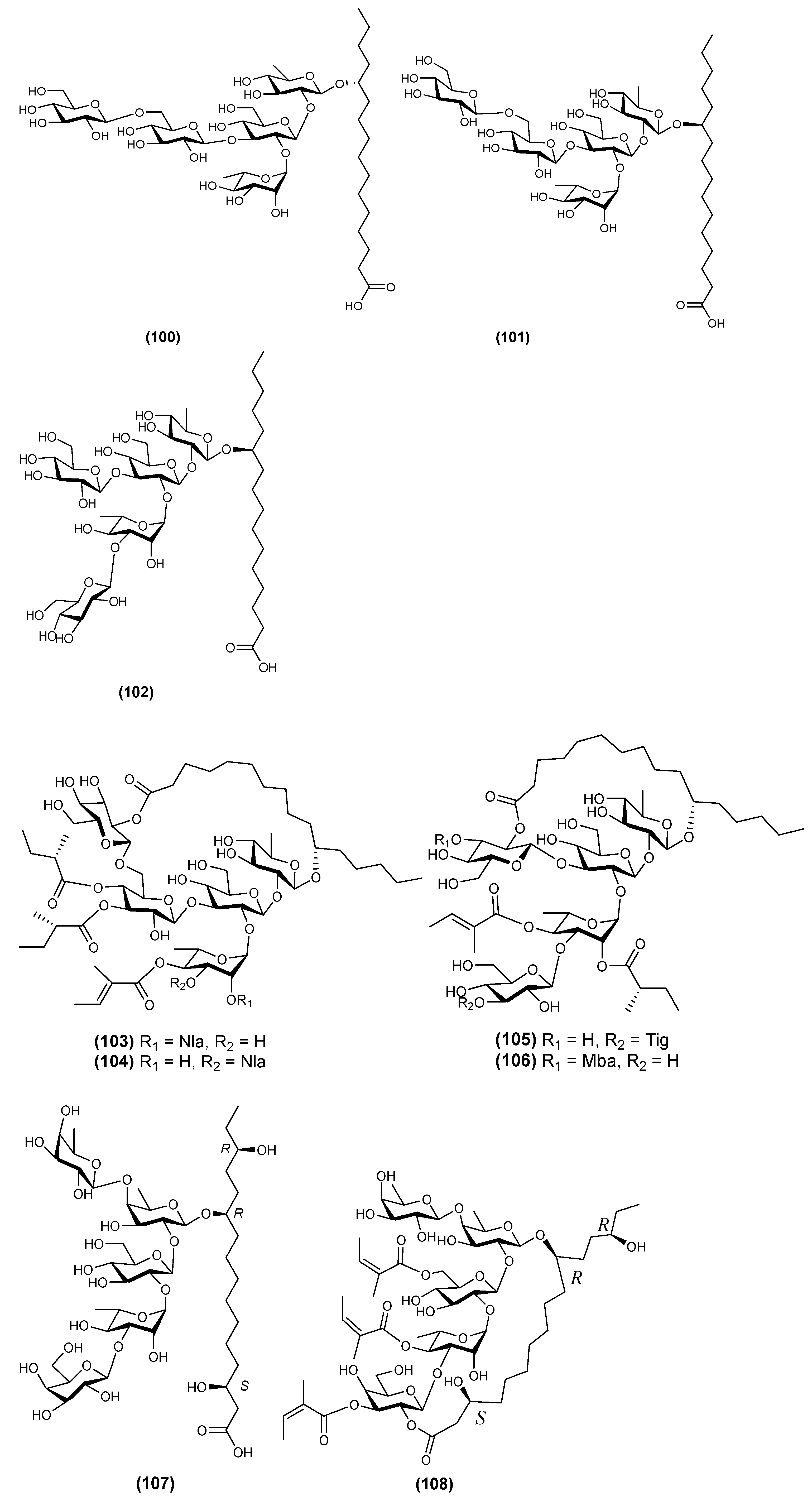
2.4. Pentasaccharides
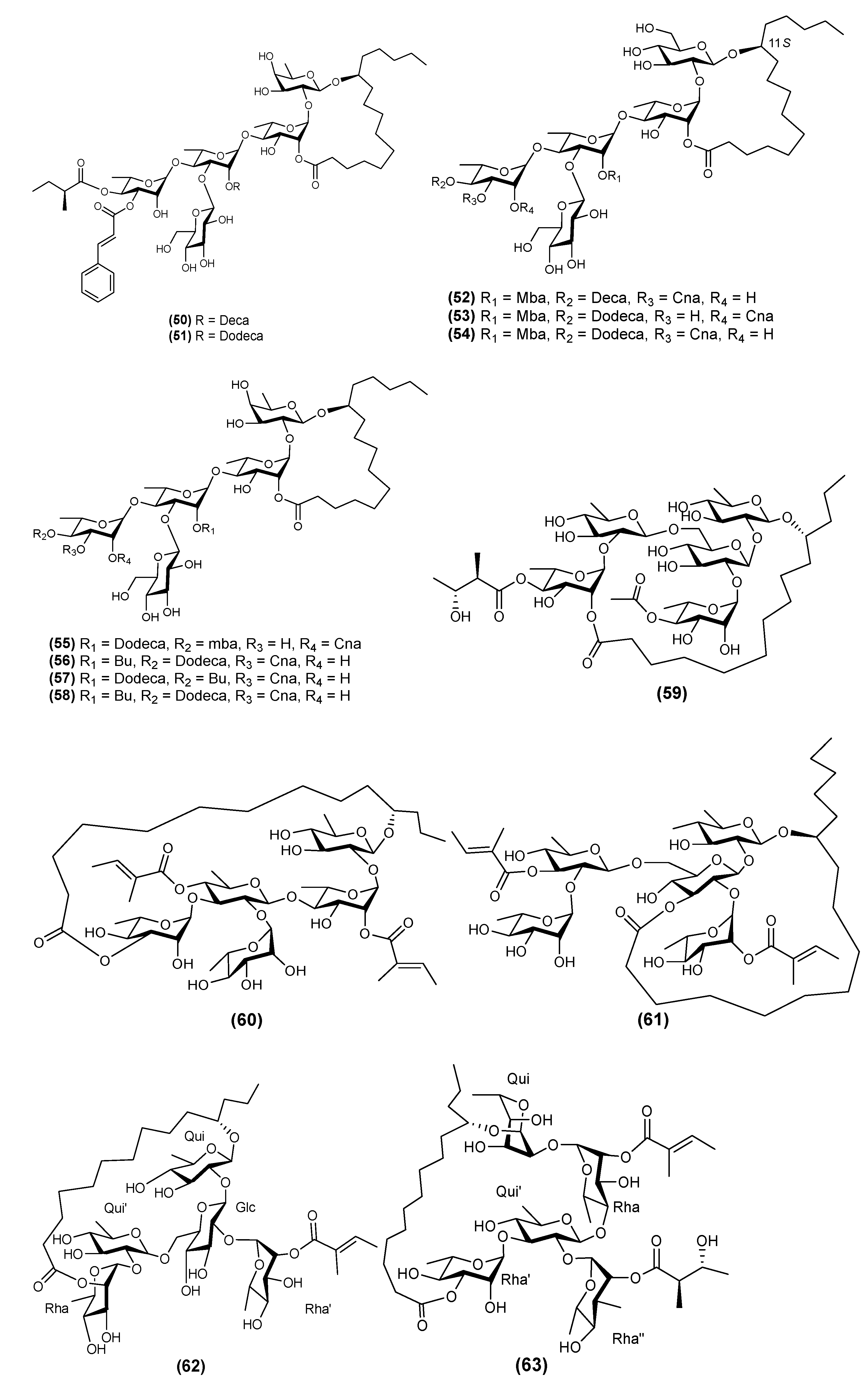
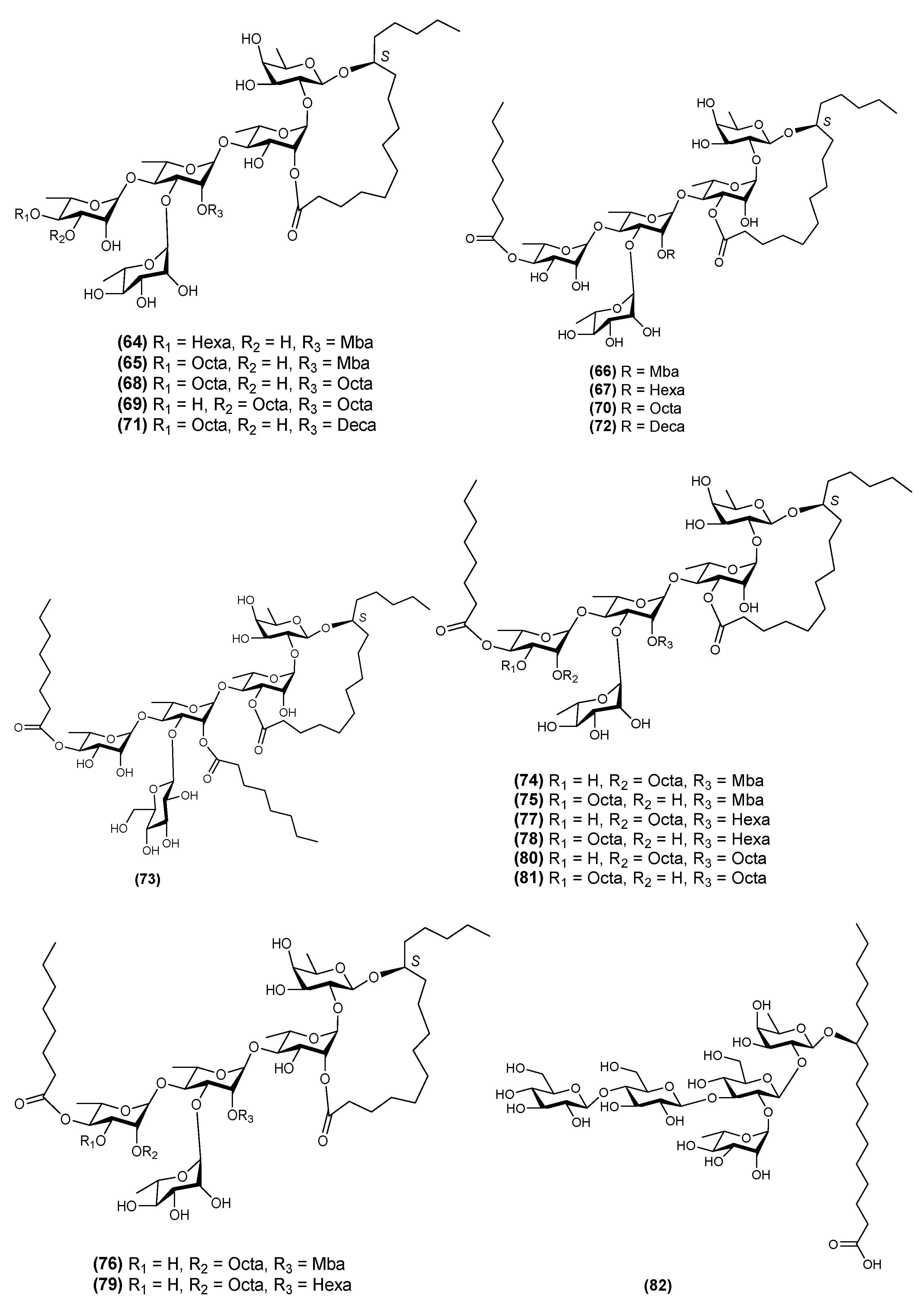
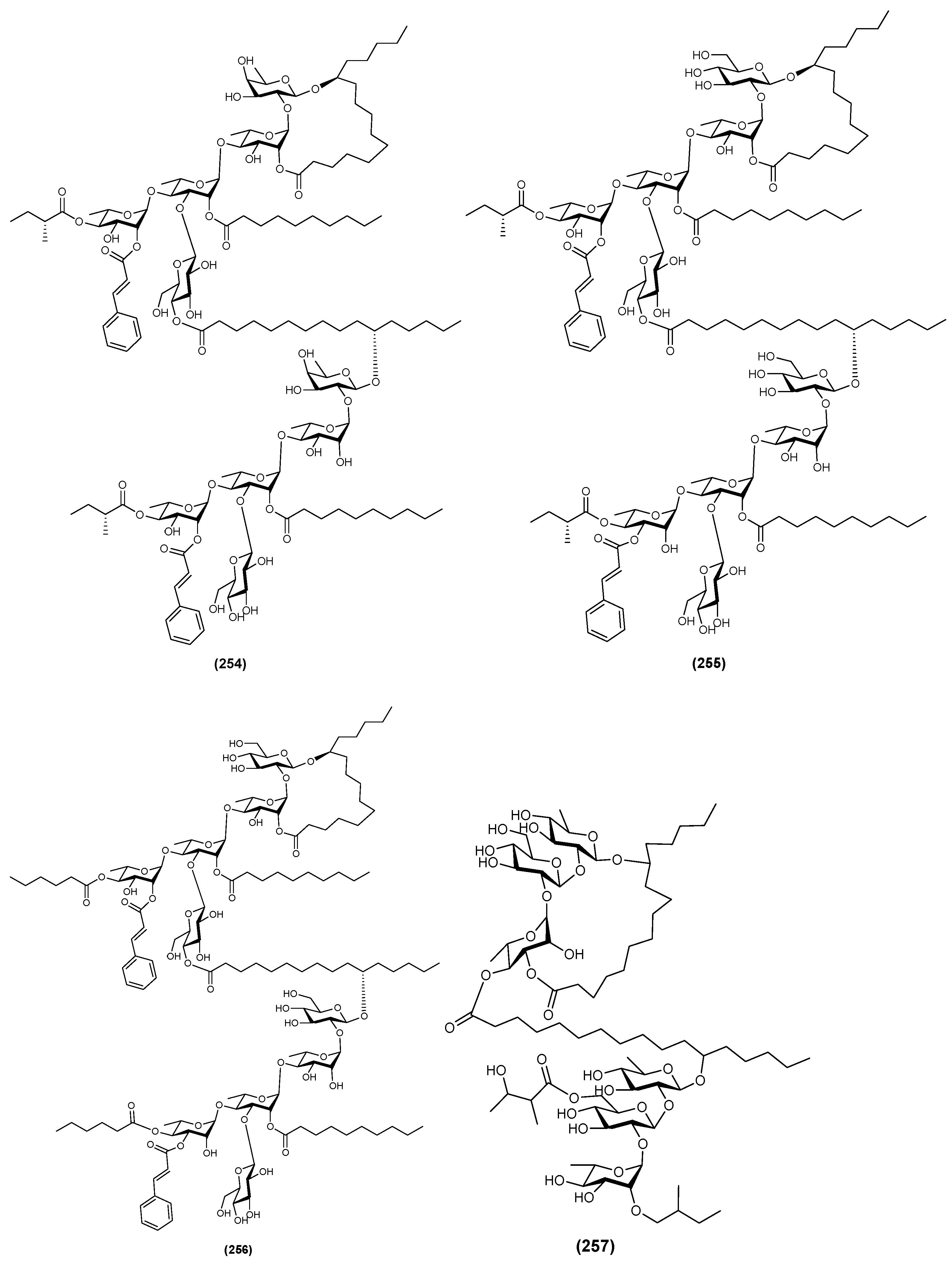
2.5. Hexasaccharides
2.6. Heptasaccharides
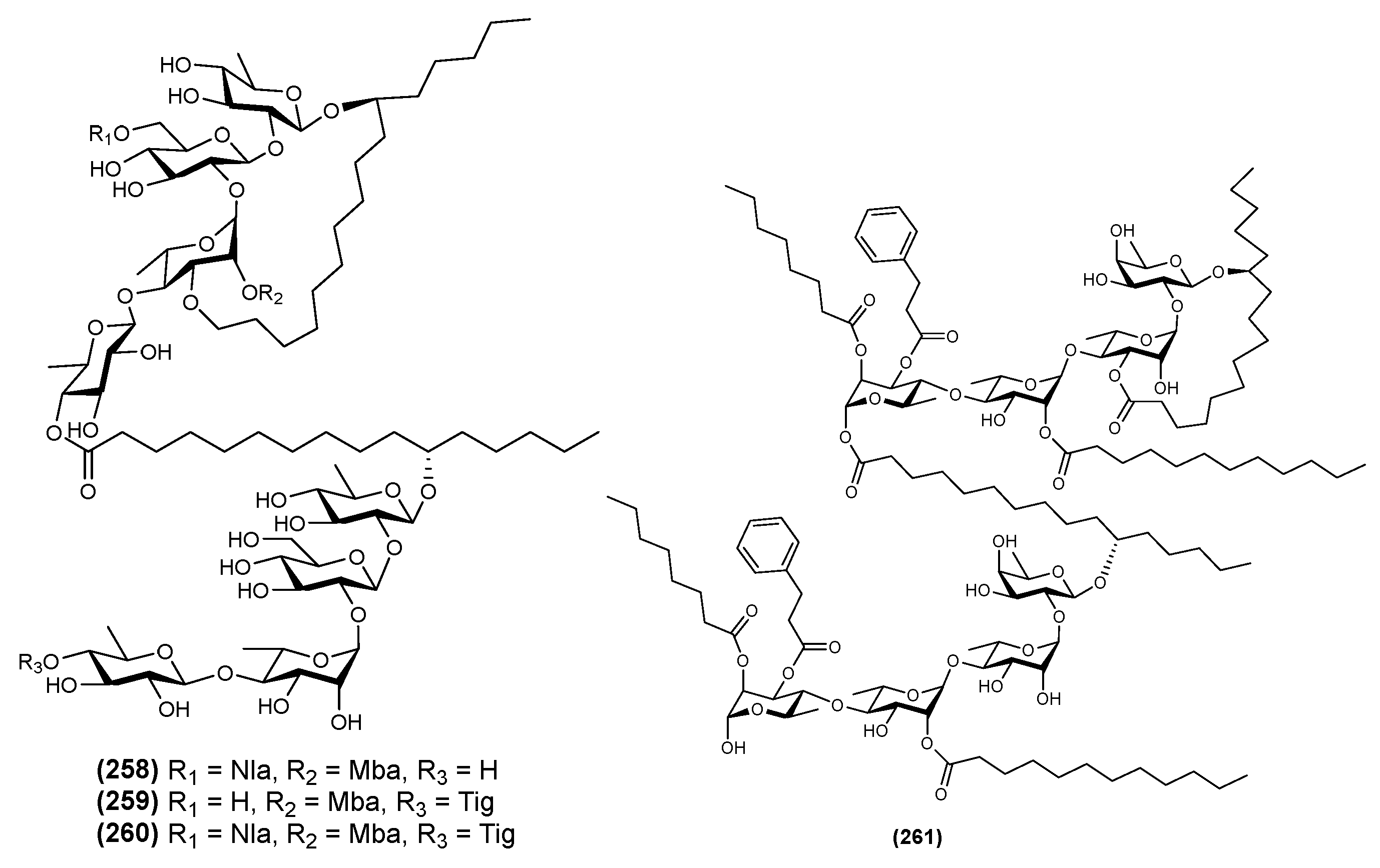
2.7. Bidesmosides
2.8. Oligosaccharide Ester-Type Dimer
3. Biological Activity
4. Isolation Techniques
5. Structural Determination
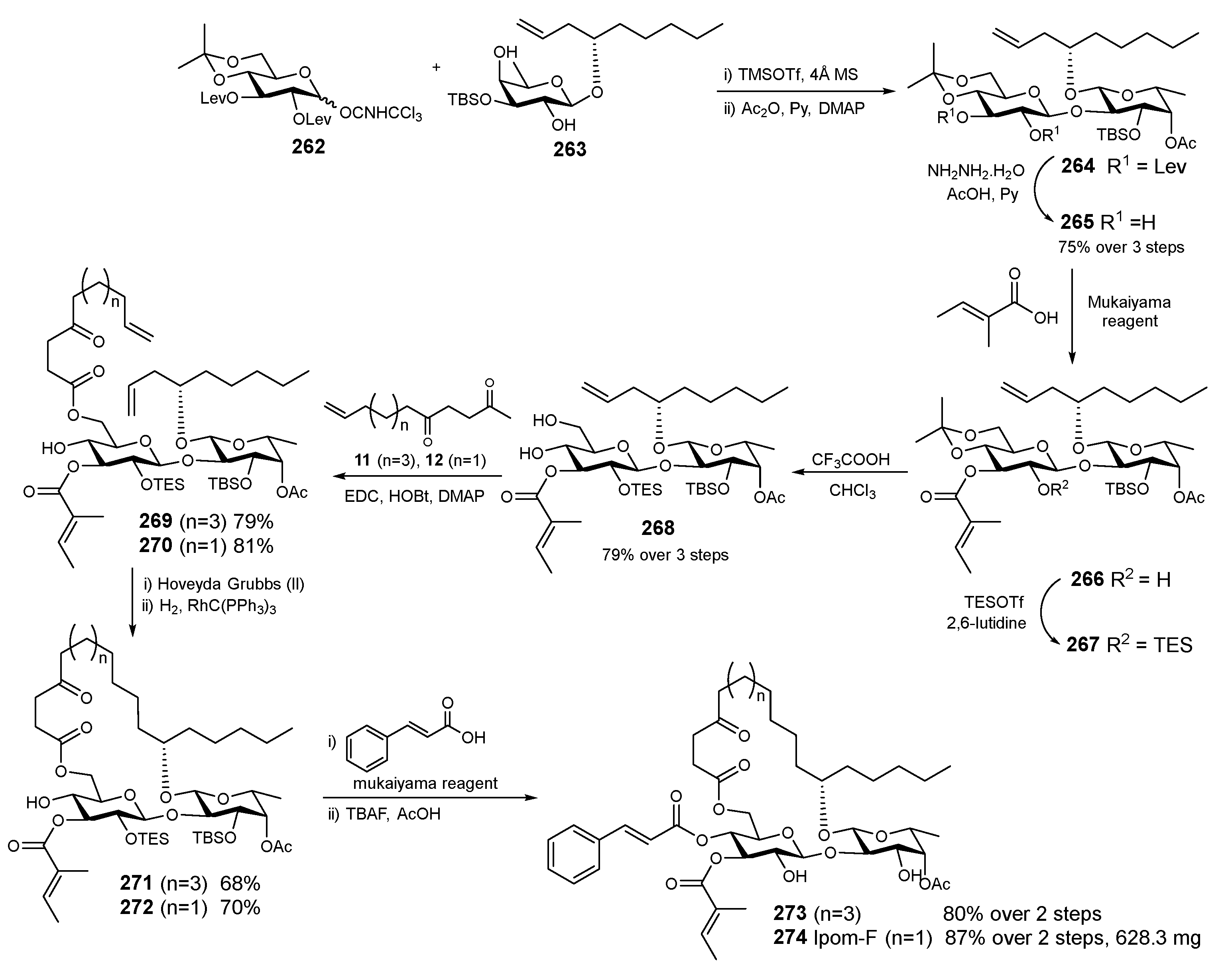
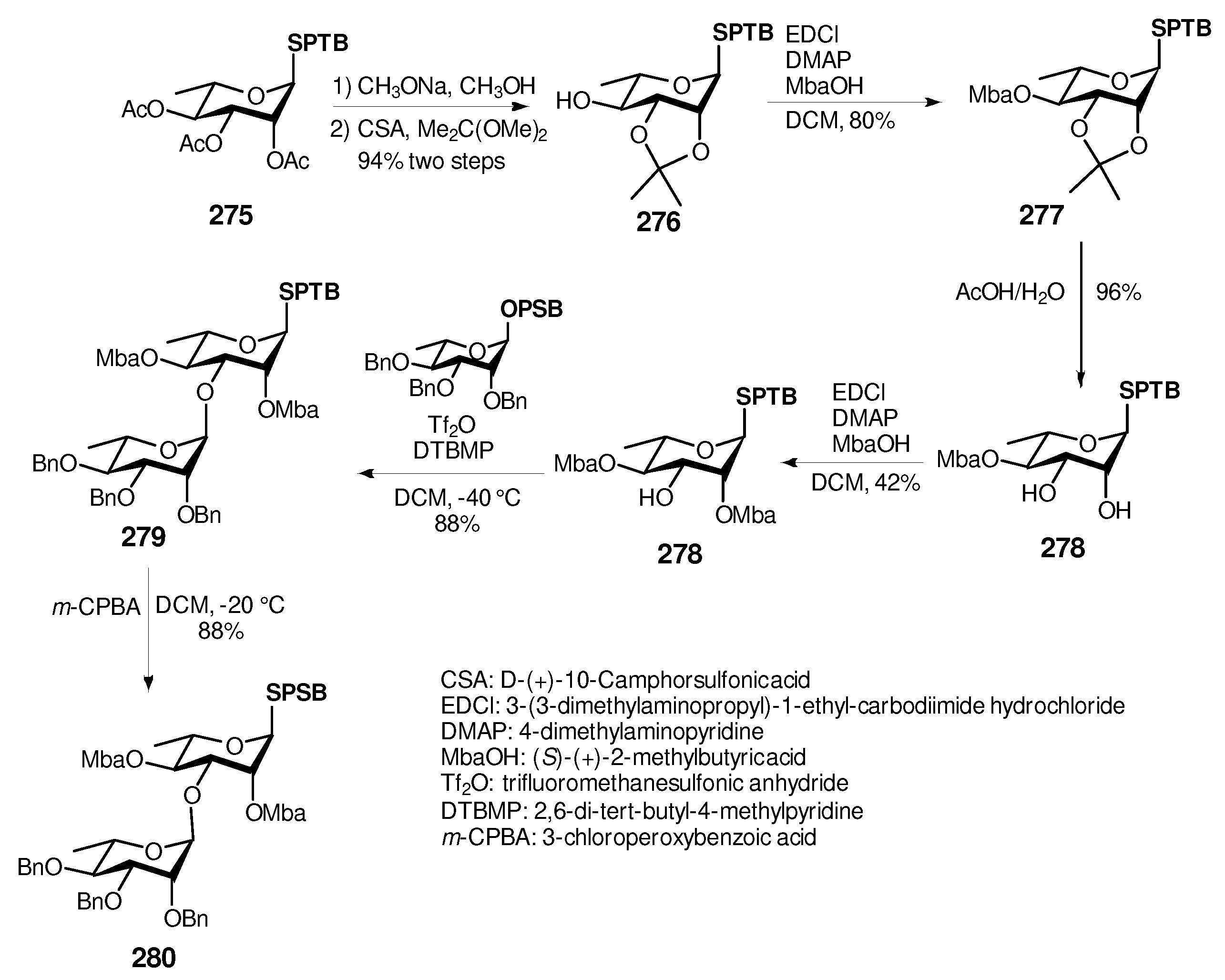
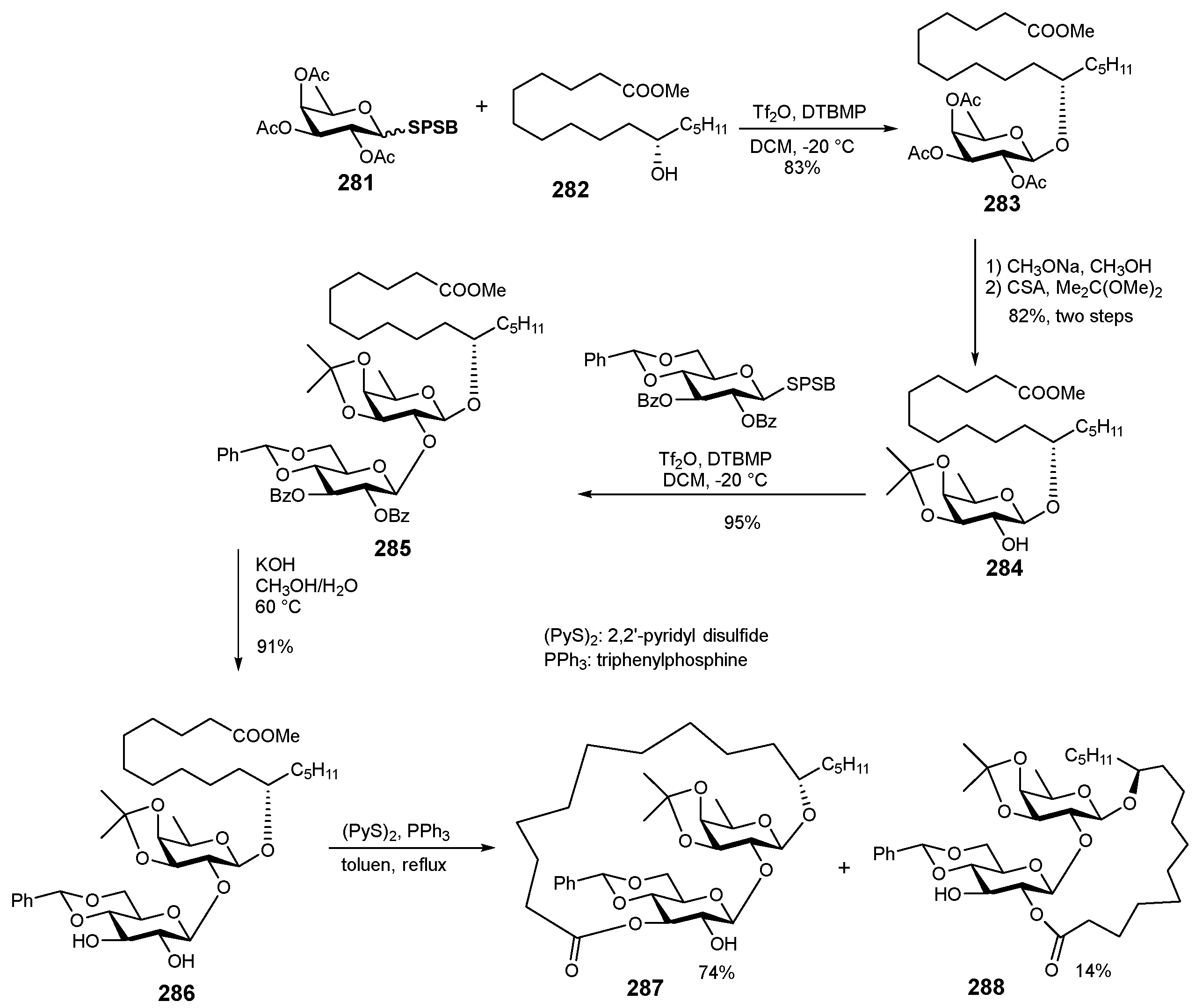
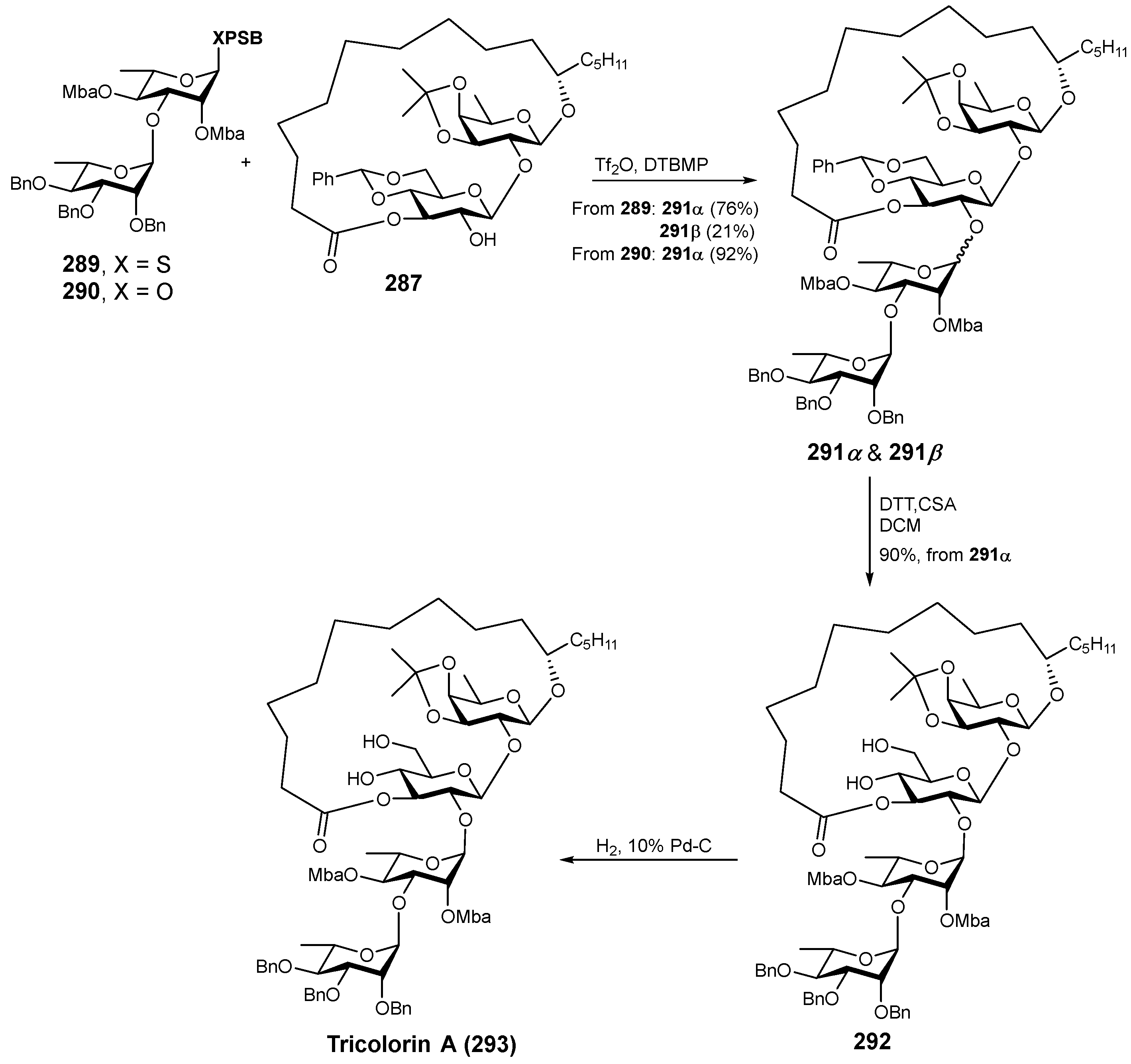
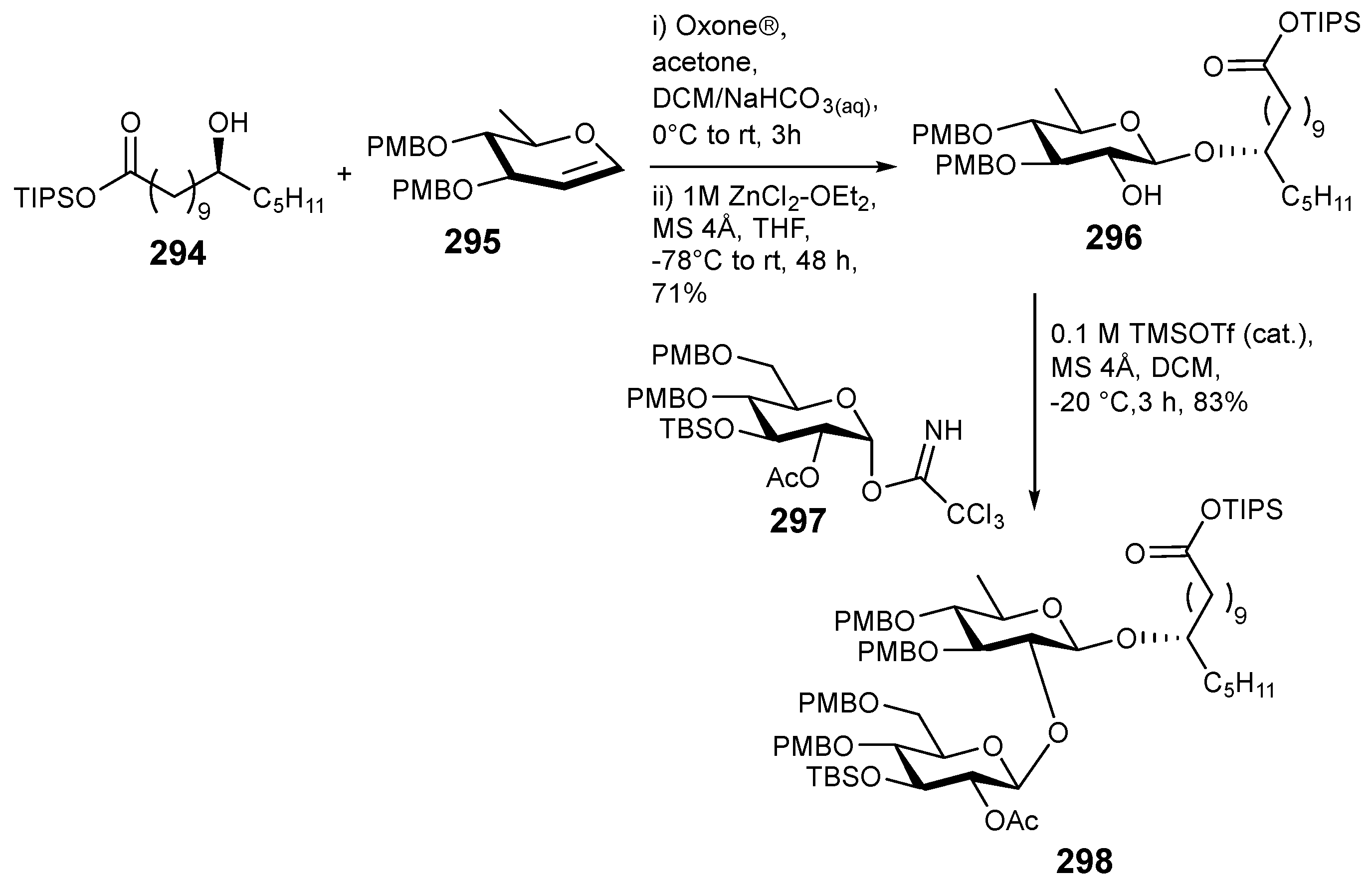
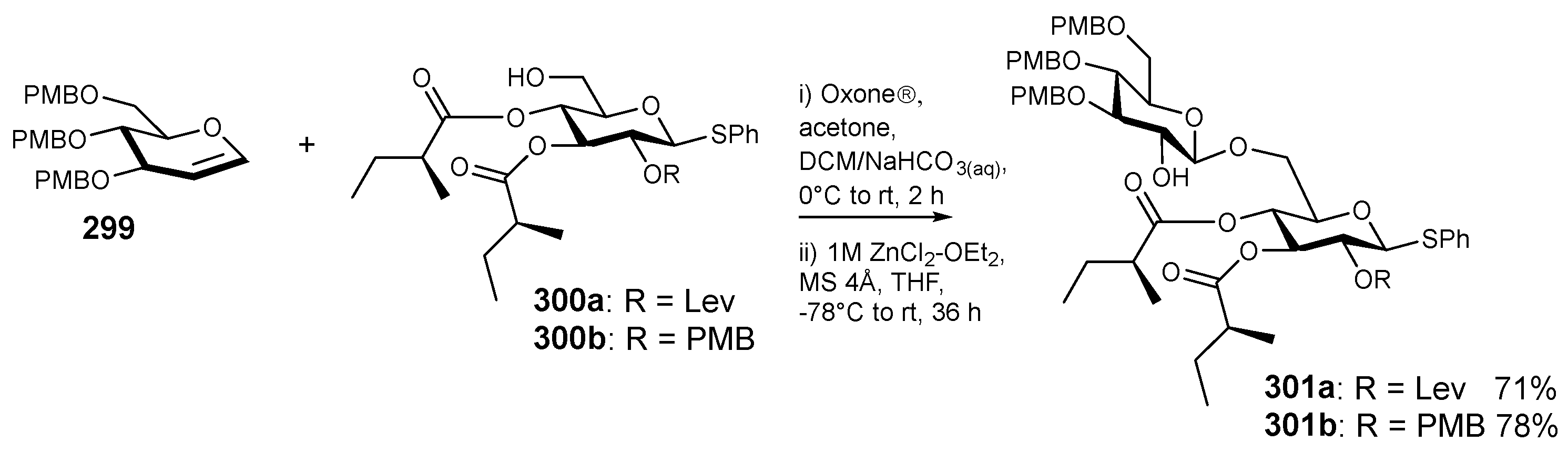
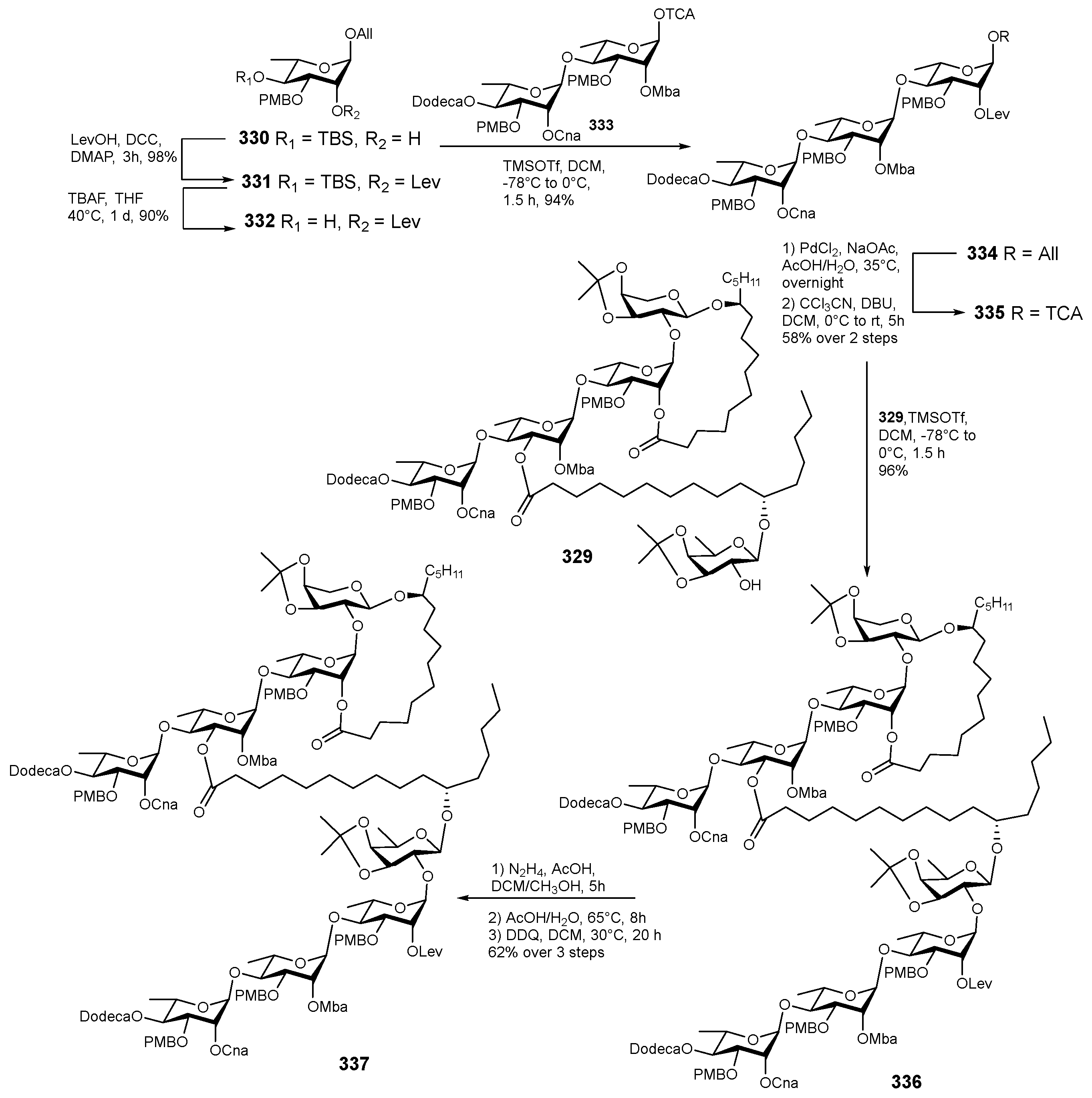
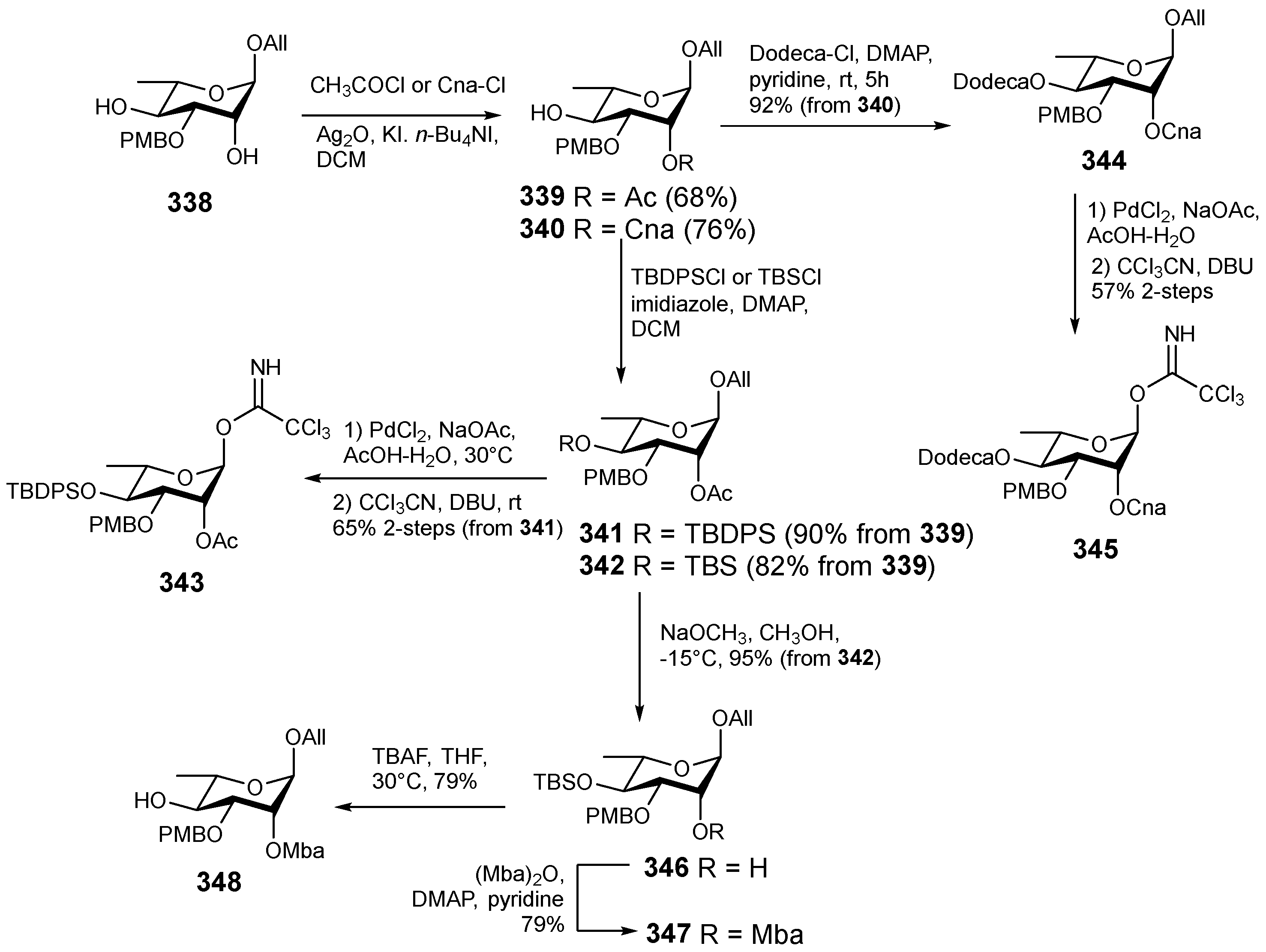
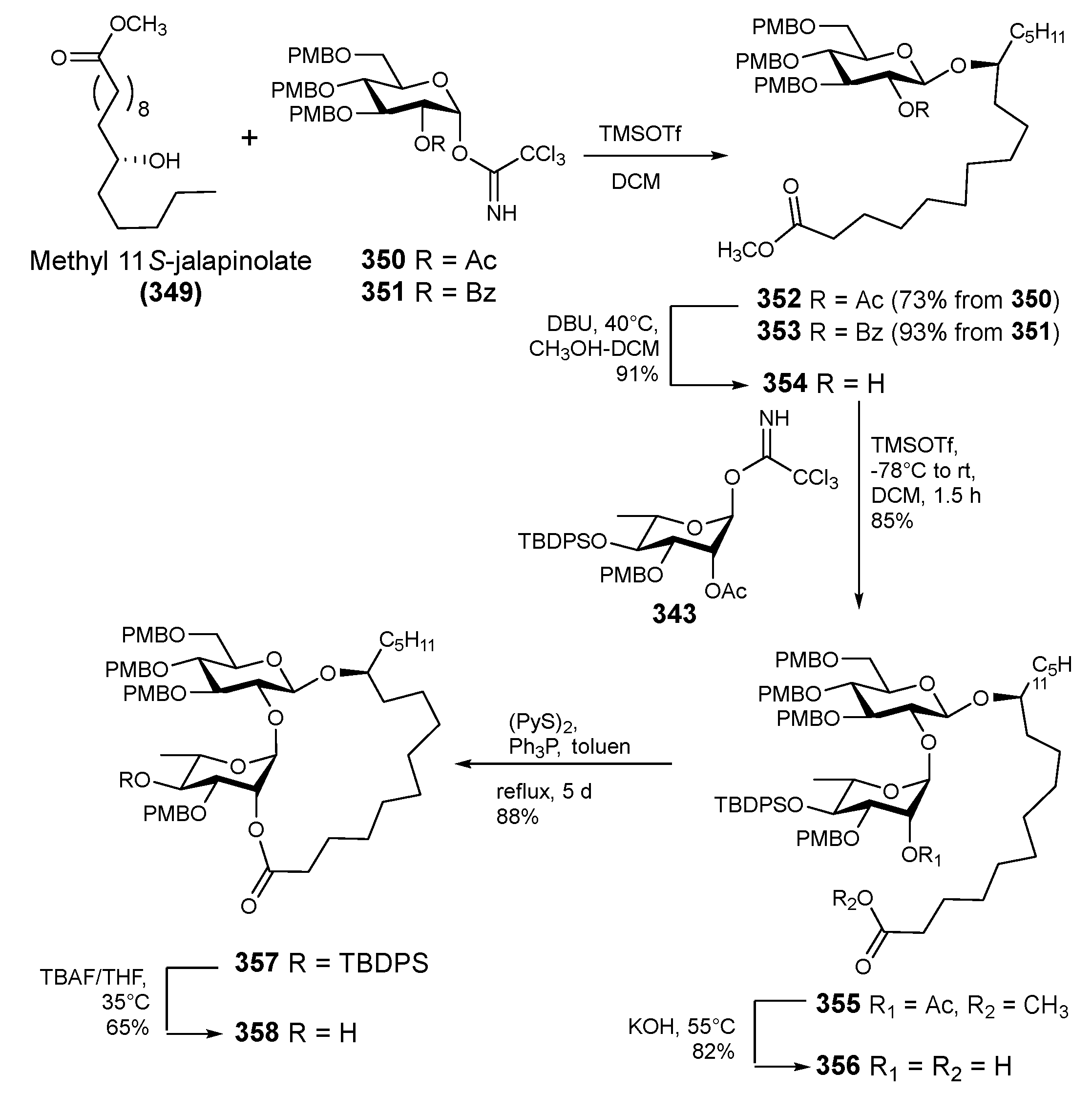
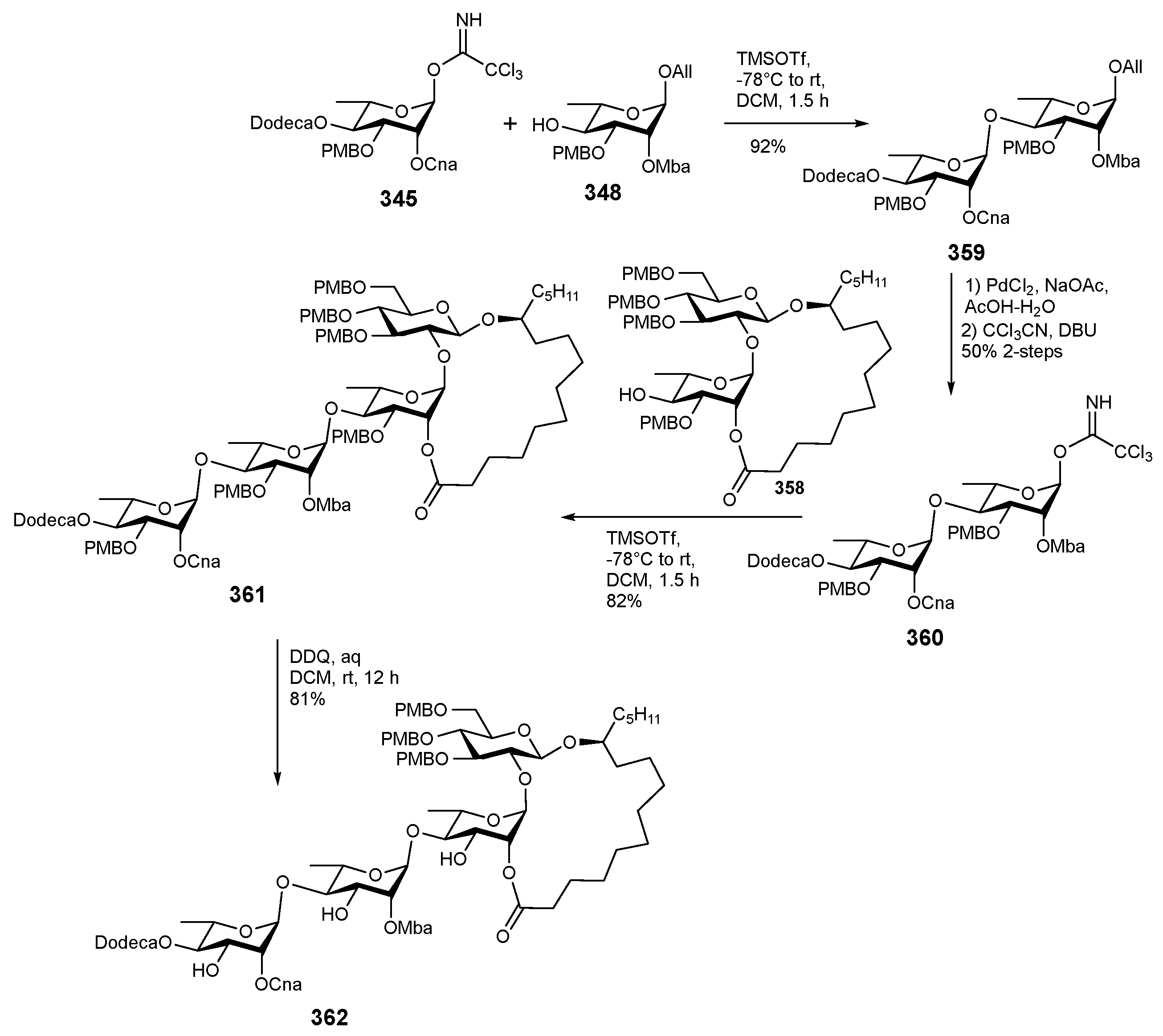
6. Synthesis
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pereda-Miranda, R.; Rosas-Ramírez, D.; Castañeda-Gómez, J. Resin Glycosides from the Morning Glory Family. In Fortschritte der Chemie Organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products; Springer: Berlin/Heidelberg, Germany, 2010; Volume 92, pp. 77–153. [Google Scholar]
- Ono, M. Resin Glycosides from Convolvulaceae Plants. J. Nat. Med. 2017, 71, 591–604. [Google Scholar] [CrossRef] [Green Version]
- Song, W.B.; Wang, W.Q.; Zhang, S.W.; Xuan, L.J. Multidrug Resistance-Reversal Effects of Resin Glycosides from Dichondra repens. Bioorg. Med. Chem. Lett. 2015, 25, 795–798. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.Y.; Luo, J.G.; Gu, Y.C.; Kong, L.Y. Unusual Ether-Type Resin Glycoside Dimers from the Seeds of Cuscuta chinensis. Tetrahedron 2014, 70, 2003–2014. [Google Scholar] [CrossRef]
- Ding, W.B.; Zhang, D.G.; Liu, C.J.; Li, G.H.; Li, Y.Z. Resin Glycosides from Porana duclouxii. J. Asian Nat. Prod. Res. 2014, 16, 135–140. [Google Scholar] [CrossRef]
- Castañeda-Gómez, J.; Figueroa-González, G.; Jacobo, N.; Pereda-Miranda, R. Purgin II, a Resin Glycoside Ester-Type Dimer and Inhibitor of Multidrug Efflux Pumps from Ipomoea purga. J. Nat. Prod. 2013, 76, 64–71. [Google Scholar] [CrossRef]
- Cruz-Morales, S.; Castañeda-Gómez, J.; Rosas-Ramírez, D.; Fragoso-Serrano, M.; Figueroa-González, G.; Lorence, A.; Pereda-Miranda, R. Resin Glycosides from Ipomoea alba Seeds as Potential Chemosensitizers in Breast Carcinoma Cells. J. Nat. Prod. 2016, 79, 3093–3104. [Google Scholar] [CrossRef]
- Yin, Y.Q.; Pan, J.T.; Yu, B.W.; Cui, H.H.; Yan, Y.S.; Chen, Y.F. Two Pentasaccharide Resin Glycosides from Argyreia acuta. Nat. Prod. Res. 2016, 30, 20–24. [Google Scholar] [CrossRef]
- Castañeda-Gómez, J.; Pereda-Miranda, R. Resin Glycosides from the Herbal Drug Jalap (Ipomoea purga). J. Nat. Prod. 2011, 74, 1148–1153. [Google Scholar] [CrossRef]
- León-Rivera, I.; Villeda-Hernández, J.; Campos-Peña, V.; Aguirre-Moreno, A.; Estrada-Soto, S.; Navarrete-Vázquez, G.; Rios, M.Y.; Aguilar-Guadarrama, B.; Castillo-España, P.; Rivera-Leyva, J.C. Evaluation of the Neuroprotective Activity of Stansin 6, a Resin Glycoside from Ipomoea stans. Bioorg. Med. Chem. Lett. 2014, 24, 3541–3545. [Google Scholar] [CrossRef]
- Zong, G.; Hu, Z.; Duah, K.B.; Andrews, L.E.; Zhou, J.; O’Keefe, S.; Whisenhunt, L.; Shim, J.S.; Du, Y.; High, S.; et al. Ring Expansion Leads to a More Potent Analogue of Ipomoeassin F. J. Org. Chem. 2020, 85, 16226–16235. [Google Scholar] [CrossRef]
- Castañeda-Gómez, J.; Lavias-Hernández, P.; Fragoso-Serrano, M.; Lorence, A.; Pereda-Miranda, R. Acylsugar Diversity in the Resin Glycosides from Ipomoea tricolor Seeds as Chemosensitizers in Breast Cancer Cells. Phytochem. Lett. 2019, 32, 77–82. [Google Scholar] [CrossRef]
- Rosas-Ramírez, D.; Escandón-Rivera, S.; Pereda-Miranda, R. Morning Glory Resin Glycosides as α-Glucosidase Inhibitors: In Vitro and in Silico Analysis. Phytochemistry 2018, 148, 39–47. [Google Scholar] [CrossRef]
- Ono, M.; Kanemaru, Y.; Yasuda, S.; Okawa, M.; Kinjo, J.; Miyashita, H.; Yokomizo, K.; Yoshimitsu, H.; Nohara, T. A New Resin Glycoside from Calystegia soldanella and Its Antiviral Activity towards Herpes. Nat. Prod. Res. 2017, 31, 2660–2664. [Google Scholar] [CrossRef]
- Chen, C.; Ma, T.; Zhang, C.; Zhang, H.; Bai, L.; Kong, L.; Luo, J. Down-Regulation of Aquaporin 5-Mediated Epithelial-Mesenchymal Transition and Anti-Metastatic Effect by Natural Product Cairicoside E in Colorectal Cancer. Mol. Carcinog. 2017, 56, 2692–2705. [Google Scholar] [CrossRef]
- León-Rivera, I.; del Río-Portilla, F.; Enríquez, R.G.; Rangel-López, E.; Villeda, J.; Rios, M.Y.; Navarrete-Vázquez, G.; Hurtado-Días, I.; Guzmán-Valdivieso, U.; Núñez-Urquiza, V.; et al. Hepta-, Hexa-, Penta-, Tetra-, and Trisaccharide Resin Glycosides from Three Species of Ipomoea and Their Antiproliferative Activity on Two Glioma Cell Lines. Magn. Reson. Chem. 2017, 55, 214–223. [Google Scholar] [CrossRef]
- León-Rivera, I.; Castro, J.M.; Mirón-López, G.; del Río-Portilla, F.; Enríquez, R.G.; Reynolds, W.F.; Estrada-Soto, S.; Rendón-Vallejo, P.; del Carmen Gutiérrez, M.; Herrera-Ruiz, M.; et al. Resin Glycosides from Ipomoea tyrianthina and Their Sedative and Vasorelaxant Effects. J. Nat. Med. 2014, 68, 655–667. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Yagi, C.; Hama, H.; Tanaka, M.; Arihara, S.; Hashimoto, T. Ipomotaosides A-D, Resin Glycosides from the Aerial Parts of Ipomoea batatas and Their Inhibitory Activity against COX-1 and COX-2. J. Nat. Prod. 2010, 73, 1763–1766. [Google Scholar] [CrossRef]
- Mirón-López, G.; Herrera-Ruiz, M.; Estrada-Soto, S.; Aguirre-Crespo, F.; Vázquez-Navarrete, L.; León-Rivera, I. Resin Glycosides from the Roots of Ipomoea tyrianthina and Their Biological Activity. J. Nat. Prod. 2007, 70, 557–562. [Google Scholar] [CrossRef]
- Zhu, D.; Chen, C.; Bai, L.; Kong, L.; Luo, J. Downregulation of Aquaporin 3 Mediated the Laxative Effect in the Rat Colon by a Purified Resin Glycoside Fraction from Pharbitis Semen. Evid.-Based Complement. Altern. Med. 2019, 2019, 9406342. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.T.; Yu, N.H.; Park, A.R.; Park, H.W.; Kim, I.S.; Kim, J.C. Antibacterial Activity of Pharbitin, Isolated from the Seeds of Pharbitis Nil, against Various Plant Pathogenic Bacteria. J. Microbiol. Biotechnol. 2017, 27, 1763–1772. [Google Scholar] [CrossRef]
- Tasdemir, D.; Brun, R.; Franzblau, S.G.; Sezgin, Y.; Çalis, I. Evaluation of Antiprotozoal and Antimycobacterial Activities of the Resin Glycosides and the Other Metabolites of Scrophularia cryptophila. Phytomedicine 2008, 15, 209–215. [Google Scholar] [CrossRef]
- Wang, W.Q.; Liu, S.S.; Song, W.B.; Li, J.; Xuan, L.J. Resin Glycosides from the Seeds of Ipomoea muricata and Their Multidrug Resistance Reversal Activities. Phytochemistry 2018, 149, 24–30. [Google Scholar] [CrossRef]
- Corona-Castañeda, B.; Rosas-Ramírez, D.; Castañeda-Gómez, J.; Aparicio-Cuevas, M.A.; Fragoso-Serrano, M.; Figueroa-González, G.; Pereda-Miranda, R. Resin Glycosides from Ipomoea wolcottiana as Modulators of the Multidrug Resistance Phenotype in Vitro. Phytochemistry 2016, 123, 48–57. [Google Scholar] [CrossRef]
- Fan, B.Y.; Jiang, X.; Li, Y.X.; Wang, W.L.; Yang, M.; Li, J.L.; Wang, A.D.; Chen, G.T. Chemistry and Biological Activity of Resin Glycosides from Convolvulaceae Species. Med. Res. Rev. 2022, 42, 2025–2066. [Google Scholar] [CrossRef]
- Ono, M.; Kishida, M.; Ikegami, Y.; Takaki, Y.; Okawa, M.; Kinjo, J.; Yoshimitsu, H.; Nohara, T.; Miyahara, K. Components of Convolvulin from Quamoclit × multifida. J. Nat. Med. 2011, 65, 95–102. [Google Scholar] [CrossRef]
- Ono, M.; Takagi-Taki, Y.; Honda-Yamada, F.; Noda, N.; Miyahara, K. Components of Ether-Insoluble Resin Glycoside (Convolvulin) from Seeds of Quamoclit pennata. Regul. Artic. Chem. Pharm. Bull. 2010, 58, 666–672. [Google Scholar] [CrossRef] [Green Version]
- Lira-Ricárdez, J.; Pereda-Miranda, R.; Castañeda-Gómez, J.; Fragoso-Serrano, M.; Simas, R.C.; Leitão, S.G. Resin Glycosides from the Roots of Operculina macrocarpa (Brazilian Jalap) with Purgative Activity. J. Nat. Prod. 2019, 82, 1664–1677. [Google Scholar] [CrossRef]
- Fan, B.Y.; Gu, Y.C.; He, Y.; Li, Z.R.; Luo, J.G.; Kong, L.Y. Cytotoxic Resin Glycosides from Ipomoea aquatica and Their Effects on Intracellular Ca2+ Concentrations. J. Nat. Prod. 2014, 77, 2264–2272. [Google Scholar] [CrossRef]
- Takigawa, A.; Muto, H.; Kabata, K.; Okawa, M.; Kinjo, J.; Yoshimitsu, H.; Nohara, T.; Ono, M. Calysolins I-IV, Resin Glycosides from Calystegia soldanella. J. Nat. Prod. 2011, 74, 2414–2419. [Google Scholar] [CrossRef]
- Ono, M.; Kawakami, G.; Takigawa, A.; Kabata, K.; Okawa, M.; Kinjo, J.; Yokomizo, K.; Yoshimitsu, H.; Nohara, T. Calysolins X-XIII, Resin Glycosides from Calystegia soldanella, and Their Antiviral Activity toward Herpes Simplex Virus. Chem. Pharm. Bull. 2014, 62, 839–844. [Google Scholar] [CrossRef]
- Xu, J.Y.; He, Y.; Zhang, A.W.; Lu, Y.; Chen, G.T.; Yang, M.; Fan, B.Y. Isolation of Evolvulic Acids B and C, Two New Components of Crude Resin Glycoside Fraction from Evolvulus alsinoides. Nat. Prod. Res. 2021, 35, 3766–3771. [Google Scholar] [CrossRef]
- Ono, M.; Taketomi, S.; Kakiki, Y.; Yasuda, S.; Okawa, M.; Kinjo, J.; Miyashita, H.; Yoshimitsu, H.; Nohara, T. A New Glycosidic Acid, Muricatic Acid D, and Resin Glycosides, Muricatins X and XI, from the Crude Resin Glycoside Fraction of the Seeds of Ipomoea muricata. Chem. Pharm. Bull. 2021, 69, 291–297. [Google Scholar] [CrossRef]
- Ono, M.; Taketomi, S.; Kakiki, Y.; Yasuda, S.; Okawa, M.; Kinjo, J.; Yoshimitsu, H.; Nohara, T. A New Resin Glycoside, Muricatin IX, from the Seeds of Ipomoea muricata. Chem. Pharm. Bull. 2016, 64, 1408–1410. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, W.F.; Yu, M.; Enriquez, R.G.; Gonzalez, H.; Leon, I.; Magos, G. Isolation and Characterization of Cytotoxic and Antibacterial Tetrasaccharide Glycosides from Ipomoea stans. J. Nat. Prod. 1995, 58, 1730–1731. [Google Scholar] [CrossRef]
- Wang, L.; Yan, Y.S.; Cui, H.H.; Yin, Y.Q.; Pan, J.T.; Yu, B.W. Three New Resin Glycosides Compounds from Argyreia acuta and Their α-Glucosidase Inhibitory Activity. Nat. Prod. Res. 2017, 31, 537–542. [Google Scholar] [CrossRef]
- Yu, B.W.; Sun, J.J.; Pan, J.T.; Wu, X.H.; Yin, Y.Q.; Yan, Y.S.; Hu, J.Y.; McPhee, D.J. Four Pentasaccharide Resin Glycosides from Argyreia acuta. Molecules 2017, 22, 440. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Morales, S.; Castañeda-Gómez, J.; Figueroa-González, G.; Mendoza-García, A.D.; Lorence, A.; Pereda-Miranda, R. Mammalian Multidrug Resistance Lipopentasaccharide Inhibitors from Ipomoea alba Seeds. J. Nat. Prod. 2012, 75, 1603–1611. [Google Scholar] [CrossRef]
- Fan, B.Y.; Li, Z.R.; Ma, T.; Gu, Y.C.; Zhao, H.J.; Luo, J.G.; Kong, L.Y. Further Screening of the Resin Glycosides in the Edible Water Spinach and Characterisation on Their Mechanism of Anticancer Potential. J. Funct. Foods 2015, 19, 141–154. [Google Scholar] [CrossRef]
- Lu, Y.; He, Y.; Yang, M.; Fan, B.Y. Arvensic Acids K and L, Components of Resin Glycoside Fraction from Convolvulus arvensis. Nat. Prod. Res. 2021, 35, 2303–2307. [Google Scholar] [CrossRef]
- Rosas-Ramírez, D.; Pereda-Miranda, R. Resin Glycosides from the Yellow-Skinned Variety of Sweet Potato (Ipomoea batatas). J. Agric. Food Chem. 2013, 61, 9488–9494. [Google Scholar] [CrossRef]
- Yu, B.; Luo, J.; Wang, J.; Zhang, D.; Yu, S.; Kong, L. Pentasaccharide Resin Glycosides from Ipomoea cairica and Their Cytotoxic Activities. Phytochemistry 2013, 95, 421–427. [Google Scholar] [CrossRef]
- Pan, J.T.; Yu, B.W.; Yin, Y.Q.; Li, J.H.; Wang, L.; Guo, L.B.; Shen, Z. Four New Pentasaccharide Resin Glycosides from Ipomoea cairica with Strong α-Glucosidase Inhibitory Activity. Molecules 2015, 20, 6601–6610. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Ichihara, Y.; Saito, N.; Yamada, M.; Yuuki, K.; Nawata, M.; Tsutsumi, S.; Yasuda, S.; Tsuchihashi, R.; Okawa, M.; et al. Identification and Characterization of Organic and Glycosidic Acids in Crude Resin Glycoside Fraction from Calystegia hederacea. J. Nat. Med. 2020, 74, 200–211. [Google Scholar] [CrossRef]
- Takigawa, A.; Setoguchi, H.; Okawa, M.; Kinjo, J.; Miyashita, H.; Yokomizo, K.; Yoshimitsu, H.; Nohara, T.; Ono, M. Identification and Characterization of Component Organic and Glycosidic Acids of Crude Resin Glycoside Fraction from Calystegia soldanella. Chem. Pharm. Bull. 2011, 59, 1163–1168. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Takigawa, A.; Kanemaru, Y.; Kawakami, G.; Kabata, K.; Okawa, M.; Kinjo, J.; Yokomizo, K.; Yoshimitsu, H.; Nohara, T. Calysolins V-IX, Resin Glycosides from Calystegia soldanella and Their Antiviral Activity toward Herpes. Chem. Pharm. Bull. 2014, 62, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.Y.; Lu, Y.; Yang, M.; Li, J.L.; Chen, G.T. Evolvulins I and II, Resin Glycosides with a Trihydroxy Aglycone Unit from Evolvulus alsinoides. Org. Lett. 2019, 21, 6548–6551. [Google Scholar] [CrossRef]
- Fan, B.Y.; Lu, Y.; Xu, J.Y.; Zhang, A.W.; Yang, M.; Chen, G.T. Evolvulin III, a New Resin Glycoside Isolated from Evolvulus alsinoides. Phytochem. Lett. 2020, 39, 132–134. [Google Scholar] [CrossRef]
- Sura, M.B.; Ponnapalli, M.G.; Annam, S.C.V.A.R.; Bobbili, V.V.P. Ipomeolides A and B, Resin Glycosides from Ipomoea pes-caprae and Combination Therapy of Ipomeolide A with Doxorubicin. J. Nat. Prod. 2019, 82, 1292–1300. [Google Scholar] [CrossRef]
- Wang, W.Q.; Song, W.B.; Lan, X.J.; Huang, M.; Xuan, L.J. Merremins A-G, Resin Glycosides from Merremia hederacea with Multidrug Resistance Reversal Activity. J. Nat. Prod. 2014, 77, 2234–2240. [Google Scholar] [CrossRef]
- Ono, M.; Azuchi, M.; Ichio, M.; Jiyoubi, Y.; Tsutsumi, S.; Yasuda, S.; Tsuchihasi, R.; Okawa, M.; Kinjo, J.; Yoshimitsu, H.; et al. Seven New Resin Glycosides from the Seeds of Quamoclit × multifida. J. Nat. Med. 2019, 73, 11–22. [Google Scholar] [CrossRef]
- Ono, M.; Teramoto, S.; Naito, S.; Takahashi, A.; Yoneda, A.; Shinkai, M.; Taga, N.; Yasuda, S.; Tsuchihasi, R.; Okawa, M.; et al. Four New Resin Glycosides, Murasakimasarins I–IV, from the Tuber of Ipomoea batatas. J. Nat. Med. 2018, 72, 784–792. [Google Scholar] [CrossRef]
- Yu, B.W.; Luo, J.G.; Wang, J.S.; Zhang, D.M.; Yu, S.S.; Kong, L.Y. Pentasaccharide Resin Glycosides from Ipomoea pes-caprae. J. Nat. Prod. 2011, 74, 620–628. [Google Scholar] [CrossRef]
- Ono, M.; Takigawa, A.; Mineno, T.; Yoshimitsu, H.; Nohara, T.; Ikeda, T.; Fukuda-Teramachi, E.; Noda, N.; Miyahara, K. Acylated Glycosides of Hydroxy Fatty Acid Methyl Esters Generated from the Crude Resin Glycoside (Pharbitin) of Seeds of Pharbitis nil by Treatment with Indium(III) Chloride in Methanol. J. Nat. Prod. 2010, 73, 1846–1852. [Google Scholar] [CrossRef]
- Ono, M.; Takaki, Y.; Takatsuji, M.; Akiyama, K.; Okawa, M.; Kinjo, J.; Miyashita, H.; Yoshimitsu, H.; Nohara, T. Three New Resin Glycosides and a New Tetrahydropyran Derivative from the Seeds of Quamoclit pennata. Chem. Pharm. Bull. 2012, 60, 1083–1087. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Jiang, Z.H.; Wu, P.; Xu, L.; Wei, X. Resin Glycosides from the Aerial Parts of Operculina turpethum. Phytochemistry 2012, 81, 165–174. [Google Scholar] [CrossRef]
- Koryudzu, K.; Arai, M.A.; Ahmed, F.; Sadhu, S.K.; Ishibashi, M. A New Resin Glycoside from Ipomoea maxima. Nat. Prod. Commun. 2012, 7, 219–220. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Saito, N.; Minamishima, H.; Yasuda, S.; Tsuchihashi, R.; Okawa, M.; Kinjo, J.; Miyashita, H.; Yoshimitsu, H.; Nohara, T. Two New Glycosidic Acids, Calyhedic Acids E and F, in Crude Resin Glycoside Fraction from Calystegia hederacea. Nat. Prod. Res. 2021, 36, 46–53. [Google Scholar] [CrossRef]
- Ono, M.; Yuhara, N.; Shimohara, T.; Matsubara, S.; Yasuda, S.; Tsuchihashi, R.; Okawa, M.; Kinjo, J.; Zhou, J.R.; Yoshimitsu, H.; et al. Calyhedins I–VI: Resin Glycosides from the Rhizomes of Calystegia hederacea. Phytochemistry 2021, 190, 112888. [Google Scholar] [CrossRef]
- Ono, M.; Shimohara, T.; Yuhara, N.; Matsubara, S.; Yasuda, S.; Tsuchihashi, R.; Okawa, M.; Kinjo, J.; Yoshimitsu, H.; Nohara, T. Four New Resin Glycosides, Calyhedins VII–X, from the Rhizomes of Calystegia hederacea. Nat. Prod. Res. 2021, 1–10. [Google Scholar] [CrossRef]
- Lv, K.Q.; Ji, H.Y.; Du, G.X.; Peng, S.; Guo, P.J.; Wang, G.; Zhu, Y.; Wang, Q.; Wang, W.Q.; Xuan, L.J. Calysepins I-VII, Hexasaccharide Resin Glycosides from Calystegia sepium and Their Cytotoxic Evaluation. J. Nat. Prod. 2022, 85, 1294–1303. [Google Scholar] [CrossRef]
- Ono, M.; Takigawa, A.; Muto, H.; Kabata, K.; Okawa, M.; Kinjo, J.; Yokomizo, K.; Yoshimitsu, H.; Nohara, T. Antiviral Activity of Four New Resin Glycosides Calysolins XIV-XVII from Calystegia soldanella against Herpes Simplex Virus. Chem. Pharm. Bull. 2015, 63, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, K.; Mineno, T.; Okawa, M.; Kinjo, J.; Miyashita, H.; Yoshimitsu, H.; Nohara, T.; Ono, M. Three Acylated Glycosidic Acid Methyl Esters and Two Acylated Methyl Glycosides Generated from the Convolvulin Fraction of Seeds of Quamoclit pennata by Treatment with Indium(III) Chloride in Methanol. Chem. Pharm. Bull. 2013, 61, 952–961. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.Y.; Lu, Y.; Yin, H.; He, Y.; Li, J.L.; Chen, G.T. Arvensic Acids A-D, Novel Heptasaccharide Glycosidic Acids as the Alkaline Hydrolysis Products of Crude Resin Glycosides from Convolvulus arvensis. Fitoterapia 2018, 131, 209–214. [Google Scholar] [CrossRef]
- Akiyama, K.; Yamamoto, K.; Mineno, T.; Okawa, M.; Kinjo, J.; Yoshimitsu, H.; Nohara, T.; Ono, M. Five New Resin Glycoside Derivatives Isolated from the Convolvulin Fraction of Seeds of Quamoclit pennata after Treatment with Indium(III) Chloride in Methanol. Chem. Pharm. Bull. 2014, 62, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Bautista, E.; Fragoso-Serrano, M.; Pereda-Miranda, R. Jalapinoside, a Macrocyclic Bisdesmoside from the Resin Glycosides of Ipomea purga, as a Modulator of Multidrug Resistance in Human Cancer Cells. J. Nat. Prod. 2015, 78, 168–172. [Google Scholar] [CrossRef]
- Bautista, E.; Fragoso-Serrano, M.; Pereda-Miranda, R. Jalapinoside II, a Bisdesmoside Resin Glycoside, and Related Glycosidic Acids from the Officinal Jalap Root (Ipomoea purga). Phytochem. Lett. 2016, 17, 85–93. [Google Scholar] [CrossRef]
- Ono, M.; Akiyama, K.; Kishida, M.; Okawa, M.; Kinjo, J.; Yoshimitsu, H.; Miyahara, K. Two New Glycosidic Acids, Multifidinic Acids F and G, of the Ether-Insoluble Resin Glycoside (Convolvulin) from the Seeds of Quamoclit × multifida. J. Nat. Med. 2013, 67, 822–826. [Google Scholar] [CrossRef]
- Ono, M.; Akiyama, K.; Yamamoto, K.; Mineno, T.; Okawa, M.; Kinjo, J.; Miyashita, H.; Yoshimitsu, H.; Nohara, T. Four New Acylated Glycosidic Acid Methyl Esters Isolated from the Convolvulin Fraction of Seeds of Quamoclit pennata after Treatment with Indium(III) Chloride in Methanol. Chem. Pharm. Bull. 2014, 62, 830–835. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Imao, M.; Miyahara, K. Two New Glycosidic Acids, Quamoclinic Acids G and H, of the ResinGlycosides (Convolvulin) from the Seeds of Quamoclit pennata. Chem. Pharm. Bull. (Tokyo) 2010, 58, 1232–1235. [Google Scholar] [CrossRef] [Green Version]
- Rosas-Ramírez, D.; Escalante-Sánchez, E.; Pereda-Miranda, R. Batatins III-VI, Glycolipid Ester-Type Dimers from Ipomoea batatas. Phytochemistry 2011, 72, 773–780. [Google Scholar] [CrossRef]
- Bah, M.; Pereda-Miranda, R. Detailed FAB-Mass Spectrometry and High Resolution NMR Investigations of Tricolorins A-E, Individual Oligosaccharides from the Resins of Ipomoea tricolor (Convolvulaceae) 1. Tetrahedron 1996, 52, 13063–13080. [Google Scholar] [CrossRef]
- Noda, N.; Tsuji, K.; Kawasaki, T.; Miyahara, K.; Hanazono, H.; Yang, C.-R. A Novel Resin Glycoside, Merremin (Tuguajalapin X Dimer), from Merremia hungaiensis. Chem. Pharm. Bull. 1995, 43, 1061–1063. [Google Scholar] [CrossRef]
- Ono, M.; Kawasaki, T.; Miyahara, K. Resin Glycosides. V. Identification and Characterization of the Component Organic and Glycosidic Acids of the Ether-Soluble Crude Resin Glycosides (“Jalapin”) from Rhizoma Jalapae Braziliensis (Roots of Ipomoea operculata). Chem. Pharm. Bull. 1989, 37, 3209–3213. [Google Scholar] [CrossRef] [Green Version]
- Chérigo, L.; Pereda-Miranda, R. Resin Glycosides from the Flowers of Ipomoea murucoides. J. Nat. Prod. 2006, 69, 595–599. [Google Scholar] [CrossRef]
- Maria Gaspar, E.M. Soldanelline B The First Acylated Nonlinear Tetrasaccharide Macrolactone from the European Convolvulaceae Calystegia soldanella. Eur. J. Org. Chem. 2001, 2001, 369–371. [Google Scholar] [CrossRef]
- Zhu, D.; Chen, C.; Xia, Y.; Kong, L.Y.; Luo, J. A Purified Resin Glycoside Fraction from Pharbitidis Semen Induces Paraptosis by Activating Chloride Intracellular Channel-1 in Human Colon Cancer Cells. Integr. Cancer Ther. 2019, 18, 1534735418822120. [Google Scholar] [CrossRef]
- Li, J.H.; Pan, J.T.; Yin, Y.Q. Two Novel Resin Glycosides Isolated from Ipomoea cairica with α-Glucosidase Inhibitory Activity. Chin. J. Nat. Med. 2016, 14, 227–231. [Google Scholar] [CrossRef]
- León-Rivera, I.; Herrera-Ruiz, M.; Estrada-Soto, S.; Gutiérrez, M.D.C.; Martínez-Duncker, I.; Navarrete-Vázquez, G.; Rios, M.Y.; Aguilar, B.; Castillo-España, P.; Aguirre-Moreno, A. Sedative, Vasorelaxant, and Cytotoxic Effects of Convolvulin from Ipomoea tyrianthina. J. Ethnopharmacol. 2011, 135, 434–439. [Google Scholar] [CrossRef]
- León-Rivera, I.; Mirón-López, G.; Molina-Salinas, G.M.; Herrera-Ruiz, M.; Estrada-Soto, S.; Gutiérrez, M.D.C.; Alonso-Cortes, D.; Navarrete-Vázquez, G.; Ríos, M.Y.; Said-Fernández, S. Tyrianthinic Acids from Ipomoea Tyrianthina and Their Antimycobacterial Activity, Cytotoxicity, and Effects on the Central Nervous System. J. Nat. Prod. 2008, 71, 1686–1691. [Google Scholar] [CrossRef]
- Ono, M.; Fukuda, H.; Murata, H.; Miyahara, K. Resin Glycosides from the Leaves and Stems of Ipomoea Digitata. J. Nat. Med. 2009, 63, 176–180. [Google Scholar] [CrossRef]
- Çaliş, I.; Sezgin, Y.; Dönmez, A.A.; Rüedi, P.; Tasdemir, D. Crypthophilic Acids A, B, and C: Resin Glycosides from Aerial Parts of Scrophularia crypthophila. J. Nat. Prod. 2007, 70, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.-Q.; Huang, X.-F.; Kong, L.-Y.; Niwa, M. Three New Pentasaccharide Resin Glycosides from the Roots of Sweet Potato (Ipomoea batatas). Chem. Pharm. Bull. 2008, 56, 1670–1674. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Hao, X.; Liu, J.; Ding, J.; Fang, Y.; Gu, Q.; Zhu, W. Resin Glycoside Constituents of Ipomoea pes-caprae (Beach Morning Glory). J. Nat. Prod. 2008, 71, 1998–2003. [Google Scholar] [CrossRef]
- Escalante-Sánchez, E.; Rosas-Ramírez, D.; Linares, E.; Bye, R.; Pereda-Miranda, R. Batatinosides II-VI, Acylated Lipooligosaccharides from the Resin Glycosides of Sweet Potato. J. Agric. Food Chem. 2008, 56, 9423–9428. [Google Scholar] [CrossRef]
- Escobedo-Martínez, C.; Pereda-Miranda, R. Resin Glycosides from Ipomoea pes-caprae. J. Nat. Prod. 2007, 70, 974–978. [Google Scholar] [CrossRef]
- Bah, M.; Chérigo, L.; Cardoso Taketa, A.T.; Fragoso-Serrano, M.; Hammond, G.B.; Pereda-Miranda, R. Intrapilosins I-VII, Pentasaccharides from the Seeds of Ipomoea intrapilosa. J. Nat. Prod. 2007, 70, 1153–1157. [Google Scholar] [CrossRef]
- Noda, N.; Horiuchi, Y. The Resin Glycosides from the Sweet Potato (Ipomoea batatas L. LAM.). Chem. Pharm. Bull. 2008, 56, 1607–1610. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.Y.; Huang, J.S.; Zheng, S.S.; Zhu, K.; Yang, J.S. First Total Synthesis of the Proposed Structure of Batatin VI. Org. Lett. 2013, 15, 4154–4157. [Google Scholar] [CrossRef]
- Nawój, M.; Grobelny, A.; Mlynarski, J. Macrolide Core Synthesis of Calysolin IX Using an Intramolecular Glycosylation Approach. Eur. J. Org. Chem. 2020, 2020, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Yang, C.S.; Zeng, L.N.; Zhang, L.L. Synthesis of Key Macrolactone Structure of Resin Glycosides Using a Keck Macrolactonization Method. J. Asian Nat. Prod. Res. 2015, 17, 289–298. [Google Scholar] [CrossRef]
- Xie, L.; Zhu, S.Y.; Shen, X.Q.; He, L.L.; Yang, J.S. Total Synthesis of Batatoside L. J. Org. Chem. 2010, 75, 5764–5767. [Google Scholar] [CrossRef]
- Sun, J.; Fang, J.; Xiao, X.; Cai, L.; Zhao, X.; Zeng, J.; Wan, Q. Total Synthesis of Tricolorin A via interrupted Pummerer Reaction-Mediated Glycosylation and One-Pot Relay Glycosylation. Org. Biomol. Chem. 2020, 18, 3818–3822. [Google Scholar] [CrossRef]
- Fang, J.; Zeng, J.; Sun, J.; Zhang, S.; Xiao, X.; Lu, Z.; Meng, L.; Wan, Q. Total Syntheses of Resin Glycosides Murucoidins IV and V. Org. Lett. 2019, 21, 6213–6216. [Google Scholar] [CrossRef]
- Son, S.H.; Yanagiya, N.; Furukawa, J.I.; Sakairi, N. Intramolecular Glycosylation Approach toward Constructing the Macrocyclic Structure of Resin Glycosides. Synlett 2009, 2009, 2957–2960. [Google Scholar]
- Nagano, T.; Pospíšil, J.; Chollet, G.; Schulthoff, S.; Hickmann, V.; Moulin, E.; Herrmann, J.; Müller, R.; Fürstner, A. Total Synthesis and Biological Evaluation of the Cytotoxic Resin Glycosides Ipomoeassin A-F and Analogues. Chem. - A Eur. J. 2009, 15, 9697–9706. [Google Scholar] [CrossRef] [Green Version]
- Noda, N.; Kobayashi, H.; Miyahara, K.; Kawasaki, T. Resin Glycosides. III. Isolation and Structural Study of the Genuine Resin Glycosides, Muricatins I-VI, from the Seeds of Ipomoea Muricata. Chem. Pharm. Bull. 1988, 36, 920–929. [Google Scholar] [CrossRef]















































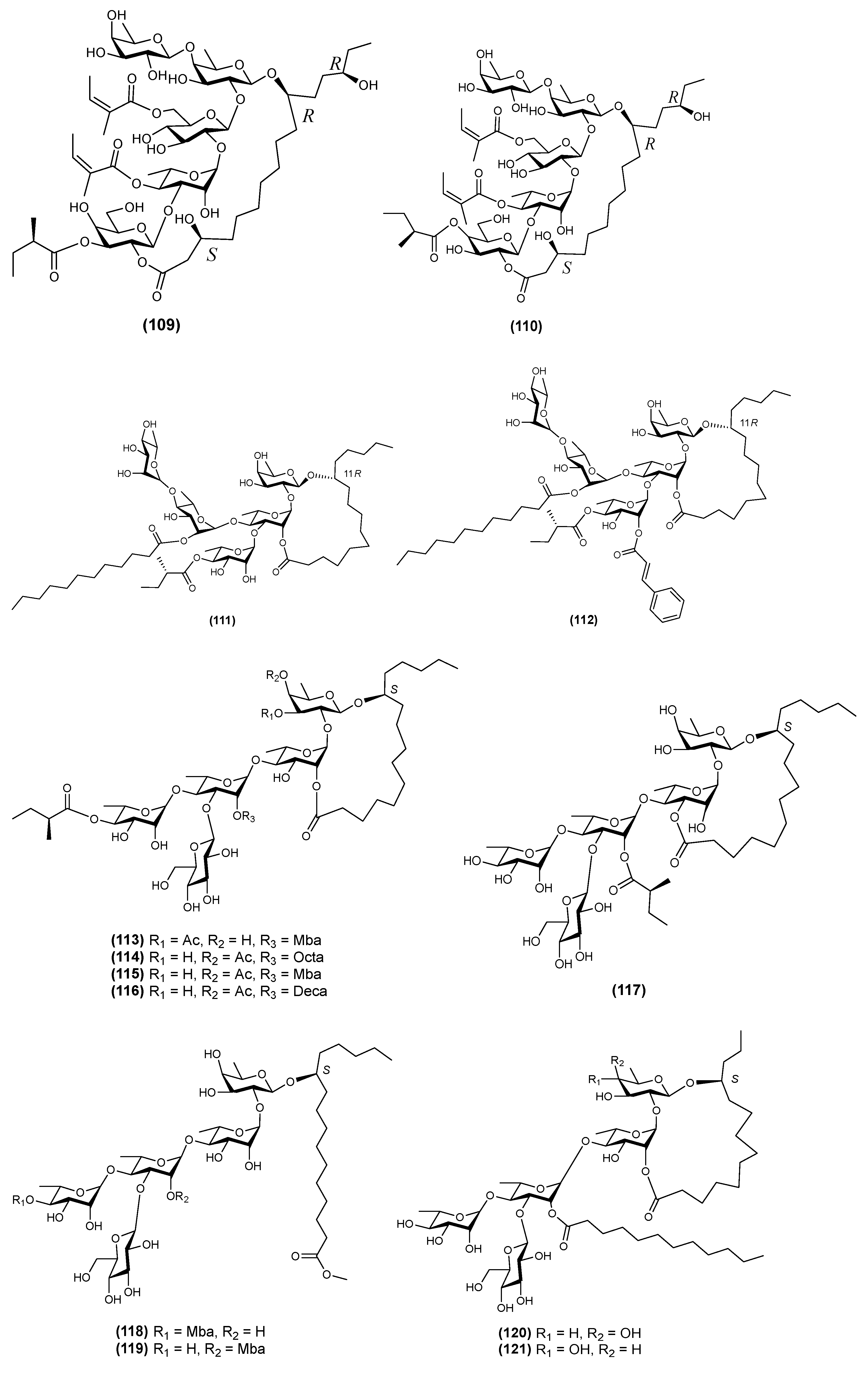
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
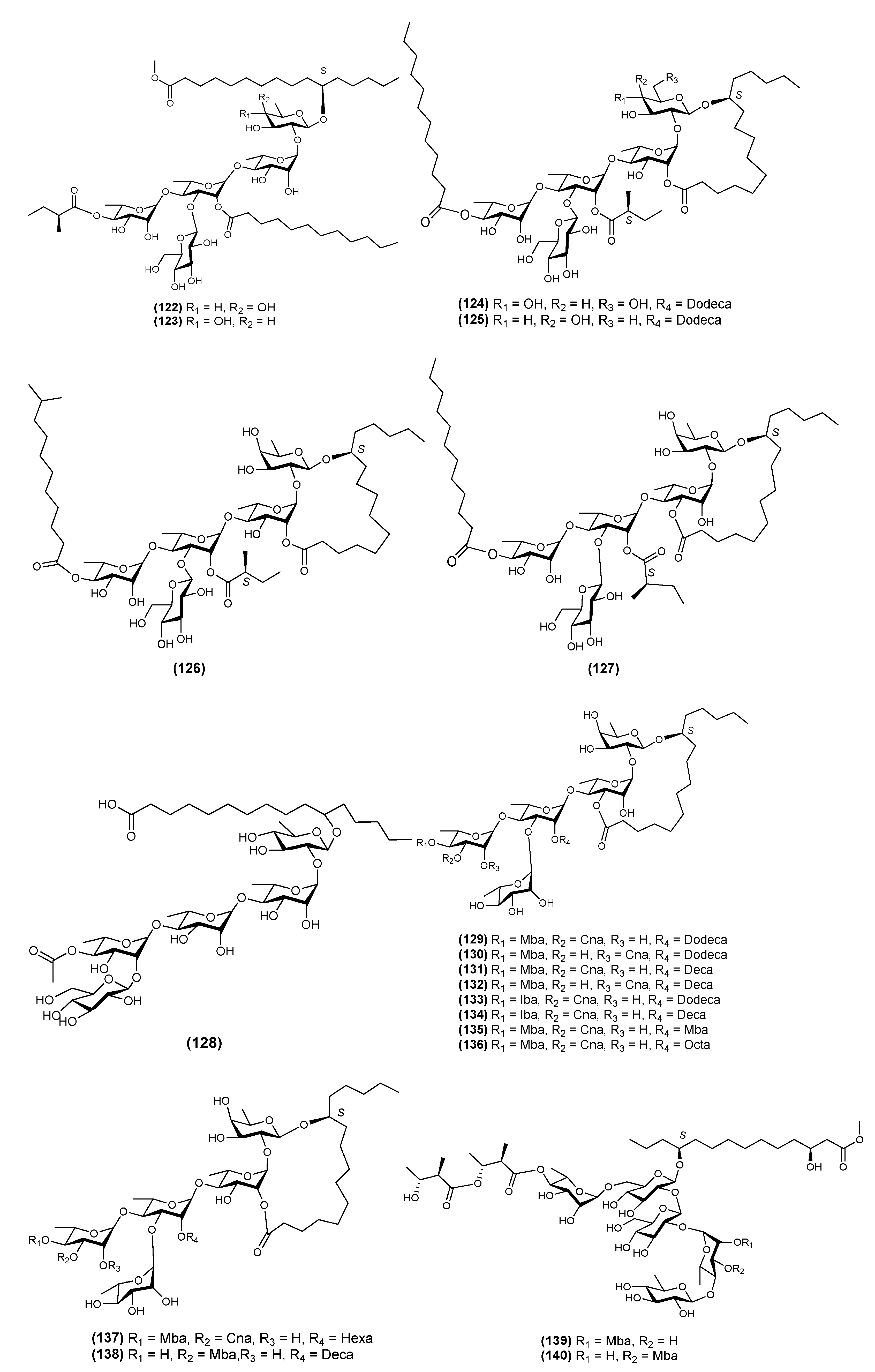{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
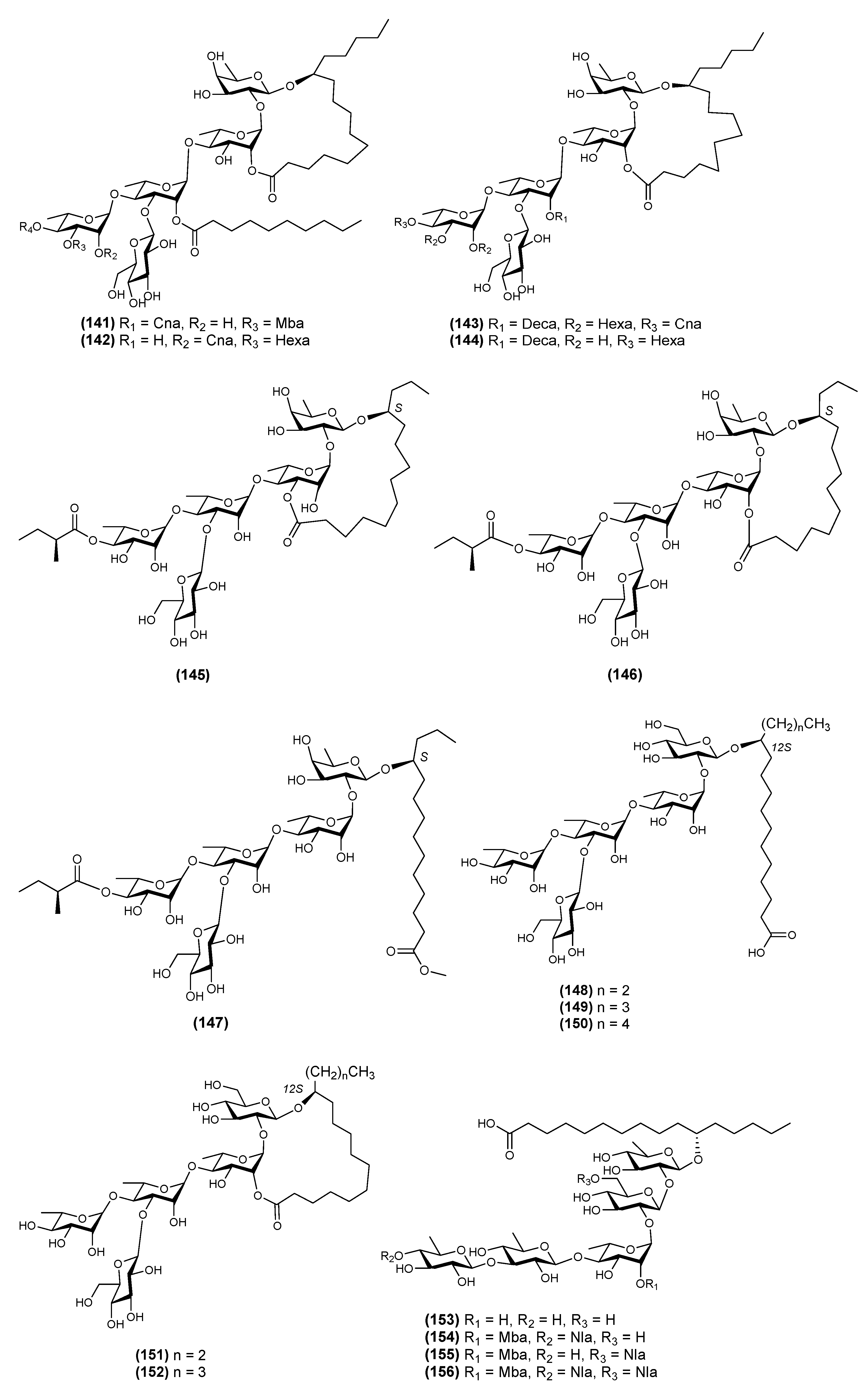
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
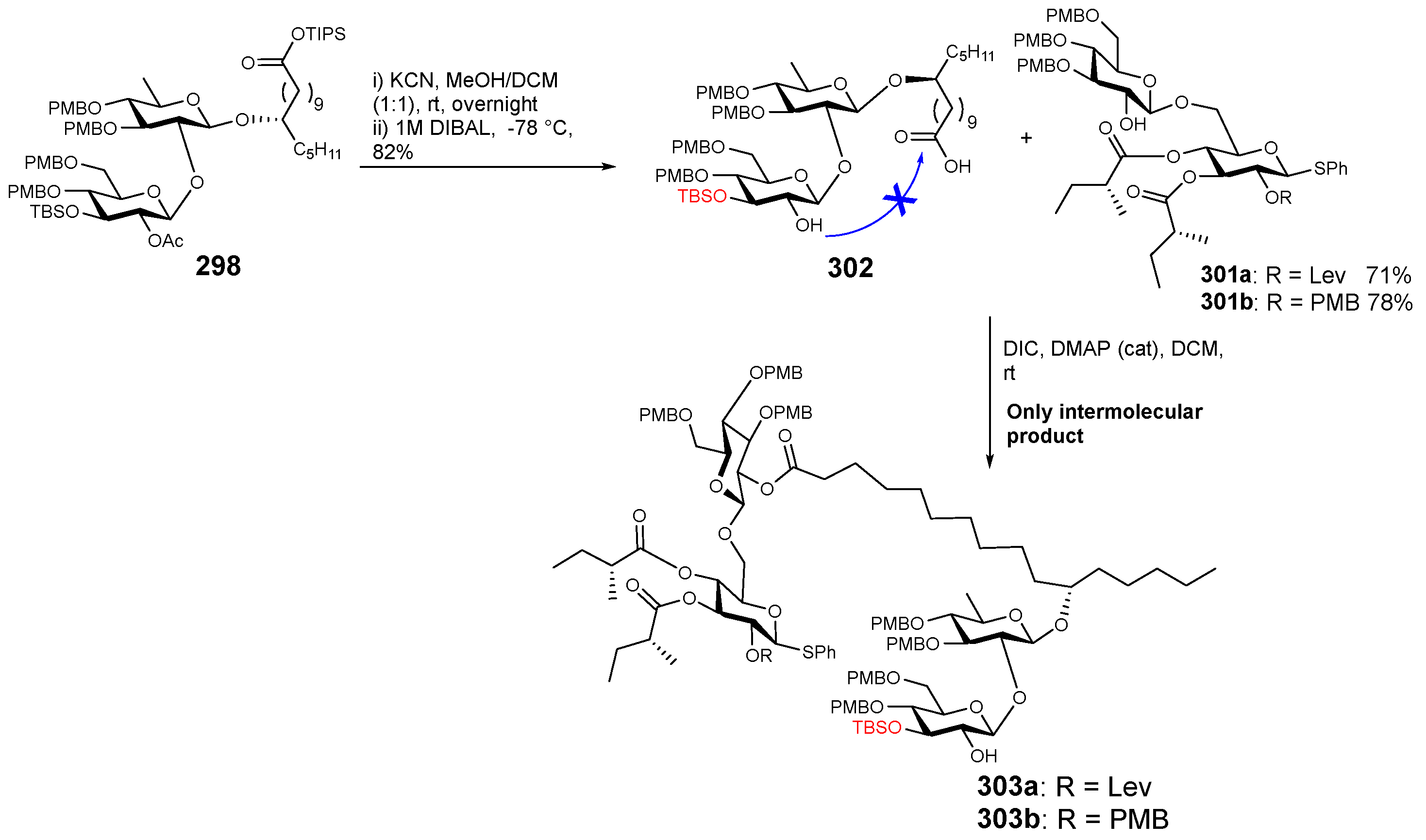{kind=link}
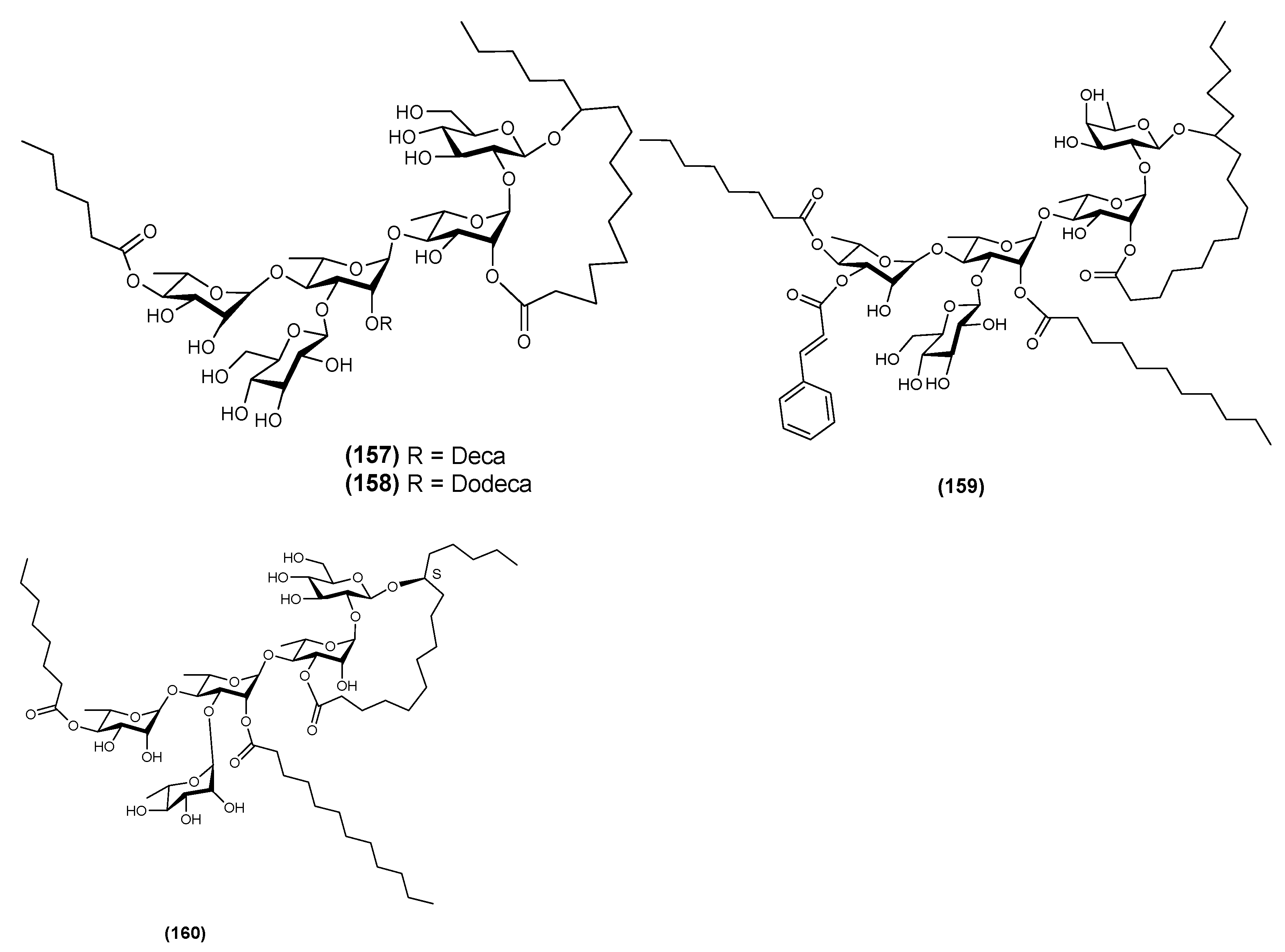
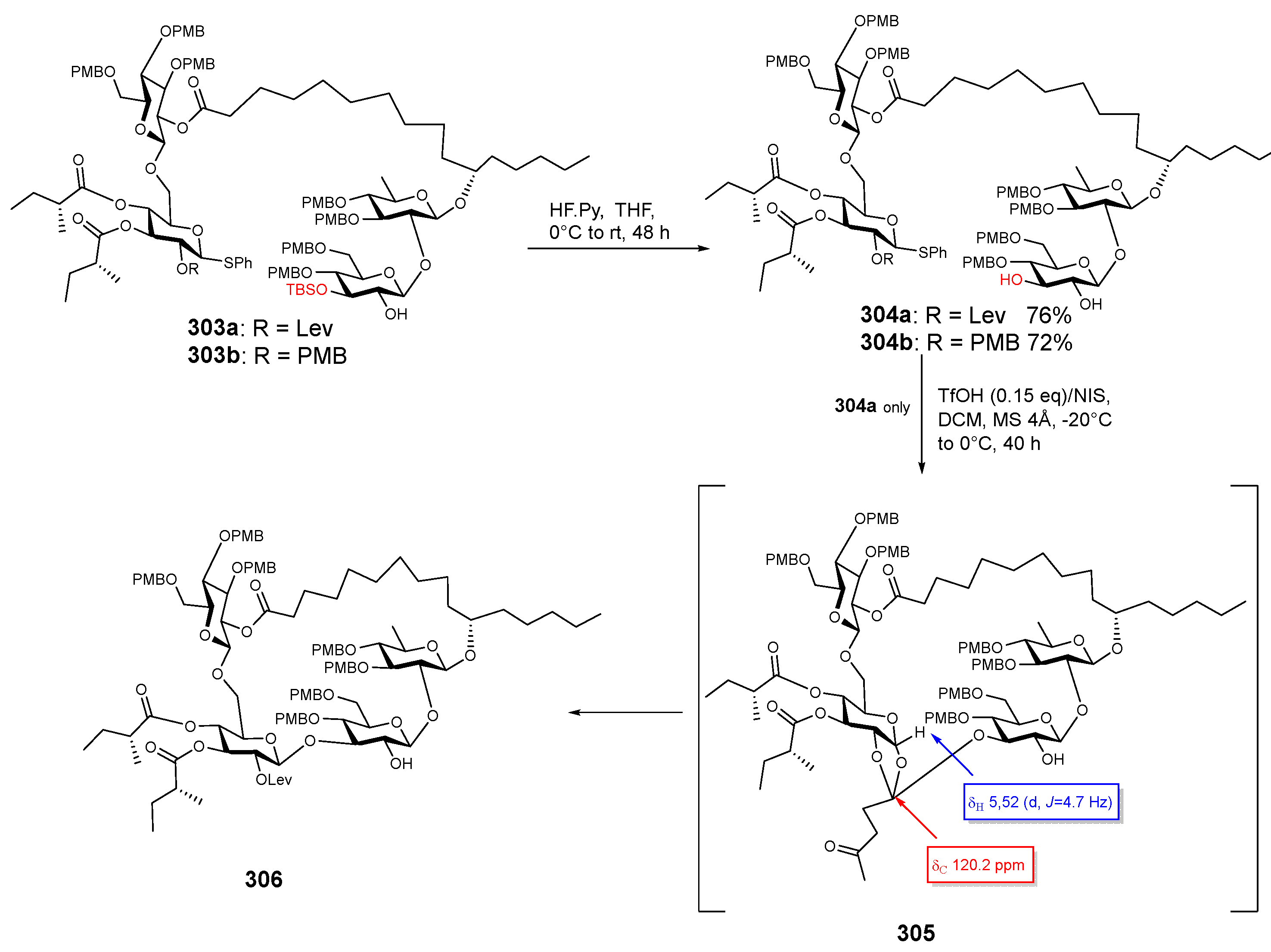{kind=link}
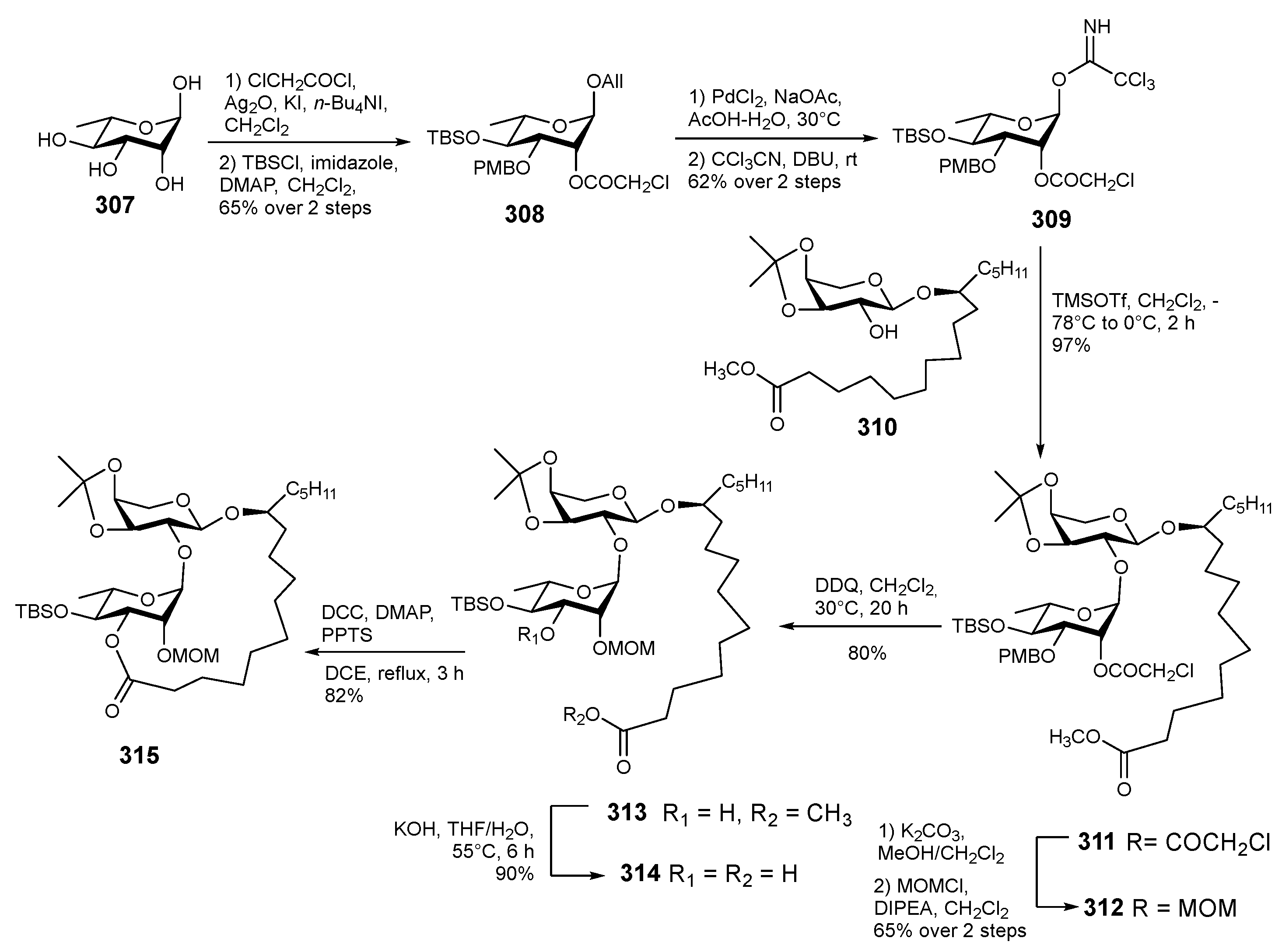{kind=link}
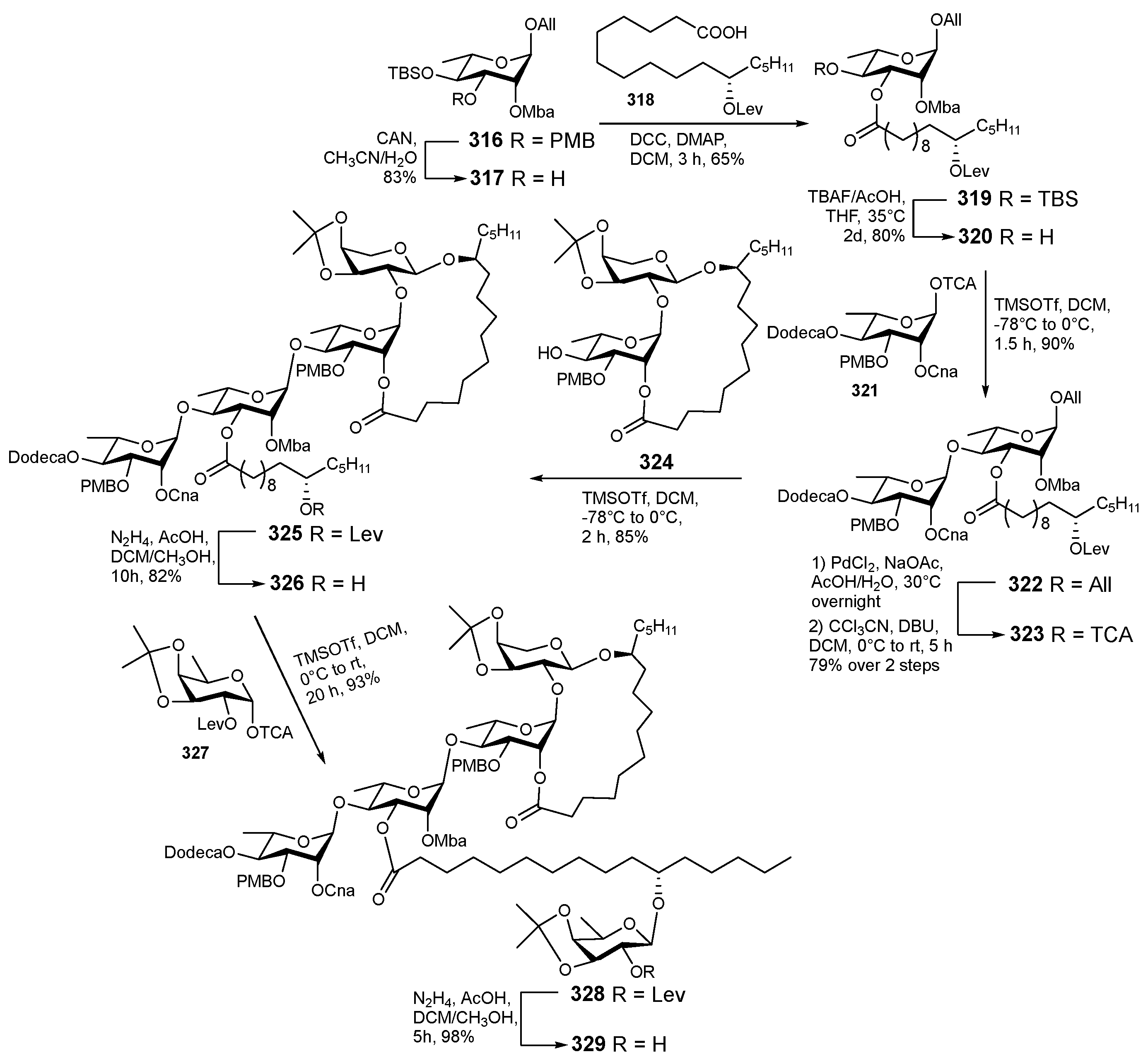{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Type | Sources | Refs. |
|---|---|---|---|
| Monosaccharides | |||
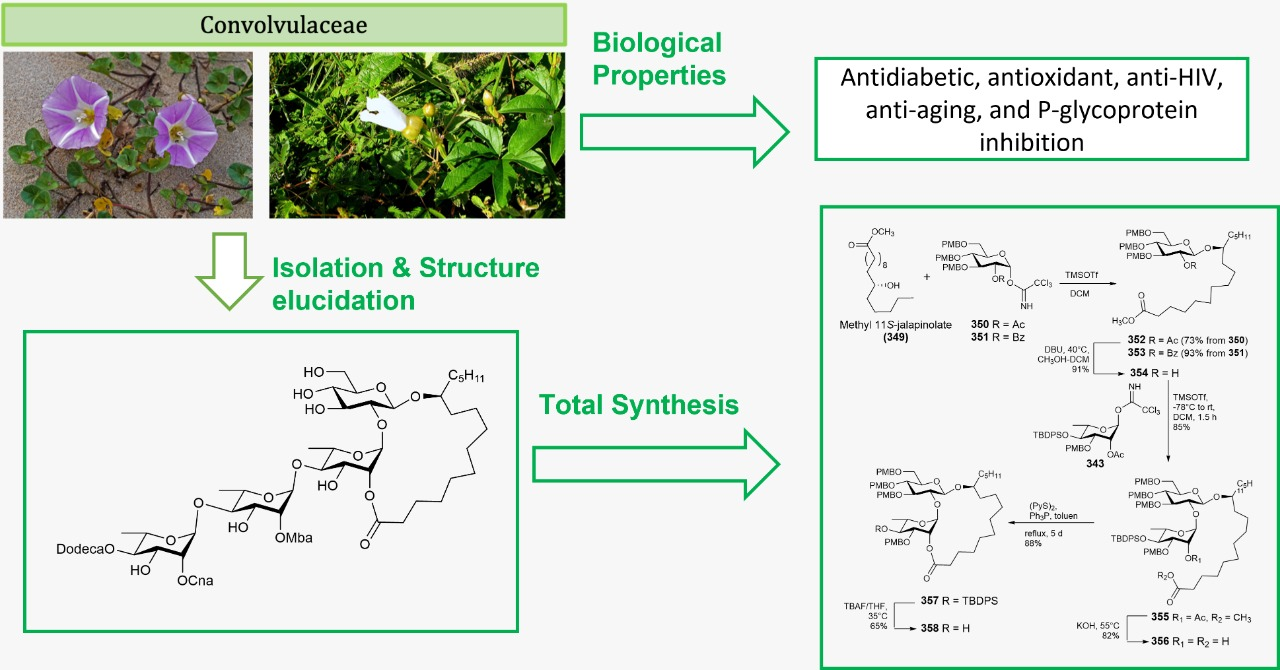
| 1 | Maltifidinic acid E (1) | Quamoclit × multifida | [26] |
| 2 | Quamoclinic acid B (2) | Quamoclit pennata | [27] |
| 3 | Operculinic acid K (3) | Operculina macrocarpa | [28] |
| Trisaccharides | |||
| 1 | Cuses 5 (4) | Cuscuta chinensis | [4] |
| 2 | Cuses 6 (5) | Cuscuta chinensis | [4] |
| 3 | Cuses 7 (6) | Cuscuta chinensis | [4] |
| 4 | Cuses 3 (7) | Cuscuta chinensis | [4] |
| 5 | Cuses 4 (8) | Cuscuta chinensis | [4] |
| 6 | Dichondrin C (9) | Dichondra repens | [3] |
| 7 | Poranic acid A (10) | Porana duclouxii | [5] |
| 8 | Poranic acid B (11) | Porana duclouxii | [5] |
| 9 | Poranaside A (12) | Porana duclouxii | [5] |
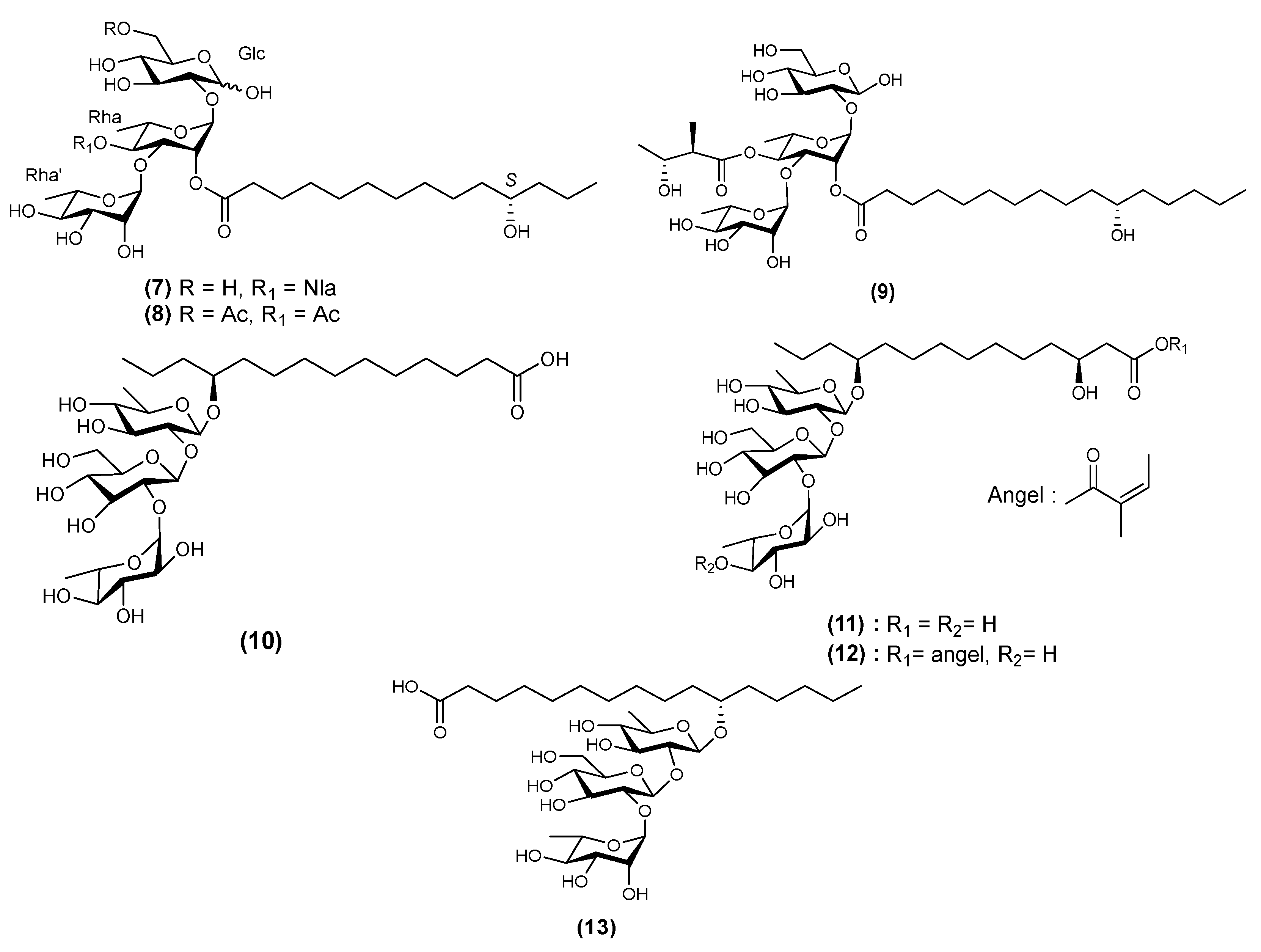
| 10 | Stansoic acid A (13) | Ipomoea stans | [16] |
| Tetrasaccharides | |||
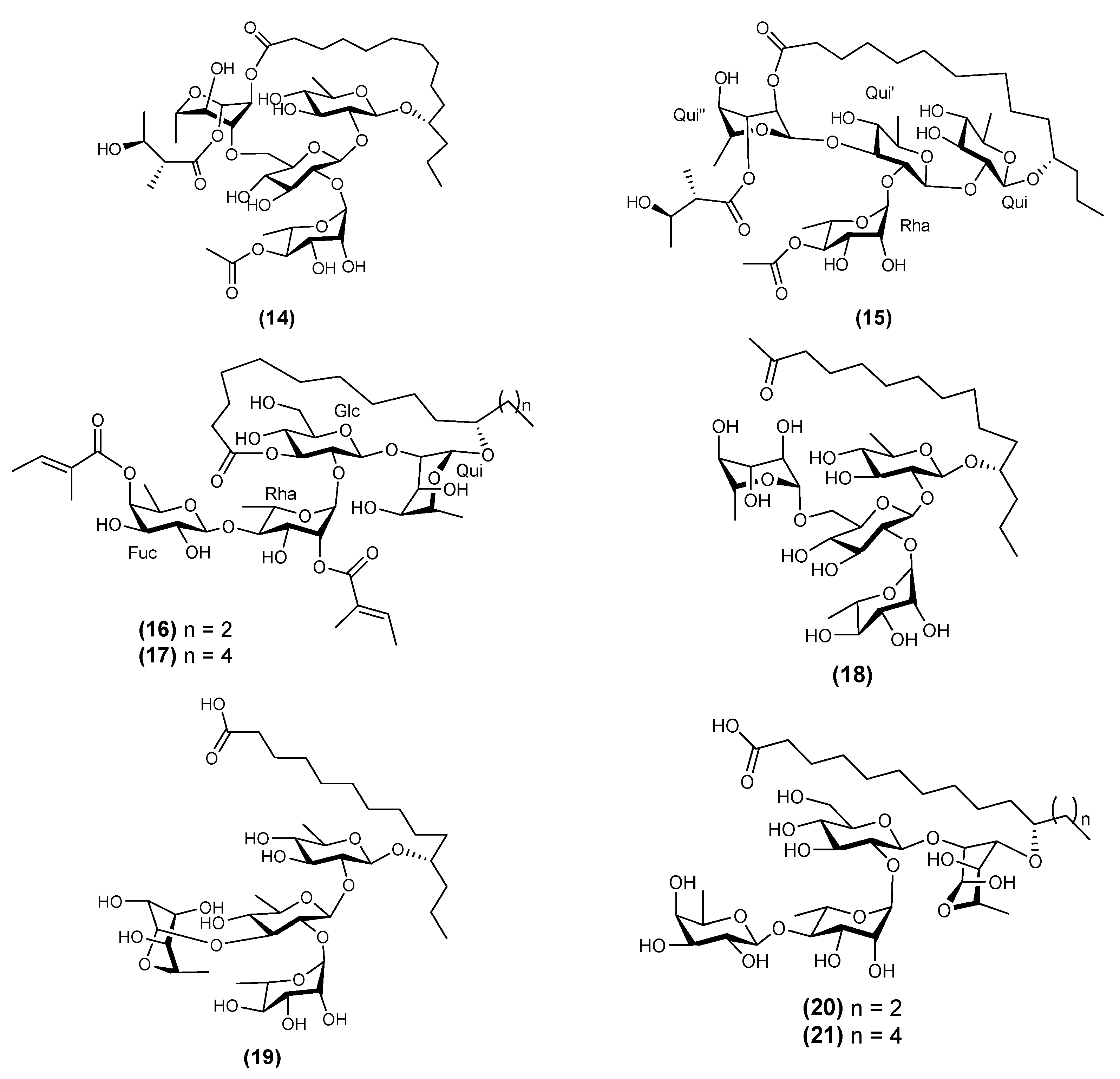
| 1 | Albinoside VI (14) | Ipomoea alba | [7] |
| 2 | Albinoside VII (15) | Ipomoea alba | [7] |
| 3 | Albinoside VIII (16) | Ipomoea alba | [7] |
| 4 | Albinoside IX (17) | Ipomoea alba | [7] |
| 5 | Albinosinic acid D (18) | Ipomoea alba | [7] |
| 6 | Albinosinic acid E (19)) | Ipomoea alba | [7] |
| 7 | Albinosinic acid F (20) | Ipomoea alba | [7] |
| 8 | Albinosinic acid G (21) | Ipomoea alba | [7] |
| 9 | Aquaterin XI (22) | Ipomoea aquatica | [29] |
| 10 | Calonyctin E (23) | Ipomoea muricata | [23] |
| 11 | Calonyctin F (24) | Ipomoea muricata | [23] |
| 12 | Calonyctin G (25) | Ipomoea muricata | [23] |
| 13 | Calonyctin H (26) | Ipomoea muricata | [23] |
| 14 | Calonyctin I (27) | Ipomoea muricata | [23] |
| 15 | Calonyctin J (28) | Ipomoea muricata | [23] |
| 16 | Calysolin I (29) | Calystegia soldanella | [30] |
| 17 | Calysolin X (30) | Calystegia soldanella | [31] |
| 18 | Dichondrins A (31) | Dichondra repens | [3] |
| 19 | Dichondrins B (32) | Dichondra repens | [3] |
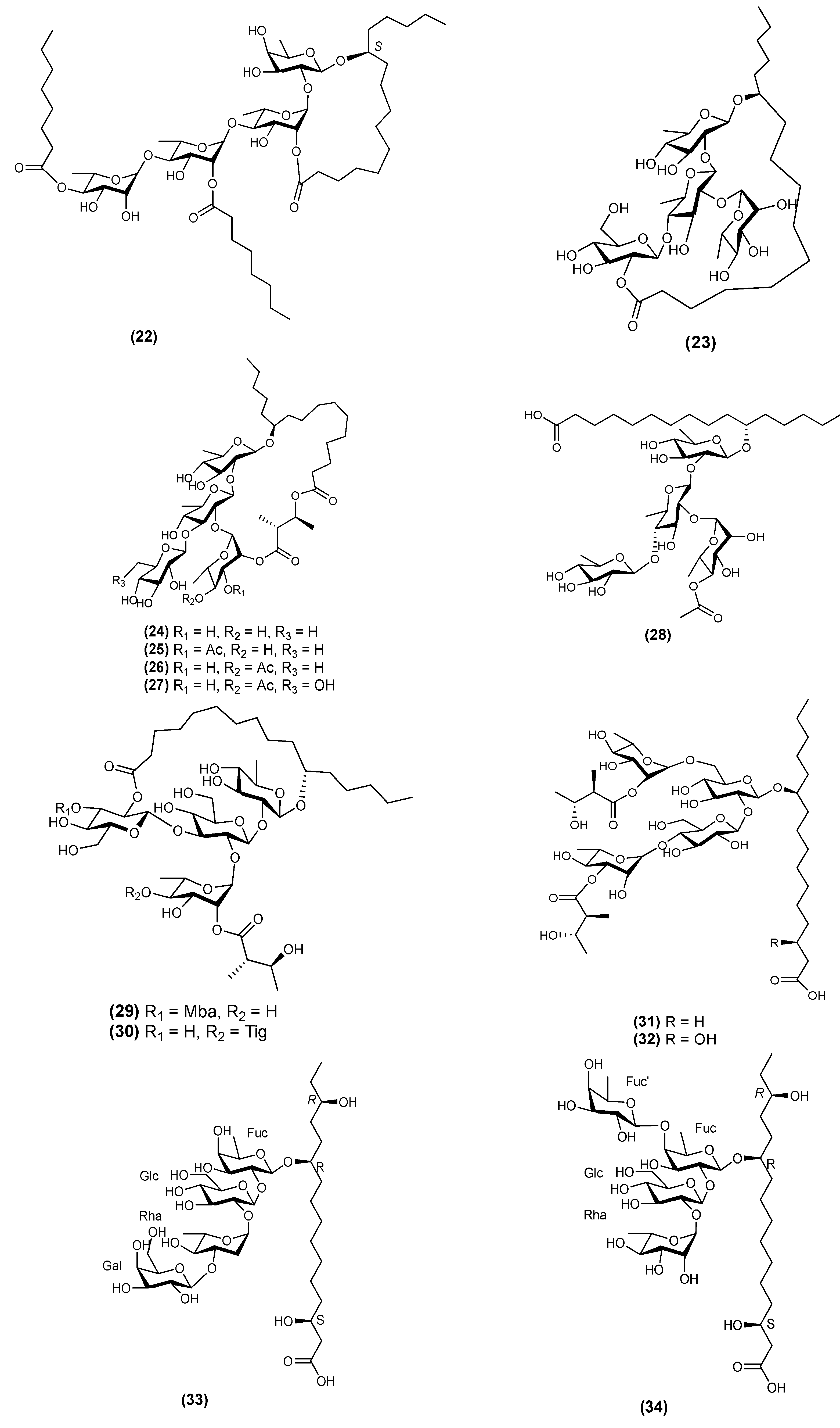
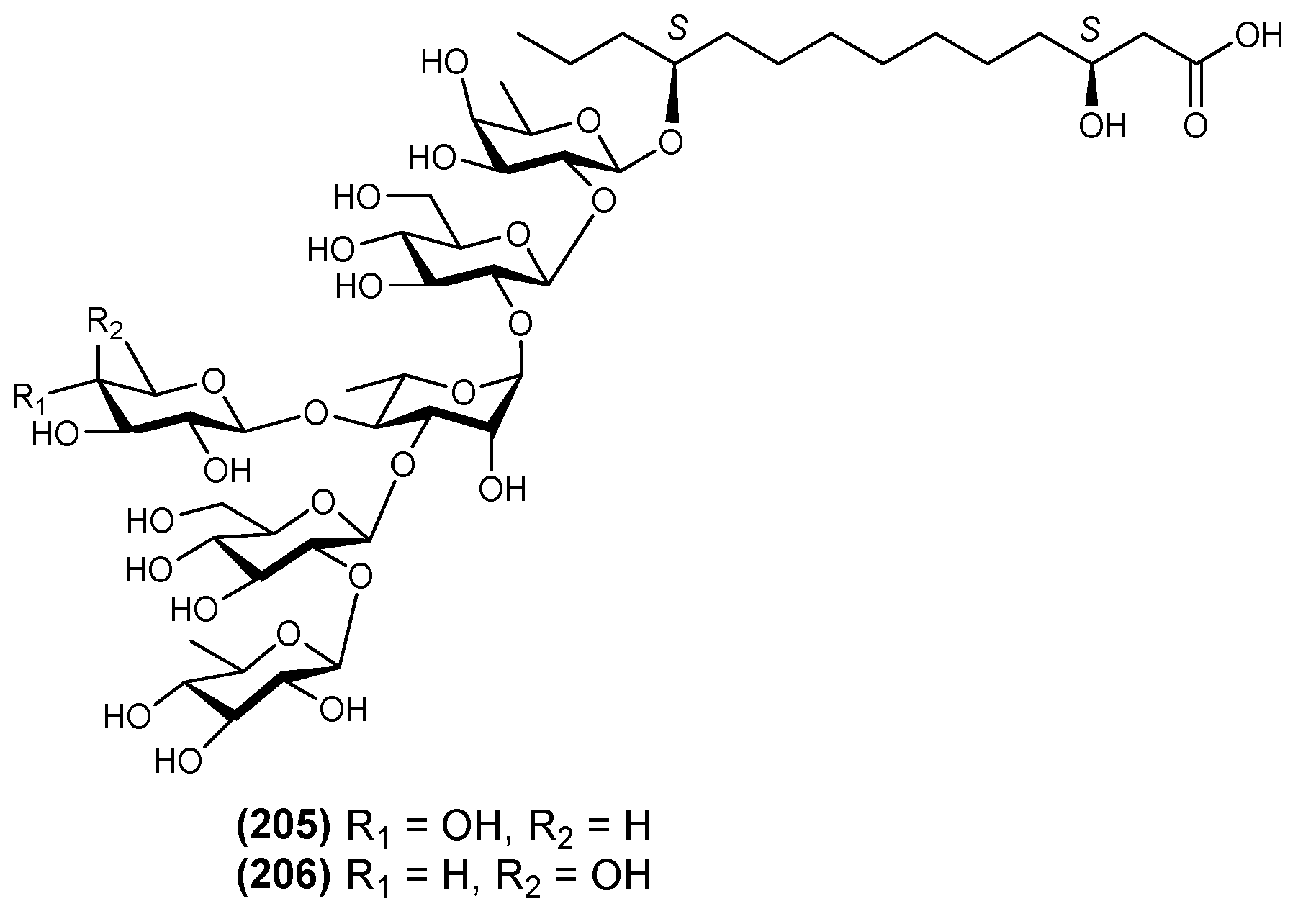
| 20 | Evolvulic acid B (33) | Evolvulus alsinoides | [32] |
| 21 | Evolvulic acid C (34) | Evolvulus alsinoides | [18] |
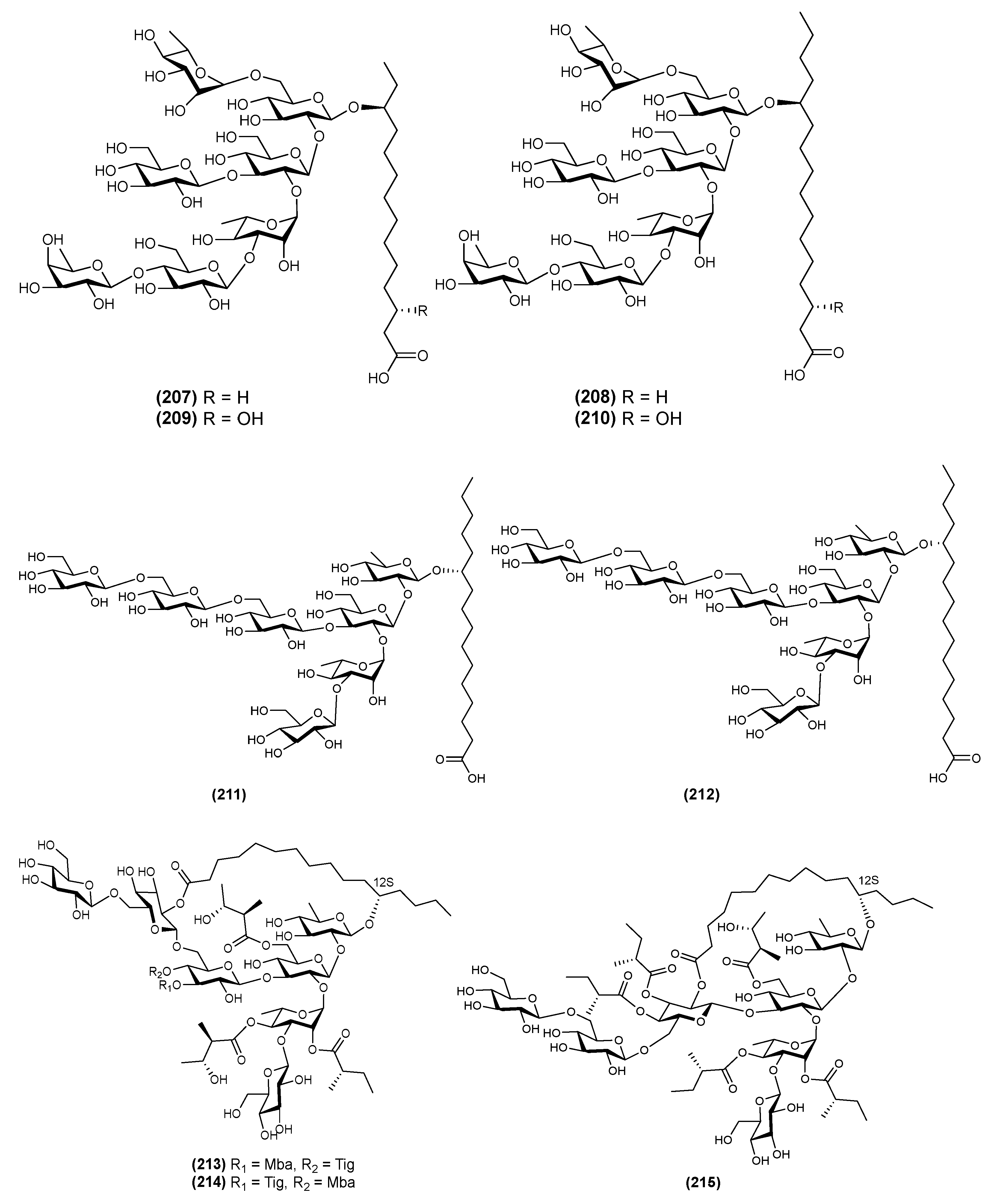
| 22 | Ipomotaoside A (35) | Ipomoea batatas | [18] |
| 23 | Ipomotaoside B (36) | Ipomoea batatas | [18] |
| 24 | Ipomotaoside C (37) | Ipomoea batatas | [18] |
| 25 | Ipomotaoside D (38) | Ipomoea batatas | [18] |
| 26 | Muricatic acid D (39) | Ipomoea muricata | [33] |
| 27 | Muricatin IX (40) | Ipomoea muricata | [34] |
| 28 | Muricatin X (41) | Ipomoea muricata | [33] |
| 29 | Muricatin XI (42) | Ipomoea muricata | [33] |
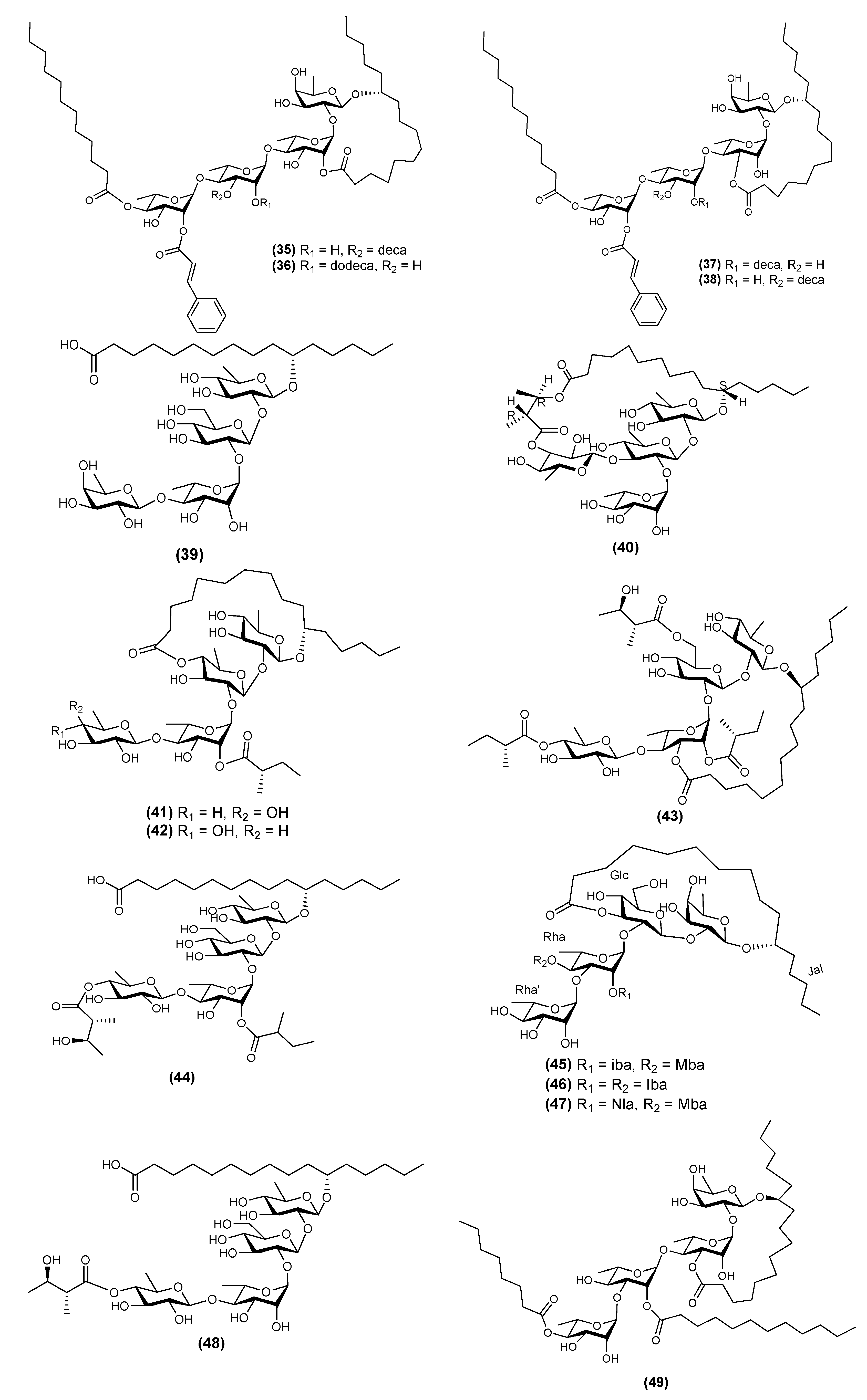
| 30 | Stansin 6 (43) | Ipomoea stans | [10,35] |
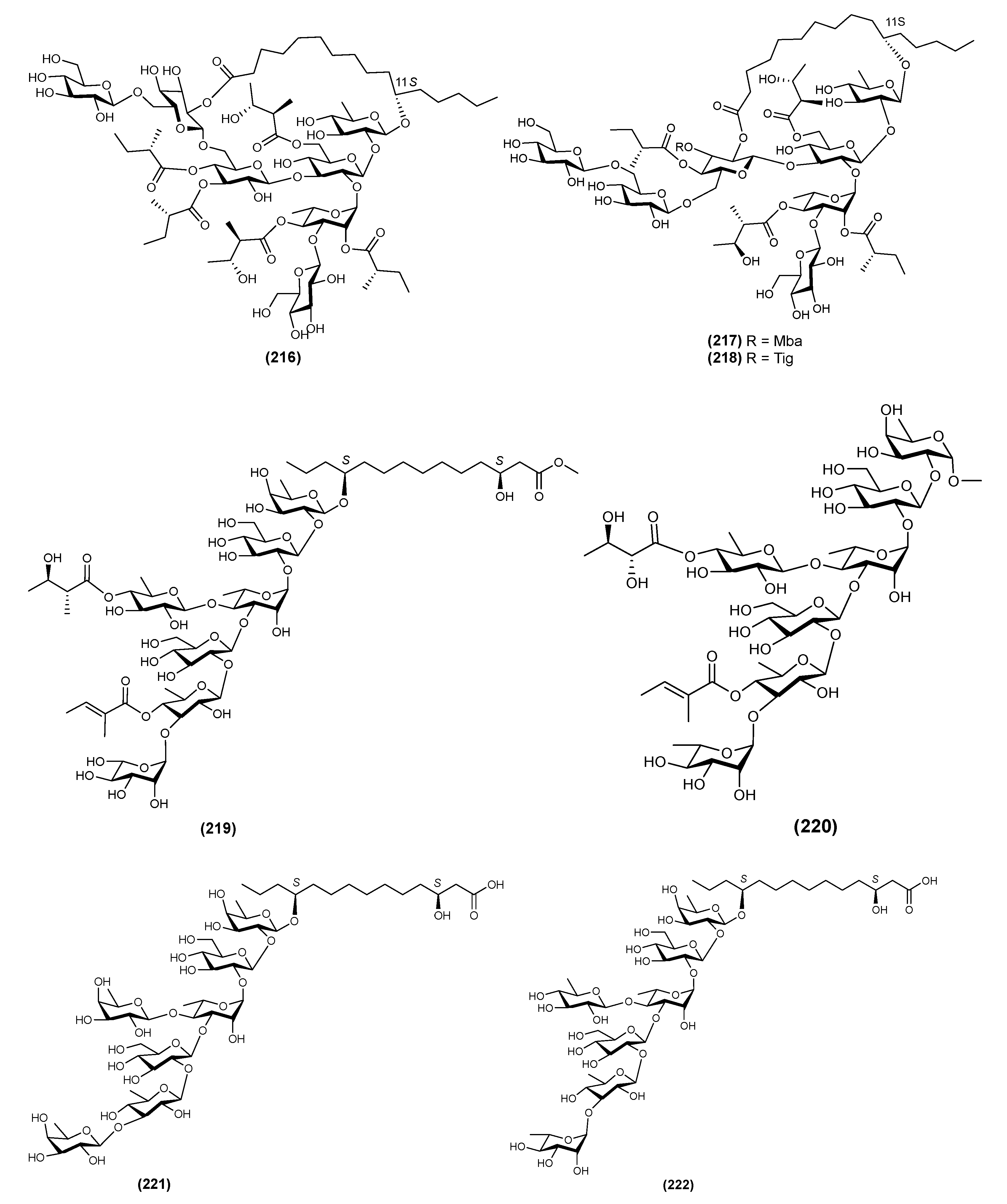
| 31 | Stansinic acid I (44) | Ipomoea stans | [16] |
| 32 | Tricolorin K (45) | Ipomoea tricolor | [12] |
| 33 | Tricolorin L (46) | Ipomoea tricolor | [12] |
| 34 | Tricolorin M (48) | Ipomoea tricolor | [12] |
| 35 | Tyrianthinic acids VI (48) | Ipomoea tyrianthina | [17] |
| 36 | Wolcottinoside I (49) | Ipomoea wolcottiana | [24] |
| Pentasaccharides | |||
| 1 | Acutacoside A (50) | Argyreia acuta | [8] |
| 2 | Acutacoside B (51) | Argyreia acuta | [8] |
| 3 | Acutacoside C (52) | Argyreia acuta | [36] |
| 4 | Acutacoside D (53) | Argyreia acuta | [36] |
| 5 | Acutacoside E (54) | Argyreia acuta | [36] |
| 6 | Acutacoside F (55) | Argyreia acuta | [37] |
| 7 | Acutacoside G (56) | Argyreia acuta | [37] |
| 8 | Acutacoside H (57) | Argyreia acuta | [37] |
| 9 | Acutacoside I (58) | Argyreia acuta | [37] |
| 10 | Albinoside I (59) | Ipomoea alba | [38] |
| 11 | Albinoside II (60) | Ipomoea alba | [38] |
| 12 | Albinoside III (61) | Ipomoea alba | [38] |
| 13 | Albinoside IV (62) | Ipomoea alba | [7] |
| 14 | Albinoside V (63) | Ipomoea alba | [7] |
| 15 | Aquaterin I (64) | Ipomoea aquatica | [29] |
| 16 | Aquaterin II (65) | Ipomoea aquatica | [29] |
| 17 | Aquaterin III (66) | Ipomoea aquatica | [29] |
| 18 | Aquaterin IV (67) | Ipomoea aquatica | [29] |
| 19 | Aquaterin V (68) | Ipomoea aquatica | [29] |
| 20 | Aquaterin VI (69) | Ipomoea aquatica | [29] |
| 21 | Aquaterin VII (70) | Ipomoea aquatica | [29] |
| 22 | Aquaterin VIII (71) | Ipomoea aquatica | [29] |
| 23 | Aquaterin IX (72) | Ipomoea aquatica | [29] |
| 24 | Aquaterin X (73) | Ipomoea aquatica | [29] |
| 25 | Aquaterin XII (74) | Ipomoea aquatica | [39] |
| 26 | Aquaterin XIII (75) | Ipomoea aquatica | [39] |
| 27 | Aquaterin XIV (76) | Ipomoea aquatica | [39] |
| 28 | Aquaterin XV (77) | Ipomoea aquatica | [39] |
| 29 | Aquaterin XVI (78) | Ipomoea aquatica | [39] |
| 30 | Aquaterin XVII (79) | Ipomoea aquatica | [39] |
| 31 | Aquaterin XVIII (80) | Ipomoea aquatica | [39] |
| 32 | Aquaterin XIX (81) | Ipomoea aquatica | [39] |
| 33 | Arvensic acid K (82) | Convolvulus arvensis | [40] |
| 34 | Arvensic acid L (83) | Convolvulus arvensis | [40] |
| 35 | Batatinoside VII (84) | Ipomoea batatas | [41] |
| 36 | Batatinoside VIII (85) | Ipomoea batatas | [41] |
| 37 | Batatinoside IX (86) | Ipomoea batatas | [41] |
| 38 | Cairicoside A (87) | Ipomoea cairica | [42] |
| 39 | Cairicoside B (88) | Ipomoea cairica | [42] |
| 40 | Cairicoside C (89) | Ipomoea cairica | [42] |
| 41 | Cairicoside D (90) | Ipomoea cairica | [42] |
| 42 | Cairicoside E (91) | Ipomoea cairica | [42] |
| 43 | Cairicoside F (92) | Ipomoea cairica | [42] |
| 44 | Cairicoside I (93) | Ipomoea cairica | [43] |
| 45 | Cairicoside II (94) | Ipomoea cairica | [43] |
| 46 | Cairicoside III (95) | Ipomoea cairica | [43] |
| 47 | Cairicoside IV (96) | Ipomoea cairica | [43] |
| 48 | Calonyctin B (97) | Ipomoea muricata | [23] |
| 49 | Calonyctin C (98) | Ipomoea muricata | [23] |
| 50 | Calonyctin D (99) | Ipomoea muricata | [23] |
| 51 | Calyhedic acid A (100) | Calystegia hederacea | [44] |
| 52 | Calysolic acid A (101) | Calystegia soldanella | [45] |
| 53 | Calysolic acid B (102) | Calystegia soldanella | [45] |
| 54 | Calysolin II (103) | Calystegia soldanella | [30] |
| 55 | Calysolin III (104) | Calystegia soldanella | [30] |
| 56 | Calysolin V (105) | Calystegia soldanella | [46] |
| 57 | Calysolin VI (106) | Calystegia soldanella | [46] |
| 58 | Evolvulic acid A (107) | Evolvulus alsinoides | [47] |
| 59 | Evolvulin I (108) | Evolvulus alsinoides | [47] |
| 60 | Evolvulin II (109) | Evolvulus alsinoides | [47] |
| 61 | Evolvulin III (110) | Evolvulus alsinoides | [48] |
| 62 | Ipomeolide A (111) | Ipomoea pes-caprae | [49] |
| 63 | Ipomeolide B (112) | Ipomoea pes-caprae | [49] |
| 64 | Merremin A (113) | Merremia hederacea | [50] |
| 65 | Merremin B (114) | Merremia hederacea | [50] |
| 66 | Merremin C (115) | Merremia hederacea | [50] |
| 67 | Merremin D (116) | Merremia hederacea | [50] |
| 68 | Merremin E (117) | Merremia hederacea | [50] |
| 69 | Merremin F (118) | Merremia hederacea | [50] |
| 70 | Merremin G (119) | Merremia hederacea | [50] |
| 71 | Multifidin III (120) | Quacmoclit × multifida | [51] |
| 72 | Multifidin IV (121) | Quacmoclit × multifida | [51] |
| 73 | Multifidin V (122) | Quacmoclit × multifida | [51] |
| 74 | Multifidin VI (123) | Quacmoclit × multifida | [51] |
| 75 | Murasakimasarin I (124) | Ipomoea batatas | [52] |
| 76 | Murasakimasarin II (125) | Ipomoea batatas | [52] |
| 77 | Murasakimasarin III (126) | Ipomoea batatas | [52] |
| 78 | Murasakimasarin IV (127) | Ipomoea batatas | [52] |
| 79 | Murucinic acid II (128) | Ipomoea murucoides | [16] |
| 80 | Pescaprein XXI (129) | Ipomoea pes-caprae | [53] |
| 81 | Pescaprein XXII (130) | Ipomoea pes-caprae | [53] |
| 82 | Pescaprein XXIII (131) | Ipomoea pes-caprae | [53] |
| 83 | Pescaprein XXIV (132) | Ipomoea pes-caprae | [53] |
| 84 | Pescaprein XXV (133) | Ipomoea pes-caprae | [53] |
| 85 | Pescaprein XXVI (134) | Ipomoea pes-caprae | [53] |
| 86 | Pescaprein XXVII (135) | Ipomoea pes-caprae | [53] |
| 87 | Pescaprein XXVIII (136) | Ipomoea pes-caprae | [53] |
| 88 | Pescaprein XXIX (137) | Ipomoea pes-caprae | [53] |
| 89 | Pescaprein XXX (138) | Ipomoea pes-caprae | [53] |
| 90 | PM6 (139) | Pharbitis nil | [54] |
| 91 | PM7 (140) | Pharbitis nil | [54] |
| 92 | Purginoside I (141) | Ipomoea purga | [9] |
| 93 | Purginoside II (142) | Ipomoea purga | [9] |
| 94 | Purginosides III (143) | Ipomoea purga | [6] |
| 95 | Purginosides IV (144) | Ipomoea purga | [6] |
| 96 | Quamoclin V (145) | Quamoclit pennata | [55] |
| 97 | Quamoclin VI (146) | Quamoclit pennata | [55] |
| 98 | Quamoclin VII (147) | Quamoclit pennata | [55] |
| 99 | Turpethic acids A (148) | Operculina turpethum | [56] |
| 100 | Turpethic acids B (149) | Operculina turpethum | [56] |
| 101 | Turpethic acids C (150) | Operculina turpethum | [56] |
| 102 | Turpethoside A (151) | Operculina turpethum | [56] |
| 103 | Turpethoside B (152) | Operculina turpethum | [56] |
| 104 | Tyrianthinoic acid (153) | Ipomoea tyrianthina | [17] |
| 105 | Tyrianthinic acid III (154) | Ipomoea tyrianthina | [17] |
| 106 | Tyrianthinic acid IV (155) | Ipomoea tyrianthina | [17] |
| 107 | Tyrianthinic acid V (156) | Ipomoea tyrianthina | [17] |
| 108 | Wolcottinoside II (157) | Ipomoea wolcottiana | [24] |
| 109 | Wolcottinoside III (158) | Ipomoea wolcottiana | [24] |
| 110 | Wolcottinoside IV (159) | Ipomoea wolcottiana | [24] |
| 111 | A new resin glycoside (160) | Ipomoea maxima | [57] |
| Hexasaccharides | |||
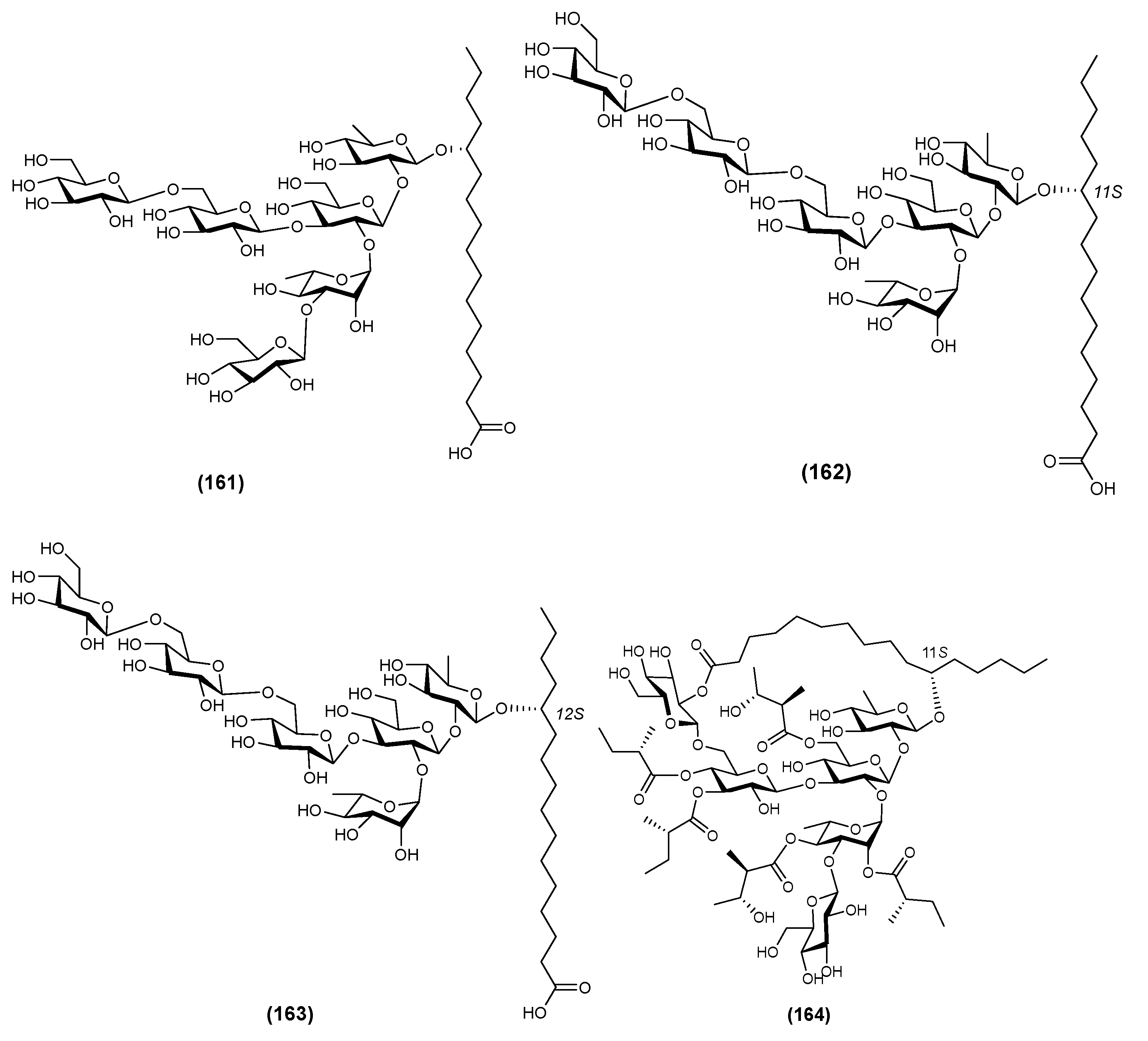
| 1 | Calyhedic acid B (161) | Calystegia hederacea | [44] |
| 2 | Calyhedic acids E (162) | Calystegia hederacea | [58] |
| 3 | Calyhedic acids F (163) | Calystegia hederacea | [58] |
| 4 | Calyhedin I (164) | Calystegia hederacea | [59] |
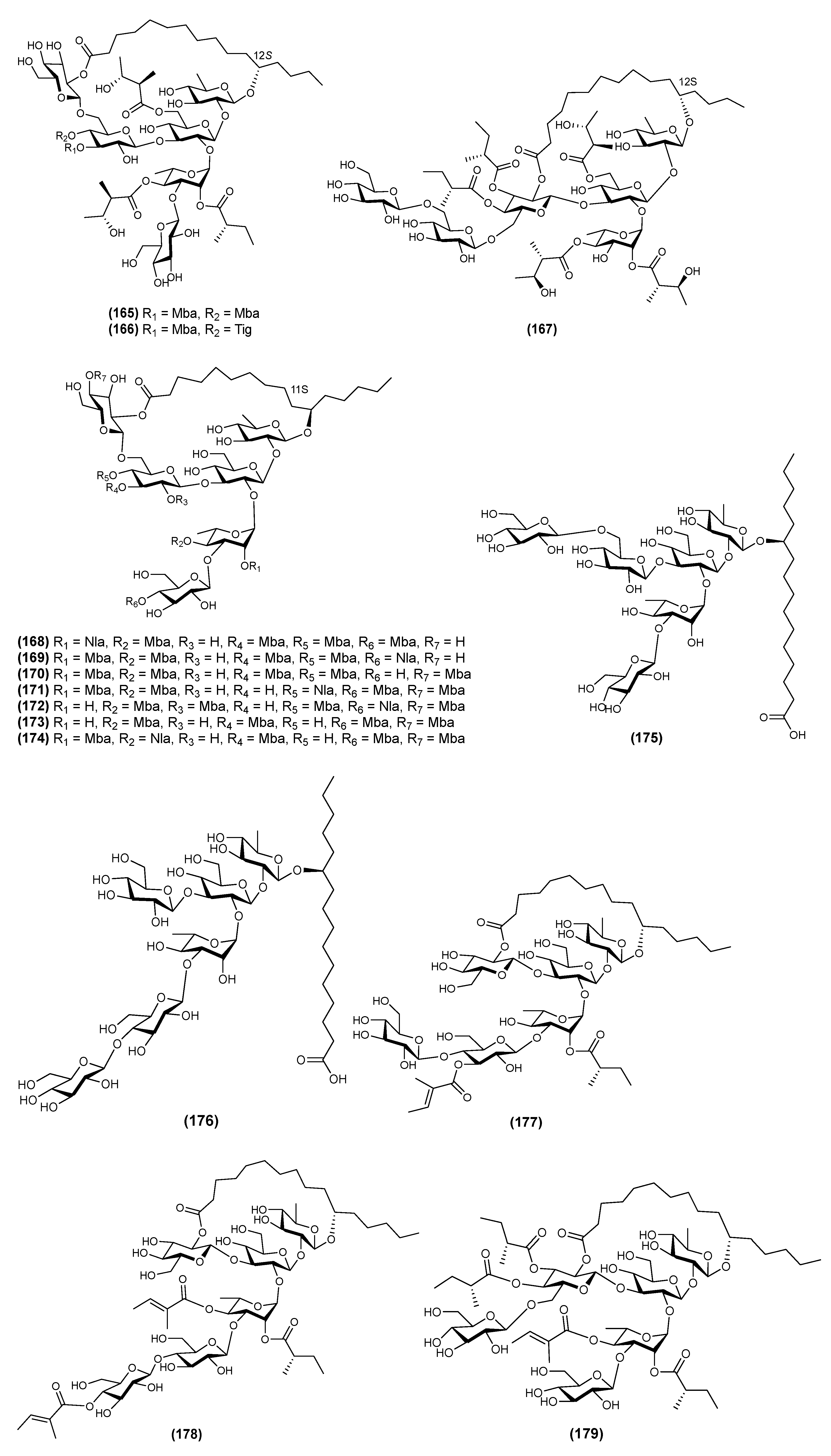
| 5 | Calyhedin II (165) | Calystegia hederacea | [59] |
| 6 | Calyhedin III (166) | Calystegia hederacea | [59] |
| 7 | Calyhedin X (167) | Calystegia hederacea | [60] |
| 8 | Calysepin I (168) | Calystegia sepium | [61] |
| 9 | Calysepin II (169) | Calystegia sepium | [61] |
| 10 | Calysepin III (170) | Calystegia sepium | [61] |
| 11 | Calysepin IV (171) | Calystegia sepium | [61] |
| 12 | Calysepin V (172) | Calystegia sepium | [61] |
| 13 | Calysepin VI (173) | Calystegia sepium | [61] |
| 14 | Calysepin VII (174) | Calystegia sepium | [61] |
| 15 | Calysolic acid C (175) | Calystegia soldanella | [45] |
| 16 | Calysolic acid D (176) | Calystegia soldanella | [45] |
| 17 | Calysolin IV (177) | Calystegia soldanella | [30] |
| 18 | Calysolin VII (178) | Calystegia soldanella | [46] |
| 19 | Calysolin VIII (179) | Calystegia soldanella | [46] |
| 20 | Calysolin IX (180) | Calystegia soldanella | [46] |
| 21 | Calysolin XI (181) | Calystegia soldanella | [31] |
| 22 | Calysolin XII (182) | Calystegia soldanella | [31] |
| 23 | Calysolin XIII (183) | Calystegia soldanella | [31] |
| 24 | Calysolin XIV (184) | Calystegia soldanella | [62] |
| 25 | Calysolin XV (185) | Calystegia soldanella | [62] |
| 26 | Calysolin XVI (186) | Calystegia soldanella | [62] |
| 27 | Calysolin XVII (187) | Calystegia soldanella | [62] |
| 28 | Calysolin XVIII (188) | Calystegia soldanella | [14] |
| 29 | Macrocarposidic acids A (189) | Operculina macrocarpa | [28] |
| 30 | Macrocarposidic acids B (190) | Operculina macrocarpa | [28] |
| 31 | Macrocarposidic acids C (191) | Operculina macrocarpa | [28] |
| 32 | Maltifidinic acid C (192) | Quamoclit × multifida | [26] |
| 33 | Maltifidinic acid D (193) | Quamoclit × multifida | [26] |
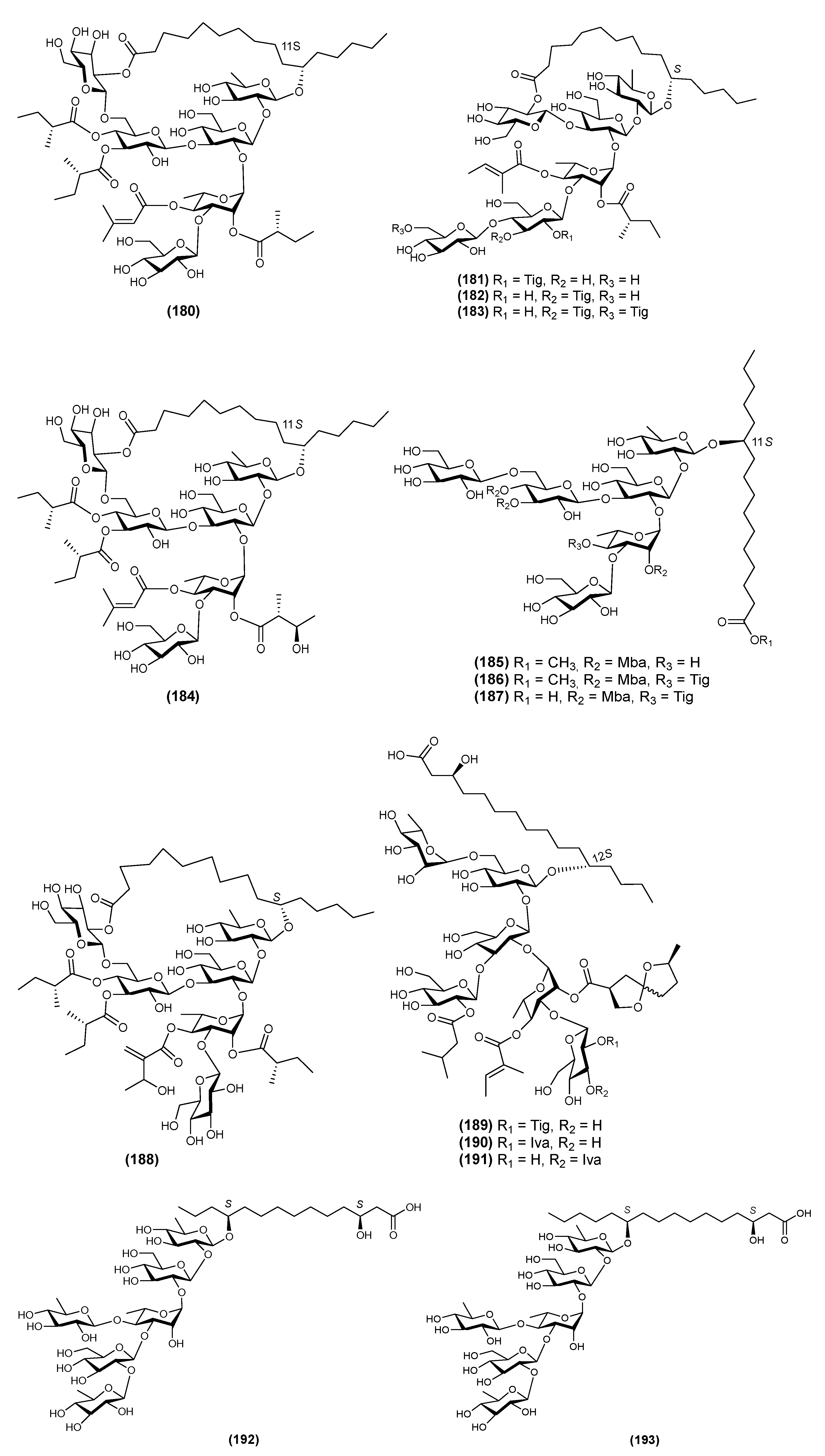
| 35 | Multifidin VII (194) | Quamoclit × multifida | [51] |
| 36 | Multifidin VIII (195) | Quamoclit × multifida | [51] |
| 37 | Operculinic acids H (196) | Operculina macrocarpa | [28] |
| 38 | Operculinic acids I (197) | Operculina macrocarpa | [28] |
| 39 | Operculinic acids J (198) | Operculina macrocarpa | [28] |
| 40 | PM1 (199) | Pharbitis nil | [54] |
| 41 | PM2 (200) | Pharbitis nil | [54] |
| 42 | PM3 (201) | Pharbitis nil | [54] |
| 43 | PM4 (202) | Pharbitis nil | [54] |
| 44 | PM5 (203) | Pharbitis nil | [54] |
| 45 | QM4 (204) | Quamoclit pennata | [63] |
| 46 | Quamoclinic acid C (205) | Quamoclit pennata | [27] |
| 47 | Quamoclinic acid D (206) | Quamoclit pennata | [27] |
| Heptasaccharides | |||
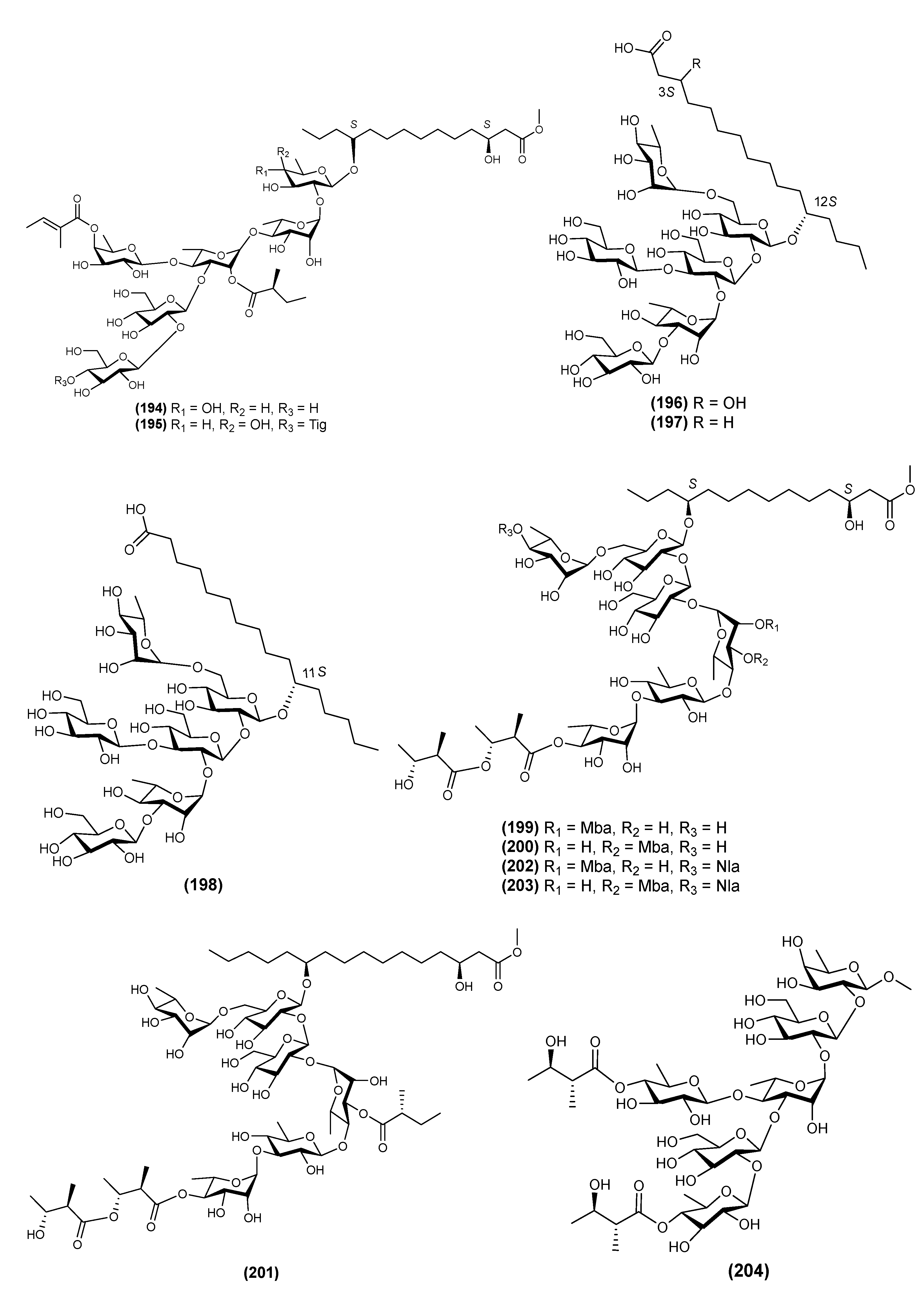
| 1 | Arvensic acid A (207) | Convolvulus arvensis | [64] |
| 2 | Arvensic acid B (208) | Convolvulus arvensis | [64] |
| 3 | Arvensic acid C (209) | Convolvulus arvensis | [64] |
| 4 | Arvensic acid D (210) | Convolvulus arvensis | [64] |
| 5 | Calyhedic acid C (211) | Calystegia hederacea | [44] |
| 6 | Calyhedic acid D (212) | Calystegia hederacea | [44] |
| 7 | Calyhedin IV (213) | Calystegia hederacea | [59] |
| 8 | Calyhedin V (214) | Calystegia hederacea | [59] |
| 9 | Calyhedin VI (215) | Calystegia hederacea | [59] |
| 10 | Calyhedin VII (216) | Calystegia hederacea | [60] |
| 11 | Calyhedin VIII (217) | Calystegia hederacea | [60] |
| 12 | Calyhedin IX (218) | Calystegia hederacea | [60] |
| 13 | QM1 (219) | Quamoclit pennata | [63] |
| 14 | QM7 (220) | Quamoclit pennata | [65] |
| 15 | Quamoclinic acid E (221) | Quamoclit pennata | [27] |
| 16 | Quamoclinic acid F (222) | Quamoclit pennata | [27] |
| Macrocyclic Bidesmoside | |||
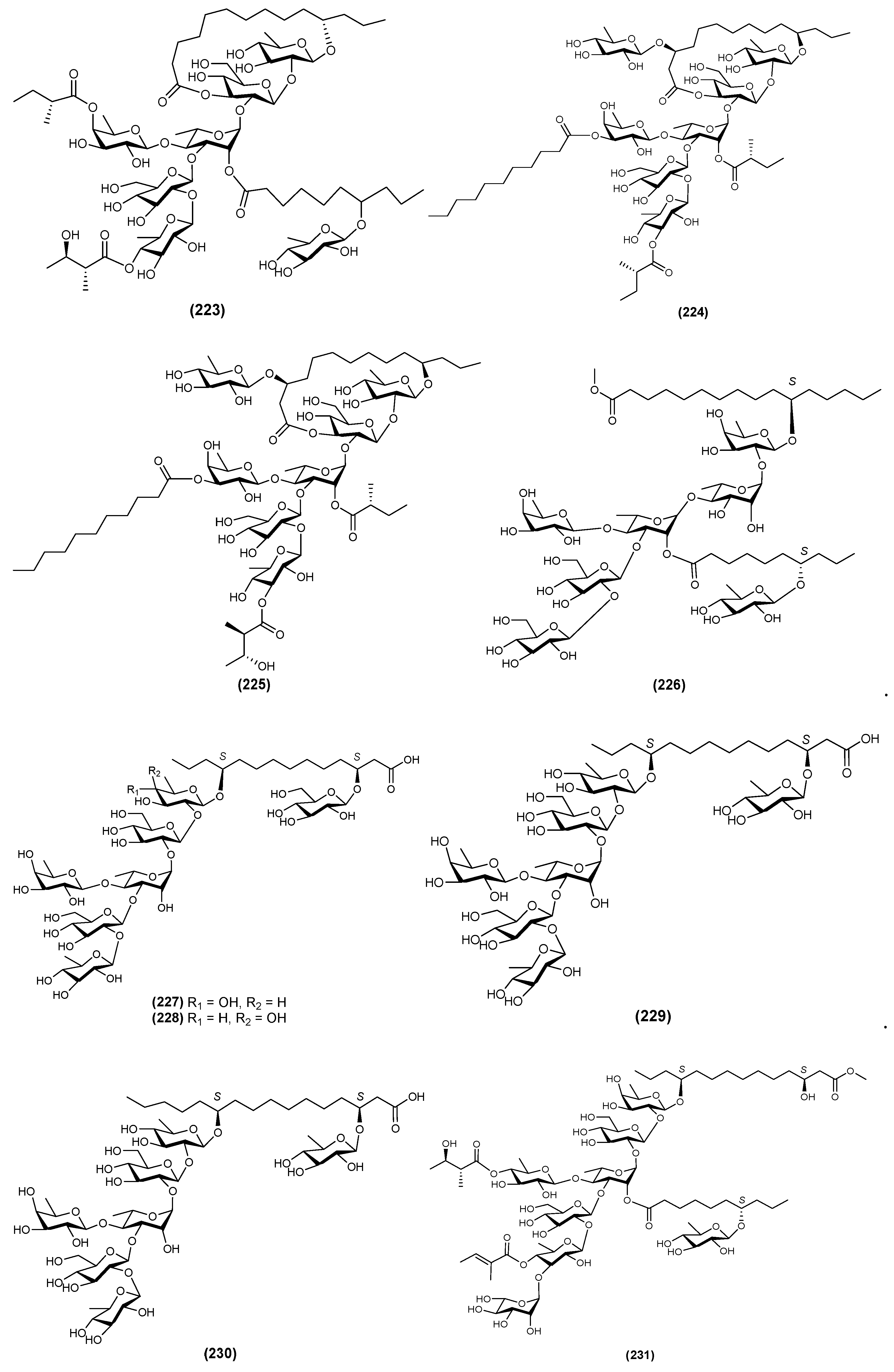
| 1 | Jalapinoside B (223) | Ipomoea purga | [16] |
| 2 | Jalapinoside I (224) | Ipomoea purga | [66] |
| 3 | Jalapinoside II (225) | Ipomoea purga | [67] |
| 4 | Multifidin IX (226) | Quamoclit × multifida | [51] |
| 5 | Multifidinic acid F (227) | Quamoclit × multifida | [68] |
| 6 | Multifidinic acid G (228) | Quamoclit × multifida | [68] |
| 7 | Purgic acid C (229) | Ipomoea purga | [67] |
| 8 | Purgic acid D (230) | Ipomoea purga | [67] |
| 9 | QM2 (231) | Quamoclit pennata | [63] |
| 10 | QM3 (232) | Quamoclit pennata | [63] |
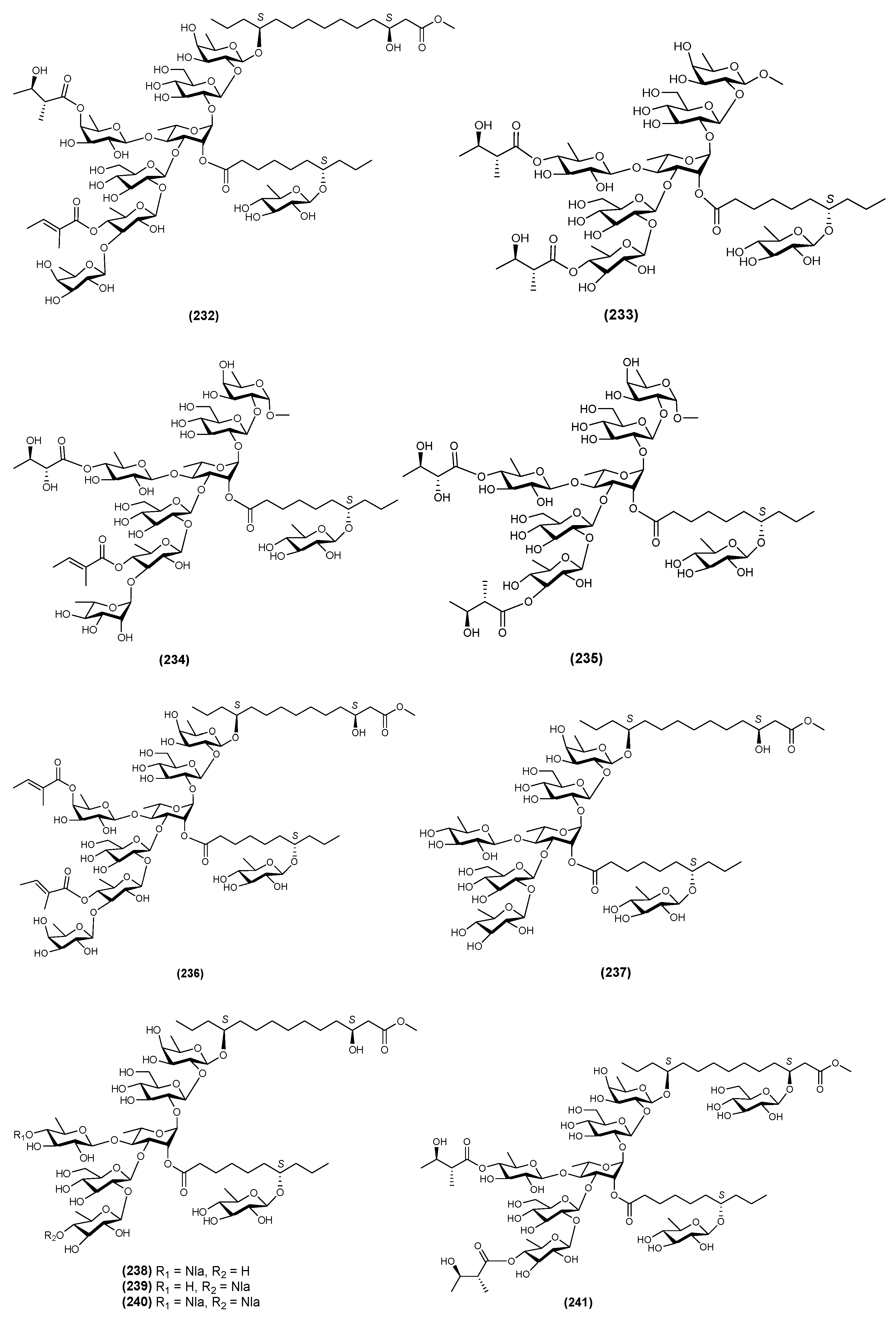
| 11 | QM5 (233) | Quamoclit pennata | [63] |
| 12 | QM6 (234) | Quamoclit pennata | [65] |
| 13 | QM8 (235) | Quamoclit pennata | [65] |
| 14 | QM9 (236) | Quamoclit pennata | [65] |
| 15 | QM10 (237) | Quamoclit pennata | [65] |
| 16 | QM11 (238) | Quamoclit pennata | [69] |
| 17 | QM12 (239) | Quamoclit pennata | [69] |
| 18 | QM13 (240) | Quamoclit pennata | [69] |
| 19 | QM14 (241) | Quamoclit pennata | [69] |
| 20 | Quamoclinic Acid G (242) | Quamoclit pennata | [70] |
| 21 | Quamoclinic Acid H (243) | Quamoclit pennata | [70] |
| Oligomers Ester-type dimer | |||
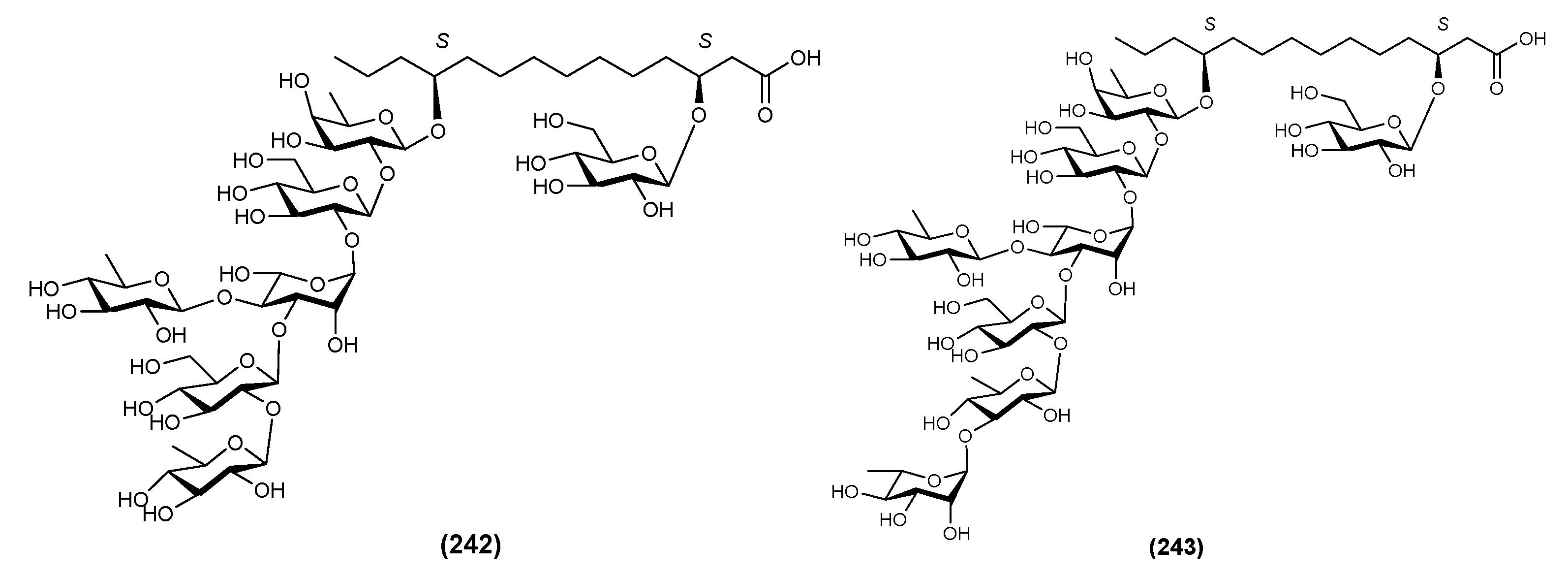
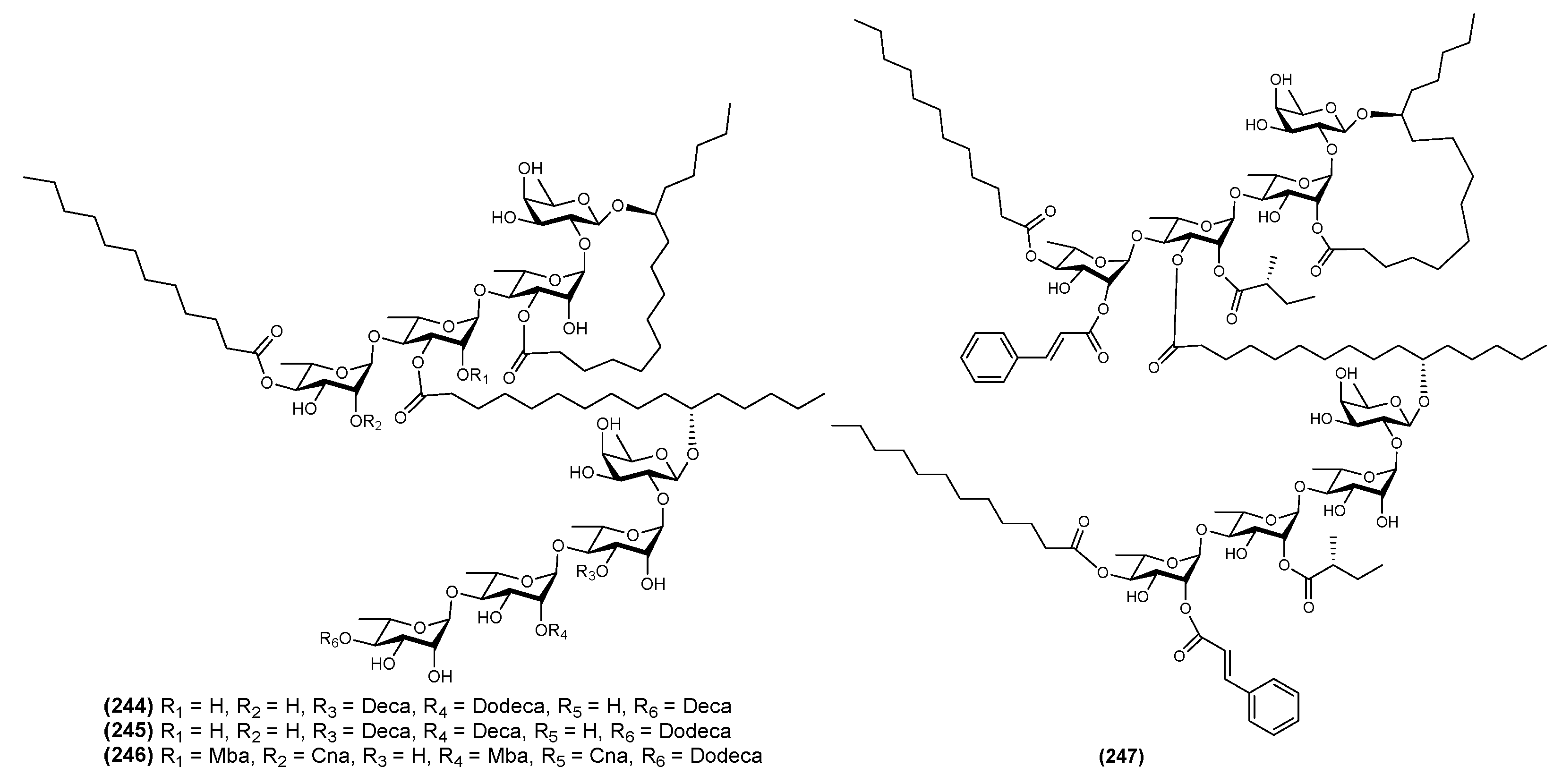
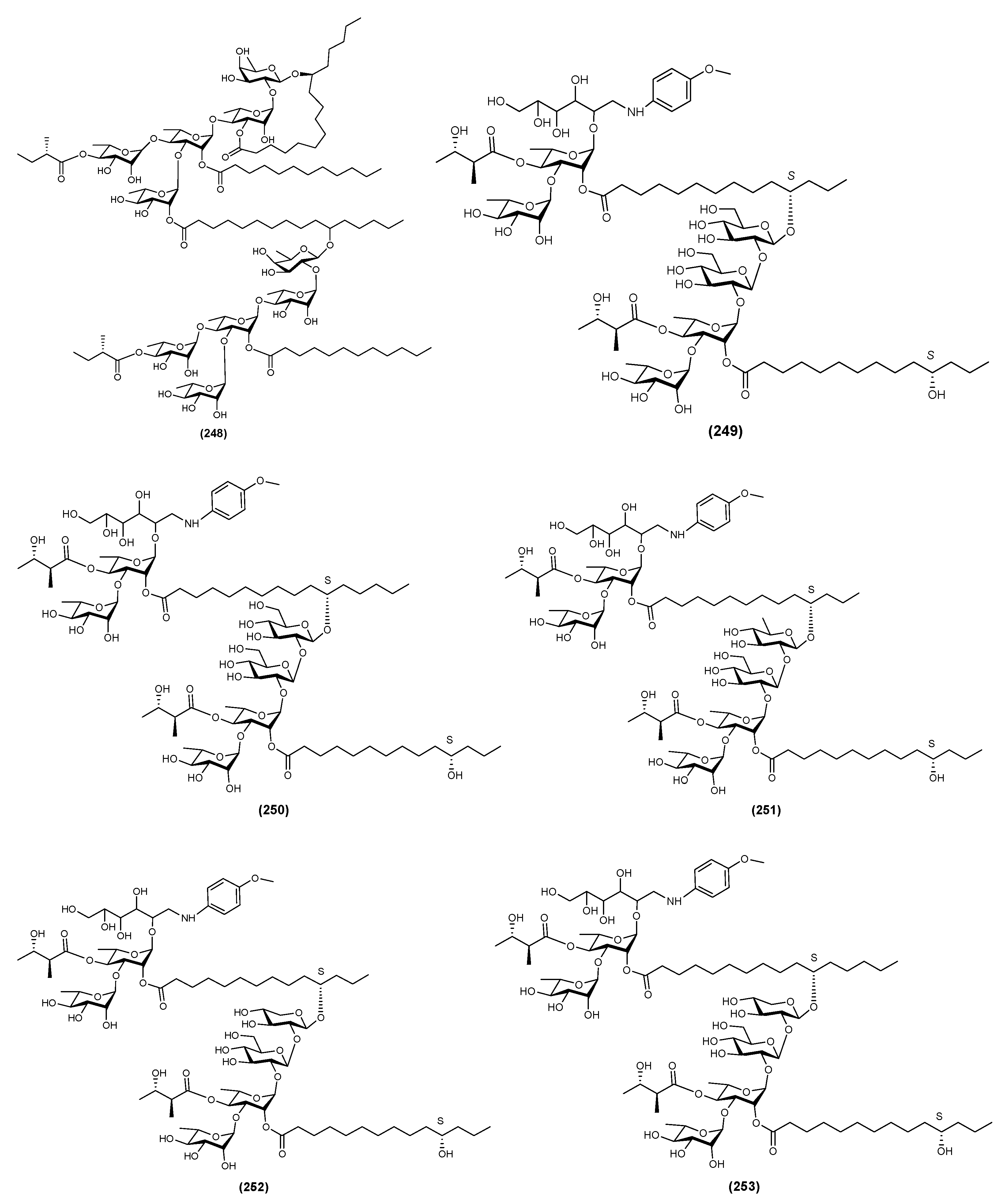
| 1 | Batatin III (244) | Ipomoea batatas | [71] |
| 2 | Batatin IV (245) | Ipomoea batatas | [71] |
| 3 | Batatin V (246) | Ipomoea batatas | [71] |
| 4 | Batatin VI (247) | Ipomoea batatas | [71] |
| 5 | Batatin VII (248) | Ipomoea batatas | [41] |
| 6 | Cuses 8 (249) | Cuscuta chinensis | [4] |
| 7 | Cuses 9 (250) | Cuscuta chinensis | [4] |
| 8 | Cuses 10 (251) | Cuscuta chinensis | [4] |
| 9 | Cuses 11 (252) | Cuscuta chinensis | [4] |
| 10 | Cuses 12 (253) | Cuscuta chinensis | [4] |
| 11 | Purgin I (254) | Ipomoea purga | [9] |
| 12 | Purgin II (255) | Ipomoea purga | [6] |
| 13 | Purgin III (256) | Ipomoea purga | [6] |
| 14 | Tyrianthin C (257) | Ipomoea tyrianthina | [17] |
| 15 | Tyrianthin D (258) | Ipomoea tyrianthina | [17] |
| 16 | Tyrianthin E (259) | Ipomoea tyrianthina | [17] |
| 17 | Stansin A (260) | Ipomoea stans | [16] |
| 18 | Wolcotinne I (261) | Ipomoea wolcottiana | [24] |
| Genus | Tri | Tetra | Penta | Hexa | Hepta | Bidesmoside | Dimer |
|---|---|---|---|---|---|---|---|
| Cuscuta | √ | √ | |||||
| Ipomoea | √ | √ | √ | √ | √ | √ | √ |
| Porana | √ | ||||||
| Evolvulus | √ | √ | |||||
| Dichondra | √ | ||||||
| Calystegia | √ | √ | √ | √ | |||
| Convolvulus | √ | √ | |||||
| Argyreia | √ | ||||||
| Merremia | √ | ||||||
| Quamoclit | √ | √ | √ | √ | |||
| Operculina | √ | ||||||
| Jalapae | √ | ||||||
| Pharbitis | √ | √ |
| No | Name of Compound | Activity | References |
|---|---|---|---|
| 1 | Ipomoeassin F and analogues (273–274) | Cytotoxic against MDA-MB 231 and Michigan Cancer Foundation-7 (MCF-7). | [11] |
| 2 | Evolvulin I (108) | Cytotoxic against MCF-7 cells (IC50 value of 3.12 μM). | [47] |
| 3 | Tricolorin A (293) | The MDR reversal factor of vinblastine cytotoxicity enhanced by 2164-fold in MCF/Vin cells. | [12] |
| 4 | Ipomeolides A (111) | Inducing apoptosis cell death in K562 cells when combined with doxorubicyn. | [49] |
| 5 | Calonyctins E (23), J (28), Muricatic acid C | The MDR reversal activity of vincristine cytotoxicity enhanced by 2.5–407.1-fold at 25 μM in KB/VCR cells (Calonyctin E was the most active). | [23] |
| 6 | Cairicosides I–IV (93–96) | α-Glucosidase inhibitory potential in vitro comparable to acarbose. | [43] |
| 7 | Cairicoside E (91) | Anti-metastatic effect through EMT inhibition via down-regulation of AQP5 in CRC cells. | [15] |
| 8 | Calysolin XVIII (188) | Anti-HSV-1 with EC50 value 2.3 µM and relatively high cytotoxicity (IC50 8.7 μM). | [14] |
| 9 | Albinosides VII-VIII (15–16) | The MDR reversal activity of vinblastine enhanced with reversal factors (RFMCF-7/Vin+) of 201 and >2517-fold, respectively, in MCF/Vin cells. | [7] |
| 10 | Jalapinoside B (223), purgic acid D (230), stansoic acid A (13), stansin A (43), murucinic acid II (128), stansinic acid I (44) |
| [16] |
| 11 | Jalapinoside II (225) | The MDR reversal activity of vinblastines enhanced with reversal factors (RFMCF-7/Vin+) of >1906 fold in MCF/Vin cells. | [67] |
| 12 | Acutacoside C-E (52–54) | A weak α-glucosidase inhibitory activity. | [3] |
| 13 | Wolcottinosides I–IV (261), (157–159) | The potentiation effect of the antibiotic susceptibility up to eightfold when combined with etracycline, kanamycin, or chloramphenicol. | [24] |
| 14 | Aquaterins XII-XIII (74–75), XV (77) | Cytotoxic against various cancer cell lines (IC50 3.0–8.9 μM). | [39] |
| 15 | Aquaterin II (65) | Inducing G0/G1 arrest regulated by related proteins CDK4/6, cyclin D/E and p21. This was caused by mitochondria-mediated apoptosis with a decrease in MMP, ROS accumulation, caspase cascade activation and Bax/Bcl-2 alteration. Additionally, the suppression of the PI3K/Akt signalling pathway was observed, suggesting that it participated in the Aquaterin II–induced cell growth inhibition (antiproliferative mechanism). | [39] |
| 16 | Dichondrins A-C (31-32), (9) | The MDR reversal activity of vincristine enhanced with reversal factors (RFMCF-7/Vin+) by 1.03–1.78-fold in KB/VCR cells. | [3] |
| 17 | Jalapinoside I (224) | The MDR reversal activity of vincristine enhanced with reversal factors (RFMCF-7/Vin+) by >1906-fold in MCF-7/Vin cells. | [66] |
| 18 | Aquaterins I−IV (64–67), Aquaterin VII (70), Aquaterin IX-X (72–73) |
| [29] |
| 19 | Merremin A (113), Merremin E-F (117–118), murucoidin V | The MDR reversal activity of vincristine enhanced with reversal factors by 2.3−142.5-fold in KB/VCR cells | [50] |
| 20 | Stansin 6 (43) | Neuroprotective and anticonvulsant activities. | [10] |
| 21 | Tyrianthins C-E (257–259) | Sedative and vasorelaxant effect. | [17] |
| 22 | Cuses 5–12 (4–6), (249–253) | Cytotoxic toward MCF-7, SMMC-7721, and MG-63 cell lines with IC50 values ranging from 8.72 to 59.35 mg/mL. | [4] |
| 23 | Cairicosides A–E (87–91) | Moderately cytotoxic against a small panel of human tumor cell lines with IC50 values in a range from 4.28 to 14.31 μM. Cairicosides A-D are more cytotoxic than cairicoside E. | [42] |
| 24 | Purgin III (256), Purginosides I–IV (141–144), Purgin I (254) | The MDR reversal activity of vinblastine enhanced with reversal factors (RFMCF-7/Vin+) by 1.4 to 6.5-fold in MCF-7/Vin cells. | [6] |
| 25 | Convolvulin | Sedative and vasorelaxant effect. Cytotoxic against nasopharyngeal carcinoma cell line KB (ED50 2.3 μg/mL). | [81] |
| 26 | Pescapreins XXI–XXX (129–138) | Multidrug resistance modulator in the human breast cancer cell line MCF-7/ADR. | [53] |
| 27 | Ipomotaosides A (35) | Anti-inflammatory activity against cyclooxygenase (COX)-1 and -2. | [18] |
| 29 | Cairicosides A-B (87–88) | Strong α-glucosidase inhibitory activity with IC50 25.3 ± 1.6 and 28.5 ± 3.3 μM, compared to acarbose. | [80] |
| 30 | Calysolin I–IX (29), 103–104, 177, 105–106, 178–180) | Antiviral activity against HSV-1 with IC50 ranging from 4.3 to 18.5 μM and EC50 from 1.9 to 19.2 μM. | [46] |
| 31 | Calysolin X–XIII (30), 181–183) | Antiviral activity against HSV-1 with IC50 from 14.6 to 32.6 μM and EC50 from 2.6 to 7.5 μM. | [31] |
| 32 | Calysolin XIV–XVII (184–187) | Antiviral activity against HSV-1 with IC50 ranging from 7.1 to 50.4 μM and EC50 ranging from 3.5 to 12.8 μM. | [62] |
| No | Plant | Part of Plant | Extraction/Separation | Refs. |
|---|---|---|---|---|
| 1 | Calystegia soldanella | Fresh leaves, stems and roots |
| [14] |
| 2 | Ipomoea tricolor | Seeds |
| [12] |
| 3 | Ipomoea muricata | Seeds |
| [23] |
| 4 | Ipomoea alba | Seeds |
| [7] |
| 5 | Ipomoea wolcottiana | Dried flower |
| [24] |
| 6 | Ipomoea batatas | Aerial part |
| [18] |
| 7 | Ipomoea tyrianthina | Root |
| [17] |
| 8 | Operculina macrocarpa | Root |
| [28] |
| No | Name of Resin Glycoside | Structural Elucidation | Refs. |
|---|---|---|---|
| 1 | Quamoclinic acid B (2) Monosaccharide Quamoclit pennata |
| [27] |
| 2 | Poranic acid A (10) Trisaccharides Porana duclouxii |
| [5] |
| 3 | Calonyctins E (23) Tetrasaccharides Ipomoea muricata |
| [23] |
| 4 | Ipomeolides A (111) Pentasaccharides Ipomoea pes-caprae |
| [49] |
| 5 | Calysolin XVIII (188) Hexasaccharides Calystegia soldanella |
| [14] |
| 6 | Arvensic acid A-D (207–210) Heptasaccharides Convolvulus arvensis |
| [64] |
| 7 | Jalapinoside I (224) Bidesmoside Ipomoea purga |
| [66] |
| 8 | Batatin VII (248) Oligomer ester type dimer Ipomoea batatas |
| [41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharani, R.; Fajar, M.; Supratman, U. Resin Glycosides from Convolvulaceae Family: An Update. Molecules 2022, 27, 8161. https://doi.org/10.3390/molecules27238161
Maharani R, Fajar M, Supratman U. Resin Glycosides from Convolvulaceae Family: An Update. Molecules. 2022; 27(23):8161. https://doi.org/10.3390/molecules27238161
Chicago/Turabian StyleMaharani, Rani, Mohamad Fajar, and Unang Supratman. 2022. "Resin Glycosides from Convolvulaceae Family: An Update" Molecules 27, no. 23: 8161. https://doi.org/10.3390/molecules27238161
APA StyleMaharani, R., Fajar, M., & Supratman, U. (2022). Resin Glycosides from Convolvulaceae Family: An Update. Molecules, 27(23), 8161. https://doi.org/10.3390/molecules27238161