
Infrared Spectra and Phototransformations of meta-Fluorophenol Isolated in Argon and Nitrogen Matrices




Abstract
:1. Introduction
2. Results and Discussion
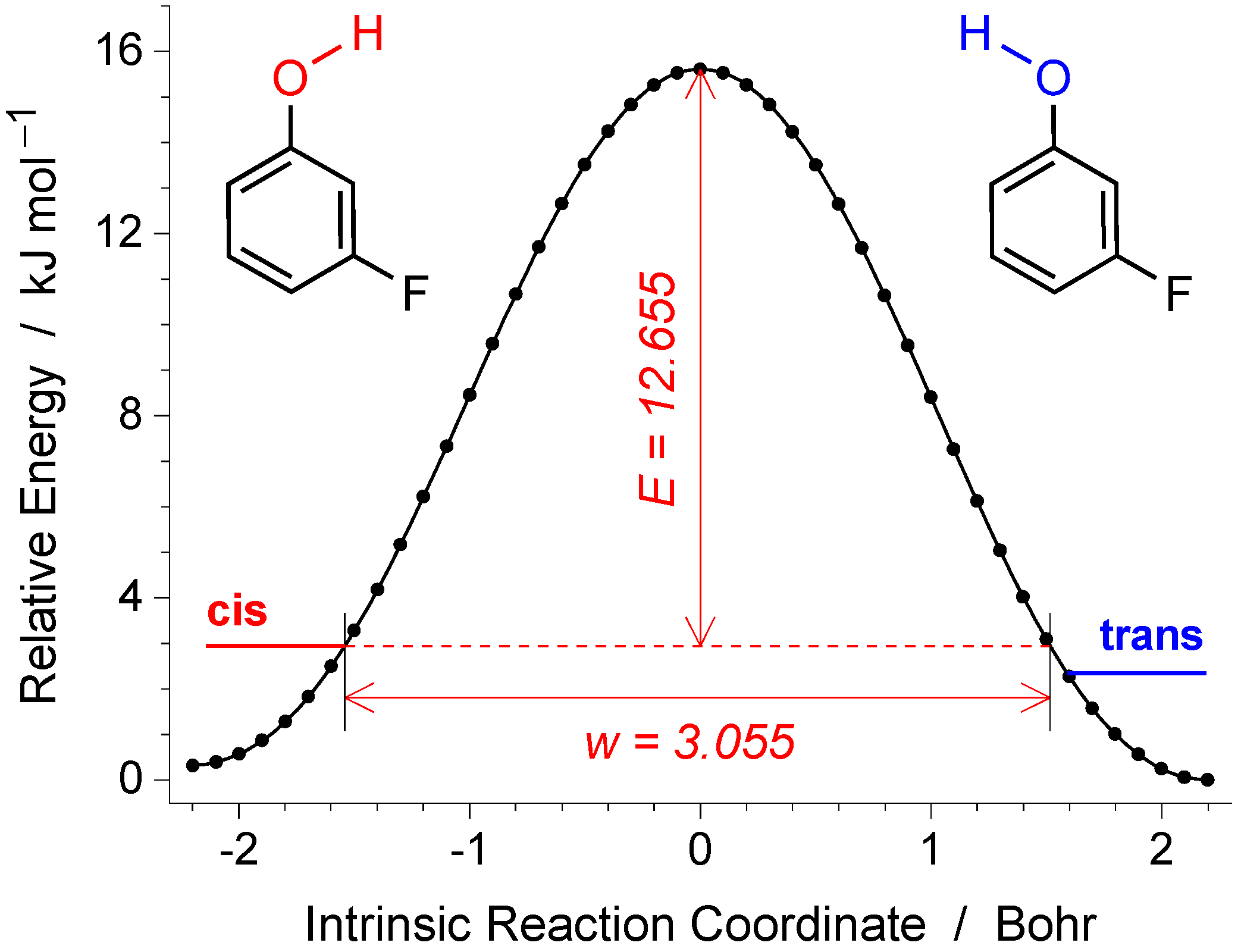
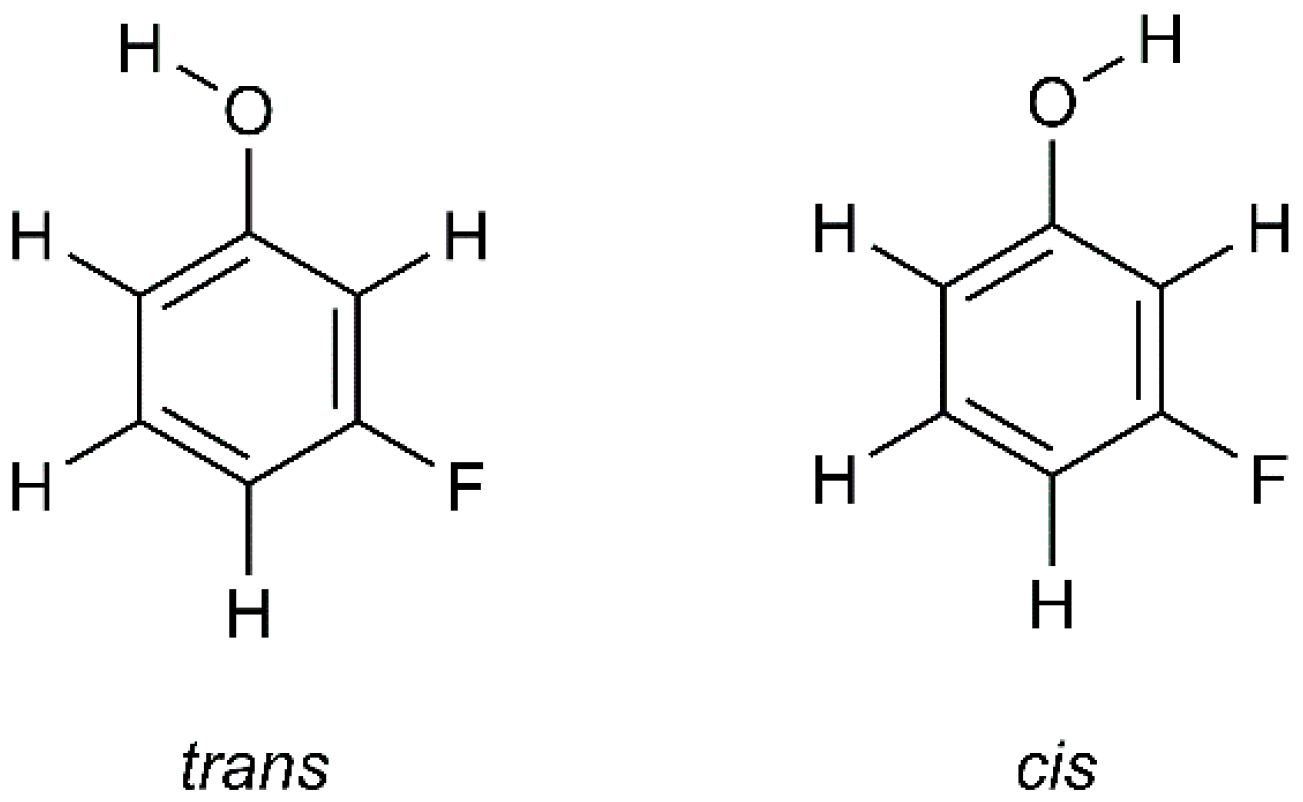
2.1. Conformers of mFP and Energy Barriers for Their Interconversion
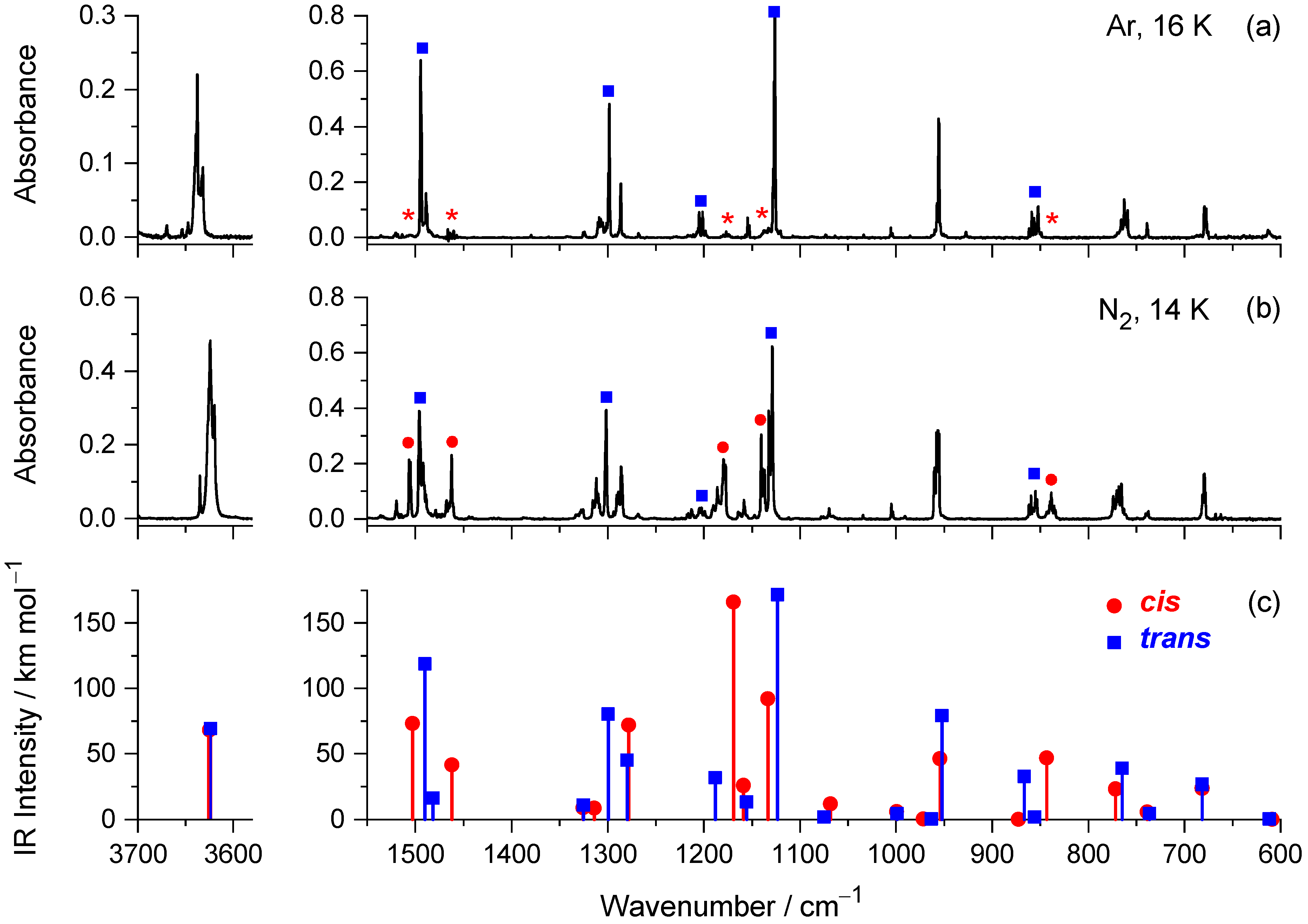
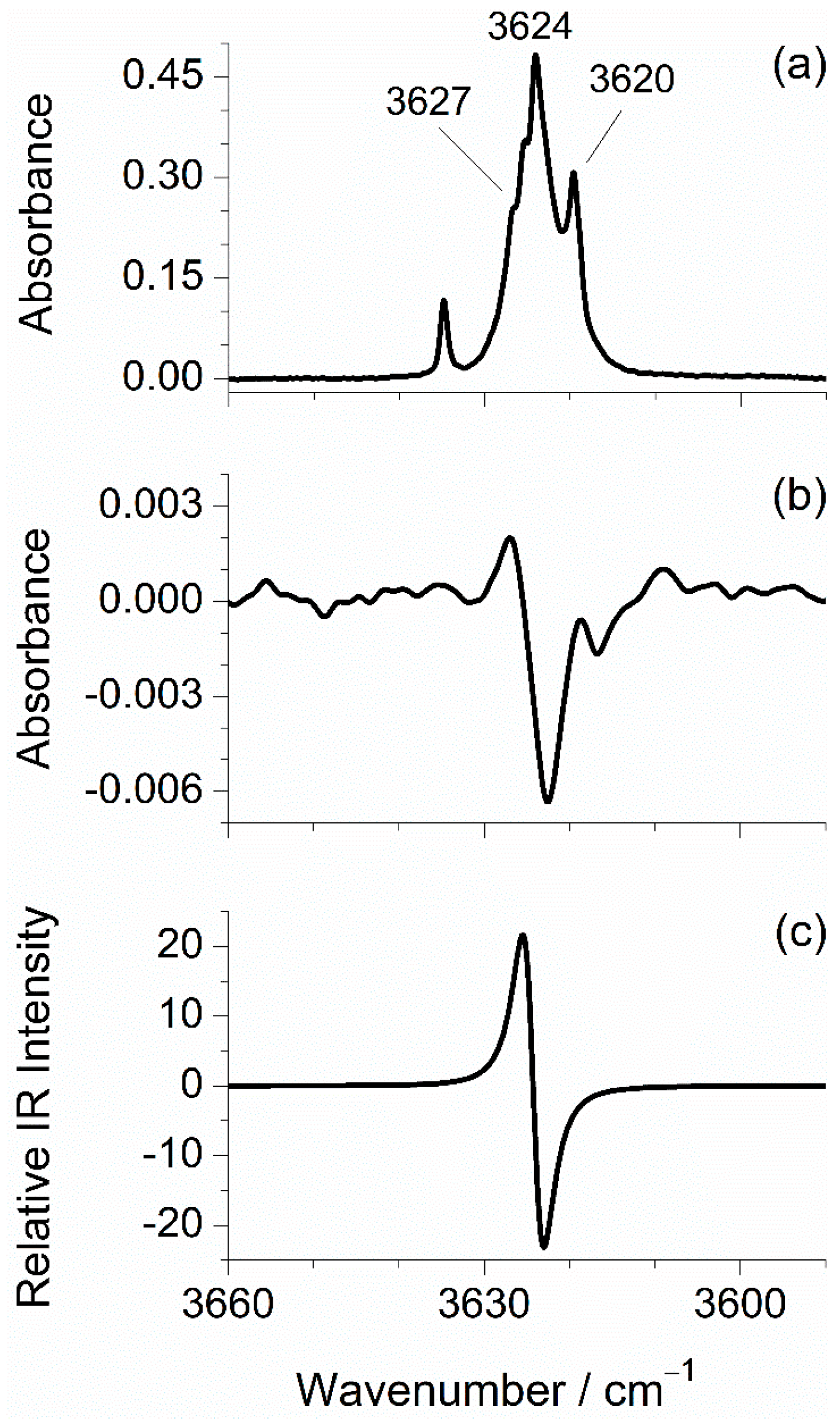
2.2. Infrared Spectra of Matrix-Isolated mFP
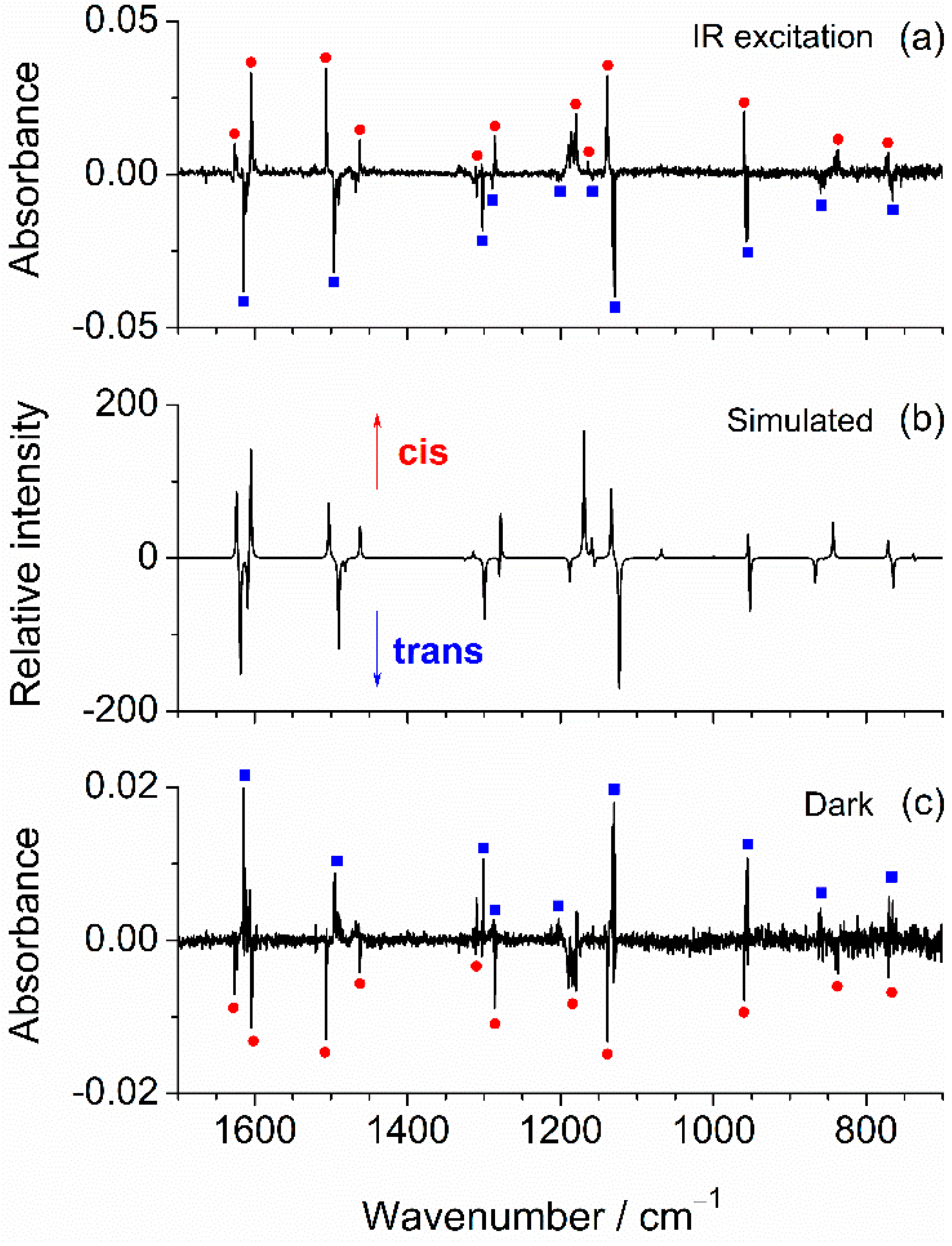
2.3. IR-Induced Conformational Changes
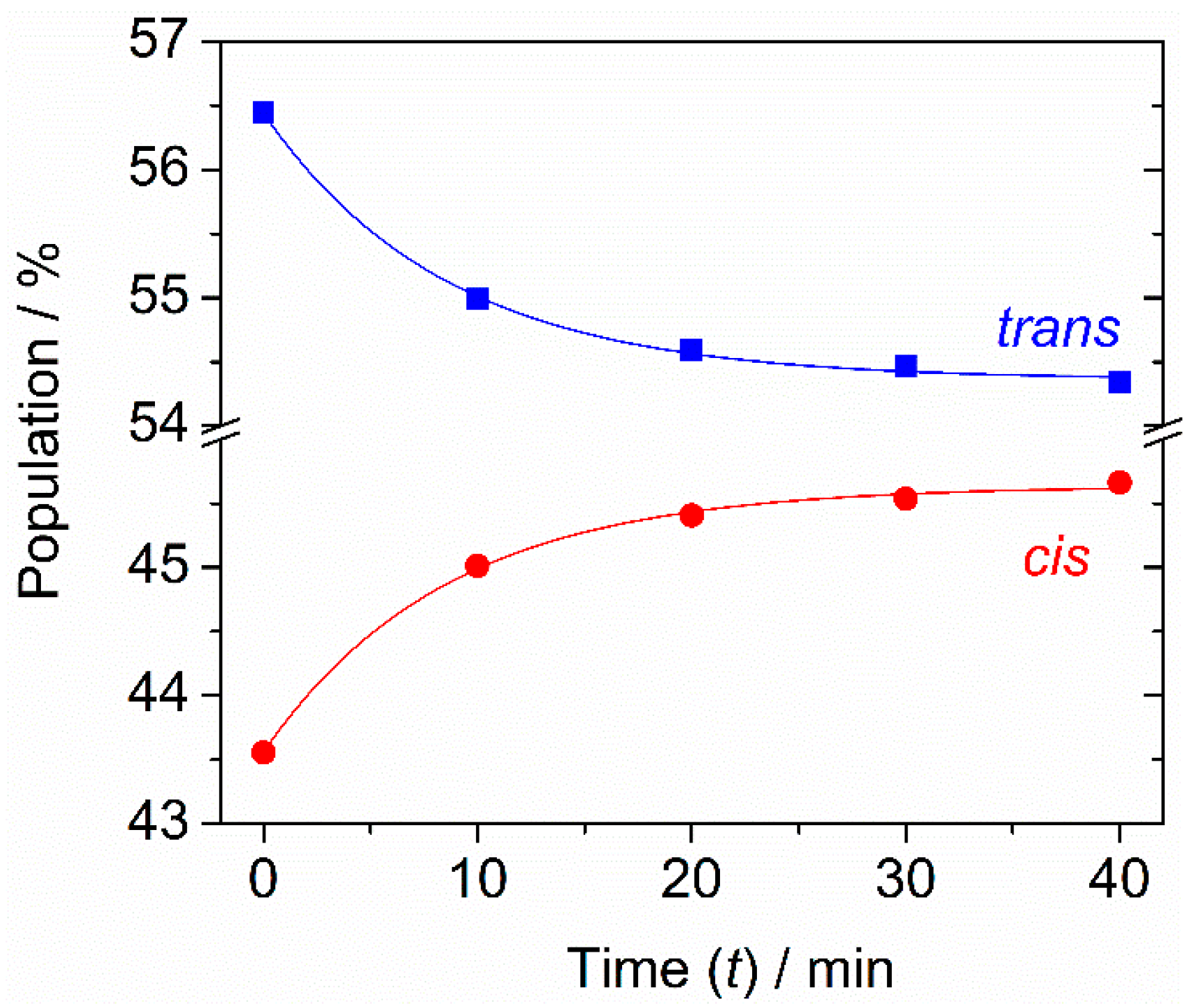
2.4. Cis-to-Trans Tunneling Decay
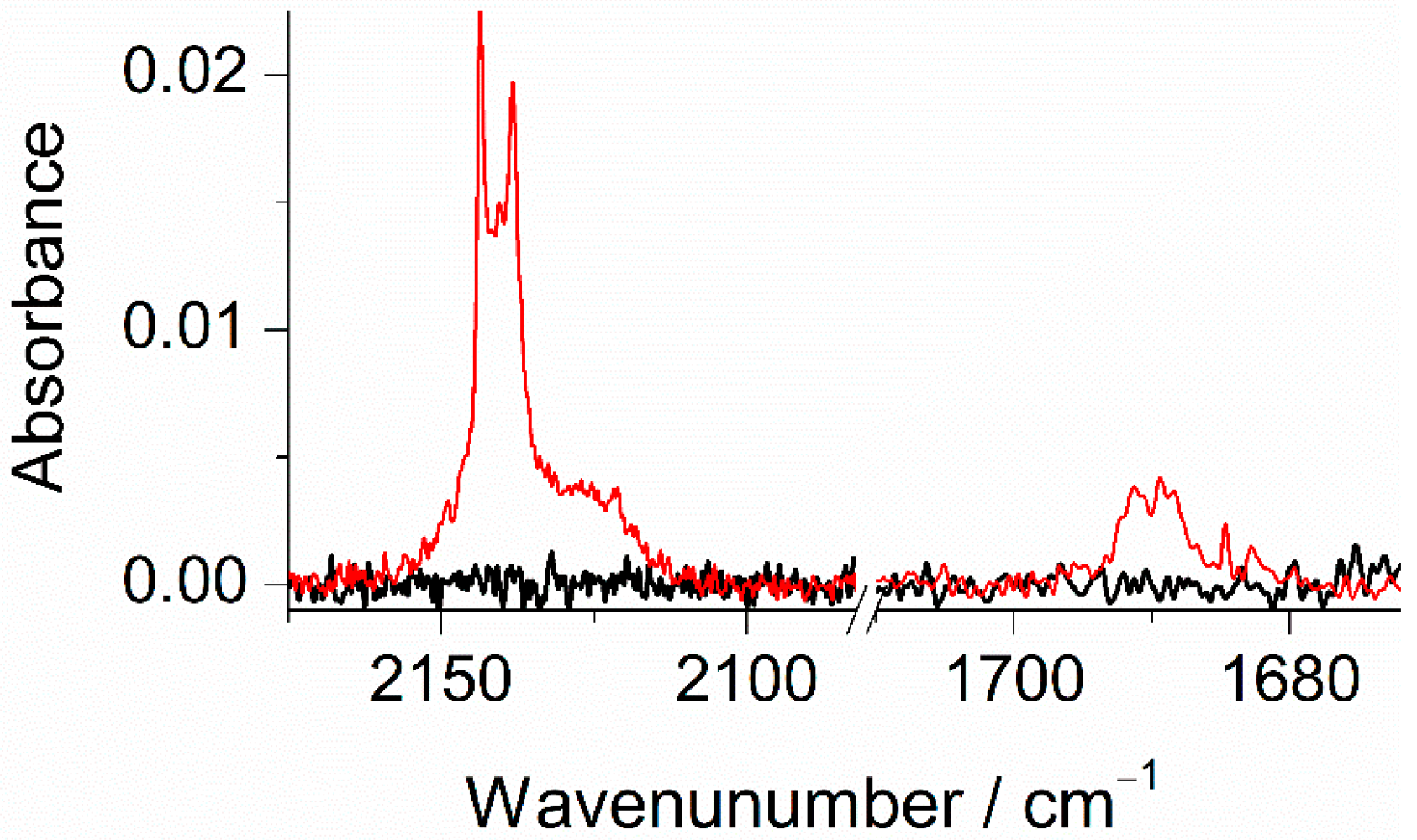
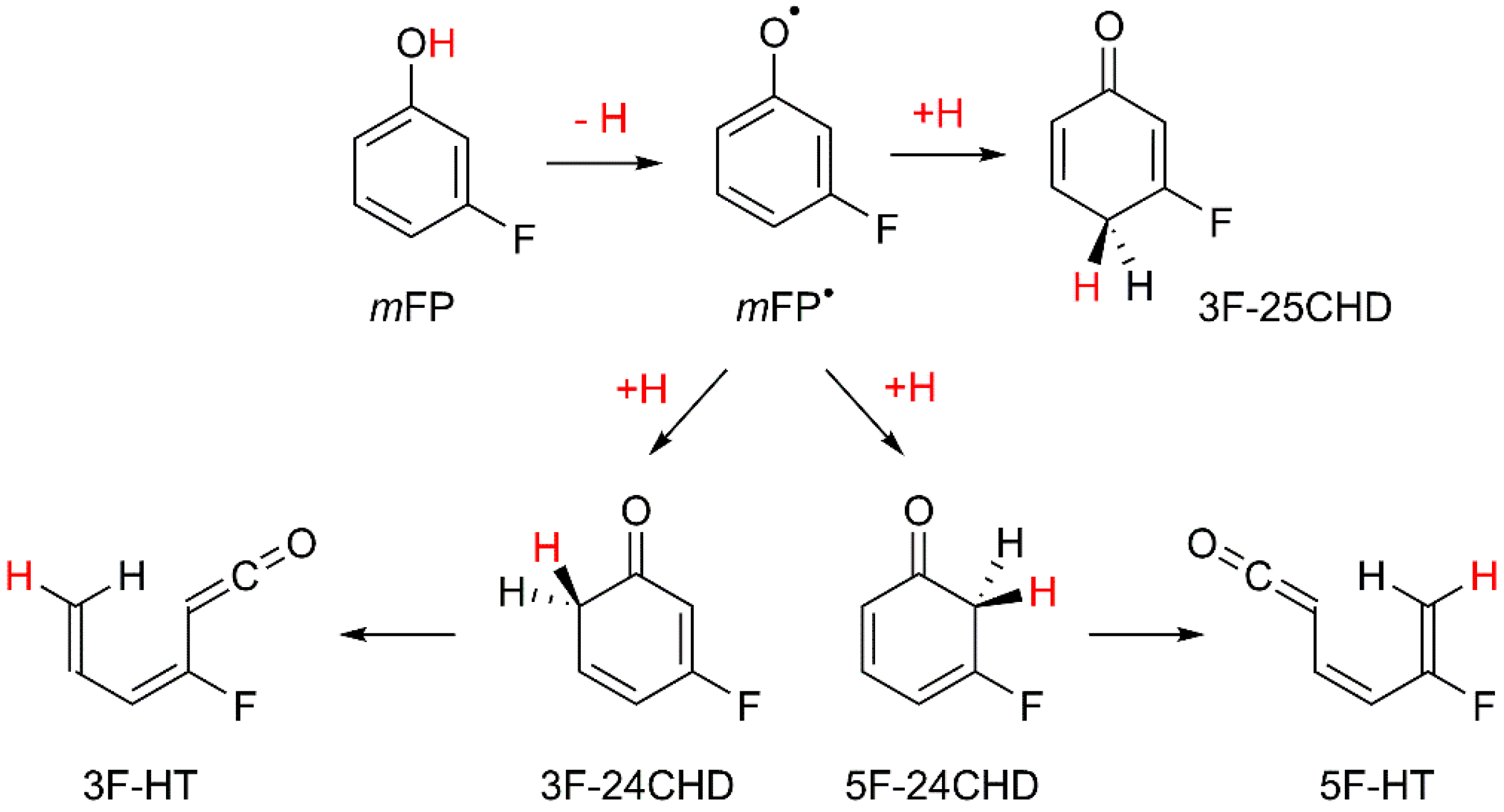
2.5. UV-Induced Transformations
3. Methods
3.1. Experimental Methods
3.2. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oswald, I.D.H.; Allan, D.R.; Motherwell, W.D.S.; Parsons, S. Structures of the monofluoro- and monochlorophenols at low temperature and high pressure. Acta Crystallogr. Sect. B 2005, 61, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, M.A.; Cormanich, R.A.; de Rezende, F.M.P.; Silla, J.M.; Tormena, C.F.; Rittner, R.; Ramalho, T.C.; Freitas, M.P. Theoretical and infrared studies on the conformations of monofluorophenols. J. Mol. Struct. 2012, 1009, 11–15. [Google Scholar] [CrossRef]
- Zeoly, L.A.; Coelho, F.; Cormanich, R.A. Intramolecular H-Bond Is Formed in 2-Fluorophenol and 2-Fluorothiophenol, but It May not Be the Main Pathway of the JFH Coupling Constant Transmission. J. Phys. Chem. A 2019, 123, 10072–10078. [Google Scholar] [CrossRef] [PubMed]
- Kierspel, T.; Horke, D.A.; Chang, Y.-P.; Küpper, J. Spatially separated polar samples of the cis and trans conformers of 3-fluorophenol. Chem. Phys. Lett. 2014, 591, 130–132. [Google Scholar] [CrossRef] [Green Version]
- Bell, A.; Singer, J.; Desmond, D.; Mahassneh, O.; van Wijngaarden, J. Rotational spectra and conformer geometries of 2-fluorophenol and 3-fluorophenol. J. Mol. Spectrosc. 2017, 331, 53–59. [Google Scholar] [CrossRef]
- Dutta, A.; Jaman, A.I. Microwave spectrum of cis 3-fluorophenol. Pramana 1985, 24, 499–502. [Google Scholar] [CrossRef]
- Jaman, A.I.; Nandi, R.N.; Ghosh, D.K. Microwave spectrum of 3-fluorophenol. J. Mol. Spectrosc. 1981, 86, 269–274. [Google Scholar] [CrossRef]
- Schaefer, T.; Salman, S.R.; Sebastian, R. Determination of the intramolecular cis-trans equilibrium for 3-fluorophenol in solution. J. Phys. Chem. 1981, 85, 499–501. [Google Scholar] [CrossRef]
- Green, J.H.S.; Harrison, D.J.; Kynaston, W. Vibrational spectra of benzene derivatives—XIV: Mono substituted phenols. Spectrochim. Acta Part A 1971, 27, 2199–2217. [Google Scholar] [CrossRef]
- Manocha, A.S.; Carlson, G.L.; Fateley, W.G. Barriers to internal rotation in some m-substituted phenols. J. Phys. Chem. 1973, 77, 2094–2098. [Google Scholar] [CrossRef]
- Fateley, W.G. Barriers and conformations. Pure Appl. Chem. 1976, 36, 109–126. [Google Scholar] [CrossRef]
- Banerjee, P.; Mukhopadhyay, D.P.; Chakraborty, T. On the origin of donor O–H bond weakening in phenol-water complexes. J. Chem. Phys. 2015, 143, 204306. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Chakraborty, T. Weak hydrogen bonds: Insights from vibrational spectroscopic studies. Int. Rev. Phys. Chem. 2018, 37, 83–123. [Google Scholar] [CrossRef]
- Gebicki, J.; Krantz, A. Substituent effects on weakly bound complexes of phenols and carbon monoxide in argon matrixes. Hydrogen bonding at cryogenic temperatures. J. Am. Chem. Soc. 1984, 106, 8097–8104. [Google Scholar] [CrossRef]
- Kovács, A.; Macsári, I.; Hargittai, I. Intramolecular Hydrogen Bonding in Fluorophenol Derivatives: 2-Fluorophenol, 2,6-Difluorophenol, and 2,3,5,6-Tetrafluorohydroquinone. J. Phys. Chem. A 1999, 103, 3110–3114. [Google Scholar] [CrossRef]
- Barnes, A.J. Matrix isolation vibrational spectroscopy as a tool for studying conformational isomerism. J. Mol. Struct. 1984, 113, 161–174. [Google Scholar] [CrossRef]
- Lopes Jesus, A.J.; Rosado, M.T.S.; Reva, I.; Fausto, R.; Eusébio, M.E.S.; Redinha, J.S. Structure of Isolated 1,4-Butanediol: Combination of MP2 Calculations, NBO Analysis, and Matrix-Isolation Infrared Spectroscopy. J. Phys. Chem. A 2008, 112, 4669–4678. [Google Scholar] [CrossRef] [Green Version]
- Rosado, M.T.S.; Lopes Jesus, A.J.; Reva, I.D.; Fausto, R.; Redinha, J.S. Conformational Cooling Dynamics in Matrix-Isolated 1,3-Butanediol. J. Phys. Chem. A 2009, 113, 7499–7507. [Google Scholar] [CrossRef] [Green Version]
- Akai, N.; Kudoh, S.; Takayanagi, M.; Nakata, M. Cis–Trans Isomerization Equilibrium in Hydroquinone in Low-Temperature Argon and Xenon Matrices Studied by FTIR Spectroscopy. Chem. Phys. Lett. 2002, 356, 133–139. [Google Scholar] [CrossRef]
- Akai, N.; Kudoh, S.; Nakata, M. Photoisomerization and Tunneling Isomerization of Tetrachlorohydroquinone in a Low-Temperature Argon Matrix. J. Phys. Chem. A 2003, 107, 3655–3659. [Google Scholar] [CrossRef]
- Nanbu, S.; Sekine, M.; Nakata, M. Hydrogen-Atom Tunneling in Isomerization Around the C–O Bond of 2-Chloro-6-Fluorophenol in Low-Temperature Argon Matrixes. J. Phys. Chem. A 2011, 115, 9911–9918. [Google Scholar] [CrossRef] [PubMed]
- Lopes Jesus, A.J.; Reva, I.; Nunes, C.M.; Roque, J.P.L.; Pinto, S.M.V.; Fausto, R. Kinetically unstable 2–isocyanophenol isolated in cryogenic matrices: Vibrational excitation, conformational changes and spontaneous tunneling. Chem. Phys. Lett. 2020, 742, 137069. [Google Scholar] [CrossRef]
- Lopes Jesus, A.J.; Nunes, C.M.; Reva, I.; Pinto, S.M.V.; Fausto, R. Effects of Entangled IR Radiation and Tunneling on the Conformational Interconversion of 2-Cyanophenol. J. Phys. Chem. A 2019, 123, 4396–4405. [Google Scholar] [CrossRef]
- Lopes Jesus, A.J.; Nunes, C.M.; Reva, I. Conformational Structure, Infrared Spectra and Light-Induced Transformations of Thymol Isolated in Noble Gas Cryomatrices. Photochem 2022, 2, 405–422. [Google Scholar] [CrossRef]
- Lopes Jesus, A.J.; Fausto, R.; Reva, I. Conformational Space, IR-Induced, and UV-Induced Chemistry of Carvacrol Isolated in a Low-Temperature Argon Matrix. J. Phys. Chem. A 2021, 125, 8215–8229. [Google Scholar] [CrossRef]
- Nanbu, S.; Sekine, M.; Nakata, M. Intramolecular hydrogen-atom tunneling and photoreaction mechanism of 4-bromo-2-chloro-6-fluorophenol in low-temperature argon matrices. J. Mol. Struct. 2012, 1025, 69–73. [Google Scholar] [CrossRef]
- Kuş, N.; Fausto, R. Effects of the matrix and intramolecular interactions on the stability of the higher-energy conformers of 2-fluorobenzoic acid. J. Chem. Phys. 2017, 146, 124305. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Reisenauer, H.P.; Schreiner, P.R. Electronic Effects on Atom Tunneling: Conformational Isomerization of Monomeric Para-Substituted Benzoic Acid Derivatives. J. Am. Chem. Soc. 2010, 132, 15902–15904. [Google Scholar] [CrossRef]
- Gerbig, D.; Schreiner, P.R. Hydrogen-Tunneling in Biologically Relevant Small Molecules: The Rotamerizations of α-Ketocarboxylic Acids. J. Phys. Chem. B 2015, 119, 693–703. [Google Scholar] [CrossRef]
- Lapinski, L.; Reva, I.; Rostkowska, H.; Halasa, A.; Fausto, R.; Nowak, M.J. Conformational Transformation in Squaric Acid Induced by Near-IR Laser Light. J. Phys. Chem. A 2013, 117, 5251–5259. [Google Scholar] [CrossRef]
- Maçôas, E.M.S.; Khriachtchev, L.; Pettersson, M.; Fausto, R.; Räsänen, M. Rotational Isomerism in Acetic Acid: The First Experimental Observation of the High-Energy Conformer. J. Am. Chem. Soc. 2003, 125, 16188–16189. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, M.; Lundell, J.; Khriachtchev, L.; Räsänen, M. IR Spectrum of the Other Rotamer of Formic Acid, cis-HCOOH. J. Am. Chem. Soc. 1997, 119, 11715–11716. [Google Scholar] [CrossRef]
- Bazsó, G.; Magyarfalvi, G.; Tarczay, G. Tunneling Lifetime of the ttc/VIp Conformer of Glycine in Low-Temperature Matrices. J. Phys. Chem. A 2012, 116, 10539–10547. [Google Scholar] [CrossRef] [PubMed]
- Bazsó, G.; Najbauer, E.E.; Magyarfalvi, G.; Tarczay, G. Near-Infrared Laser Induced Conformational Change of Alanine in Low-Temperature Matrixes and the Tunneling Lifetime of Its Conformer VI. J. Phys. Chem. A 2013, 117, 1952–1962. [Google Scholar] [CrossRef]
- Nunes, C.M.; Lapinski, L.; Fausto, R.; Reva, I. Near-IR laser generation of a high-energy conformer of L-alanine and the mechanism of its decay in a low-temperature nitrogen matrix. J. Chem. Phys. 2013, 138, 125101. [Google Scholar] [CrossRef] [PubMed]
- Marushkevich, K.; Räsänen, M.; Khriachtchev, L. Interaction of Formic Acid with Nitrogen: Stabilization of the Higher-Energy Conformer. J. Phys. Chem. A 2010, 114, 10584–10589. [Google Scholar] [CrossRef]
- Lopes, S.; Domanskaya, A.V.; Fausto, R.; Räsänen, M.; Khriachtchev, L. Formic and Acetic Acids in a Nitrogen Matrix: Enhanced Stability of the Higher-Energy Conformer. J. Chem. Phys. 2010, 133, 144507. [Google Scholar] [CrossRef] [Green Version]
- Halasa, A.; Lapinski, L.; Reva, I.; Rostkowska, H.; Fausto, R.; Nowak, M.J. Near-Infrared Laser-Induced Generation of Three Rare Conformers of Glycolic Acid. J. Phys. Chem. A 2014, 118, 5626–5635. [Google Scholar] [CrossRef]
- Araujo-Andrade, C.; Reva, I.; Fausto, R. Tetrazole acetic acid: Tautomers, conformers, and isomerization. J. Chem. Phys. 2014, 140, 064306. [Google Scholar] [CrossRef]
- Justino, L.L.G.; Reva, I.; Fausto, R. Thermally and vibrationally induced conformational isomerizations, infrared spectra, and photochemistry of gallic acid in low-temperature matrices. J. Chem. Phys. 2016, 145, 014304. [Google Scholar] [CrossRef]
- Reva, I.; Nunes, C.M.; Biczysko, M.; Fausto, R. Conformational Switching in Pyruvic Acid Isolated in Ar and N2 Matrixes: Spectroscopic Analysis, Anharmonic Simulation, and Tunneling. J. Phys. Chem. A 2015, 119, 2614–2627. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Andrijchenko, N.; Ahola, A.-E.; Domanskaya, A.; Räsänen, M.; Ermilov, A.; Nemukhin, A.; Khriachtchev, L. Interaction of phenol with xenon and nitrogen: Spectroscopic and computational characterization. J. Chem. Phys. 2012, 137, 134305. [Google Scholar] [CrossRef]
- Tylli, H.; Konschin, H. The self-association of phenol under cryogenic matrix conditions. J. Mol. Struct. 1986, 142, 571–574. [Google Scholar] [CrossRef]
- Giuliano, B.M.; Reva, I.; Lapinski, L.; Fausto, R. Infrared spectra and ultraviolet-tunable laser induced photochemistry of matrix-isolated phenol and phenol-d5. J. Chem. Phys. 2012, 136, 024505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapinski, L.; Reva, I.; Rostkowska, H.; Fausto, R.; Nowak, M.J. Near-IR-Induced, UV-Induced, and Spontaneous Isomerizations in 5-Methylcytosine and 5-Fluorocytosine. J. Phys. Chem. B 2014, 118, 2831–2841. [Google Scholar] [CrossRef]
- Truhlar, D.G. Tunneling in enzymatic and nonenzymatic hydrogen transfer reactions. J. Phys. Org. Chem. 2010, 23, 660–676. [Google Scholar] [CrossRef]
- Kästner, J. Theory and simulation of atom tunneling in chemical reactions. WIREs Comput. Mol. Sci. 2014, 4, 158–168. [Google Scholar] [CrossRef]
- Schreiner, P.R. Tunneling Control of Chemical Reactions: The Third Reactivity Paradigm. J. Am. Chem. Soc. 2017, 139, 15276–15283. [Google Scholar] [CrossRef]
- Wentzel, G. Eine Verallgemeinerung der Quantenbedingungen für die Zwecke der Wellenmechanik. Z. Phys. 1926, 38, 518–529. [Google Scholar] [CrossRef]
- Kramers, H.A. Wellenmechanik und halbzahlige Quantisierung. Z. Phys. 1926, 39, 828–840. [Google Scholar] [CrossRef]
- Brillouin, L. La mécanique ondulatoire de Schrödinger; une méthode générale de resolution par approximations successives. Comptes Rendus L’academie Sci. 1926, 183, 24–26. [Google Scholar]
- Borden, W.T. Reactions that involve tunneling by carbon and the role that calculations have played in their study. WIREs Comput. Mol. Sci. 2016, 6, 20–46. [Google Scholar] [CrossRef]
- Cooper, G.A.; Cobbin, M.R.; Ashfold, M.N.R. Effects of Ring Fluorination on the Ultraviolet Photodissociation Dynamics of Phenol. J. Phys. Chem. A 2020, 124, 9698–9709. [Google Scholar] [CrossRef]
- Yosida, K.; Suzuki, K.; Ishiuchi, S.-I.; Sakai, M.; Fujii, M.; Dessent, C.E.H.; Müller-Dethlefs, K. The PFI-ZEKE photoelectron spectrum of m-fluorophenol and its aqueous complexes: Comparing intermolecular vibrations in rotational isomers. Phys. Chem. Chem. Phys. 2002, 4, 2534–2538. [Google Scholar] [CrossRef]
- Fujimaki, E.; Fujii, A.; Ebata, T.; Mikami, N. Autoionization-detected infrared spectroscopy of intramolecular hydrogen bonds in aromatic cations. I. Principle and application to fluorophenol and methoxyphenol. J. Chem. Phys. 1999, 110, 4238–4247. [Google Scholar] [CrossRef]
- Reva, I.; Jesus, A.J.L.; Nunes, C.M.; Roque, J.P.L.; Fausto, R. UV-Induced Photochemistry of 1,3-Benzoxazole, 2-Isocyanophenol, and 2-Cyanophenol Isolated in Low-Temperature Ar Matrixes. J. Org. Chem. 2021, 86, 6126–6137. [Google Scholar] [CrossRef]
- Akai, N.; Kudoh, S.; Takayanagi, M.; Nakata, M. Photoreaction mechanisms of 2-bromophenols studied by low-temperature matrix-isolation infrared spectroscopy and density-functional-theory calculation. Chem. Phys. Lett. 2002, 363, 591–597. [Google Scholar] [CrossRef]
- Akai, N.; Kudoh, S.; Takayanagi, M.; Nakata, M. Photoreaction mechanisms of 2-chlorophenol and its multiple chloro-substituted derivatives studied by low-temperature matrix-isolation infrared spectroscopy and density-functional-theory calculations. J. Photochem. Photobiol. A 2001, 146, 49–57. [Google Scholar] [CrossRef]
- Kuş, N.; Sagdinc, S.; Fausto, R. Infrared Spectrum and UV-Induced Photochemistry of Matrix-Isolated 5-Hydroxyquinoline. J. Phys. Chem. A 2015, 119, 6296–6308. [Google Scholar] [CrossRef]
- Krupa, J.; Wierzejewska, M. UV-tunable laser induced photolysis of matrix isolated anisole. Chem. Phys. Lett. 2015, 618, 219–224. [Google Scholar] [CrossRef]
- Samanta, A.K.; Pandey, P.; Bandyopadhyay, B.; Chakraborty, T. Keto–enol tautomers of 1,2-cyclohexanedione in solid, liquid, vapour and a cold inert gas matrix: Infrared spectroscopy and quantum chemistry calculation. J. Mol. Struct. 2010, 963, 234–239. [Google Scholar] [CrossRef]
- Breda, S.; Reva, I.; Lapinski, L.; Fausto, R. Matrix isolation FTIR and theoretical study of α-pyrone photochemistry. Phys. Chem. Chem. Phys. 2004, 6, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, B.M.; Reva, I.; Fausto, R. Infrared Spectra and Photochemistry of Matrix-Isolated Pyrrole-2-carbaldehyde. J. Phys. Chem. A 2010, 114, 2506–2517. [Google Scholar] [CrossRef] [Green Version]
- Dubost, H. Infrared absorption spectra of carbon monoxide in rare gas matrices. Chem. Phys. 1976, 12, 139–151. [Google Scholar] [CrossRef]
- Miyazaki, J.; Toh, S.Y.; Moore, B.; Djuricanin, P.; Momose, T. UV photochemistry of 1,3-cyclohexadiene isolated in solid parahydrogen. J. Mol. Struct. 2021, 1224, 128986. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Reva, I.; Nowak, M.J.; Lapinski, L.; Fausto, R. Hydrogen atom transfer reactions in thiophenol: Photogeneration of two new thione isomers. Phys. Chem. Chem. Phys. 2015, 17, 4888–4898. [Google Scholar] [CrossRef] [PubMed]
- Schachtschneider, J.H.; Mortimer, F.S. Vibrational Analysis of Polyatomic Molecules. VI. FORTRAN IV Programs for Solving the Vibrational Secular Equation and for the Least-Squares Refinement of Force Constants. Project No. 31450. Structural Interpretation of Spectra; Shell Development Co.: Emeryville, CA, USA, 1969. [Google Scholar]
- Pulay, P.; Fogarasi, G.; Pang, F.; Boggs, J.E. Systematic ab initio gradient calculation of molecular geometries, force constants, and dipole moment derivatives. J. Am. Chem. Soc. 1979, 101, 2550–2560. [Google Scholar] [CrossRef]
- Schleif, T.; Prado Merini, M.; Henkel, S.; Sander, W. Solvation Effects on Quantum Tunneling Reactions. Acc. Chem. Res. 2022, 55, 2180–2190. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| trans | cis | |
|---|---|---|
| B3LYP/aug-cc-pVTZ | ||
| ΔEel | 0.00 | 0.58 |
| ΔE0 | 0.00 | 0.69 |
| ΔG | 0.00 | 0.70 |
| Pop. | 57.0 | 43.0 |
| µ | 2.75 | 0.83 |
| ΔE‡el | 16.5 | 15.9 |
| MP2/aug-cc-pVTZ (QCISD/aug-cc-pVDZ) | ||
| ΔEel | 0.00 (0.00) | 0.70 (0.63) |
| ΔE0 | 0.00 | 0.75 |
| ΔG | 0.00 | 0.73 |
| Pop. | 57.3 | 42.7 |
| µ | 2.70 (3.11) | 0.73 (0.78) |
| ΔE‡el | 15.0 | 14.3 |
| Experimental a | Calculated | Assignment, PED (%) c | ||
|---|---|---|---|---|
| N2, 14 K | Ar, 16 K | b | I | |
| A′ modes | ||||
| 3624/3620 (vs) | 3639/3638/3632 (vs) | 3623.7 | 69.2 | νOH (100) |
| n.o. | n.o. | 3147.8 | <0.1 | νC6H (100) |
| n.o. | n.o. | 3146.9 | 1.0 | νC4H (92) |
| 3064 (vw) | 3069/3064 (vw) | 3122.0 | 7.8 | νC3H (81), νC2H (11) |
| 3047 (vw) | 3047 (vw) | 3098.7 | 9.5 | νC2H (89), νC3H (11) |
| 1614 (vs) | 1613 (vs) | 1618.5 | 155.7 | νcCC (66) |
| 1612/1611 (s) | 1611 (s) | 1609.2 | 71.3 | νbCC (70) |
| 1496/1495 (s) | 1495/1494 (vs) | 1490.1 | 118.7 | νdCC (38), δC3H (26), δC6H (13), δC2H + δC4H (11) |
| 1492/1489 (w) | 1489/1488 (w) | 1481.6 | 16.1 | νeCC (39), δC2H − δC4H (31), δOH (10) |
| 1328/1326 (w) | 1325/1324 (w) | 1325.4 | 11.1 | νfCC (78), δC2H + δC4H (13) |
| 1302 (s) | 1299/1298 (s) | 1299.3 | 80.2 | δC2H + δC4H (21), δC3H (16), δC6H (15), νCO (15), δOH (12) |
| 1291/1289 (m) | 1286 (m) | 1279.7 | 45.0 | νCO (21), νC5F (17), νaCC (15), δC2H − δC4H (12), δaR (12) |
| 1202 (m) | 1202 (m) | 1188.0 | 31.6 | δOH (41), δC3H (16), νC5F (14) |
| 1158 (w) | 1155/1153 (w) | 1155.4 | 13.5 | δC2H + δC4H (38), δC3H (25), νfCC (15), δOH (10) |
| 1132/1130/1129 (vs) | 1128/1127/1126 (vs) | 1123.1 | 171.8 | δC6H (43), νC5F (18), νCO (13) |
| n.o. | n.o. | 1074.9 | 1.8 | δC2H − δC4H (42), νeCC (39) |
| 1005/1004 (vw) | 1006/1004 (vw) | 998.9 | 4.5 | δaR (58), νaCC (40) |
| 958/957/955 (s) | 958/956/955 (s) | 952.3 | 78.4 | νdCC (40), νC5F (14), νCO (13) |
| 740/738 (vw) | 739/738 (vw) | 736.4 | 4.4 | νaCC (25), δcR (20), δaR (19), νC5F (12), νCO (11) |
| 532/530 (vw) | 534/531 (vw) | 528.1 | 3.8 | δbR (36), δcR (28), νCO (11) |
| 515 (vw) | 516/515 (vw) | 511.3 | 5.8 | δbR (37), δcR (35), νC5F (10) |
| 477 (vw) | 474/473 (vw) | 468.8 | 9.7 | δCO + δCF (83) |
| n.i. | n.i. | 324.5 | 1.4 | δCO − δCF (80), νeCC (12) |
| A″ modes | ||||
| n.o. | n.o. | 963.3 | 0.2 | γcCH (84) |
| 862/860/855/854 (w) | 862/859/857/853 (m) | 866.8 | 32.6 | γbCH (42), γaCH (34), γCF (15) |
| n.o. | n.o. | 855.9 | 1.7 | γdCH (70), γbCH (27) |
| 771/769/766 (m) | 766/763/760 (m) | 764.7 | 38.9 | γaCH (39), γcCH (17), γbCH (15), γdCH (13) |
| 681/680/678 (w) | 679/677 (w) | 681.7 | 26.7 | τaR (87) |
| n.o. | n.o. | 611.8 | 0.3 | γCF (40), γCO (39), τbR (14) |
| n.o. | n.o. | 459.5 | 2.4 | τcR (59), τbR (17), γCF (13), γCO (10) |
| n.i. | n.i. | 343.9 | 94.7 | τOH (99) |
| n.i. | n.i. | 236.0 | <0.1 | τbR (60), τcR (33) |
| n.i. | n.i. | 220.5 | 2.2 | τaR (23), γCO (22), τbR (22), τcR (18), γCF (16) |
| Experimental a | Calculated | Assignment, PED (%) c | |
|---|---|---|---|
| N2, 14 K | b | I | |
| A′ modes | |||
| 3627 (vs) | 3624.8 | 68.1 | νOH (100) |
| n.o. | 3147.0 | 0.8 | νC4H (85), νC3H (10) |
| n.o. | 3138.3 | 3.7 | νC2H (83) |
| n.o. | 3117.8 | 0.1 | νC6H (71), νC3H (24) |
| 3064 (vw) | 3116.4 | 8.0 | νC3H (60), νC6H (29) |
| 1625 (s) | 1623.6 | 91.5 | νbCC (56), νcCC (16) |
| 1605/1602 (vs) | 1604.8 | 145.8 | νcCC (52), νbCC (16) |
| 1506 (s) | 1502.9 | 73.2 | νdCC (36), δC3H (21), δC6H (14) |
| 1463 (m) | 1462.0 | 41.3 | νeCC (41), δC2H − δC4H (32) |
| 1328/1326 (vw) | 1325.6 | 8.8 | νfCC (78), δOH (12) |
| 1312 (vw) | 1314.1 | 8.6 | δC2H + δC4H (25), δC3H (17), δOH (14), νCO (13), δC6H (12) |
| 1286/1285 (s) | 1278.3 | 72.1 | νCO (20), νC5F (19), νaCC (15), δaR (12), δC6H (11) |
| 1190/1186/1179 (vs) | 1169.2 | 166.0 | δOH (29), δC3H (26), νcCC (11), νCO (10) |
| 1164 (w) | 1158.9 | 25.6 | δOH (26), νfCC (19), δC2H + δC4H (18), δC6H (15) |
| 1140/1139/1137 (s) | 1133.5 | 92.0 | δC6H (29), δC2H + δC4H (28), νC5F (17) |
| 1070 (vw) | 1068.1 | 11.8 | δC2H − δC4H (43), νeCC (40) |
| 1005/1004 (vw) | 999.4 | 5.8 | δaR (57), νaCC (41) |
| 960 (m) | 954.3 | 46.2 | νdCC (39), νC5F (15), νCO (14) |
| 740/738 (vw) | 738.7 | 5.6 | νaCC (25), δcR (20), δaR (19), νC5F (12), νCO (11) |
| 532/530 (vw) | 527.0 | 4.1 | δbR (38), δcR (26), νCO (11) |
| 515 (vw) | 511.0 | 7.5 | δcR (37), δbR (37), νC5F (10) |
| 477 (vw) | 472.3 | 3.9 | δCO + δCF (85) |
| n.i. | 323.2 | 7.6 | δCO − δCF (80), νeCC (12) |
| A″ modes | |||
| n.o. | 971.8 | 0.4 | γcCH (84) |
| n.o. | 872.5 | <0.1 | γbCH (60), γdCH (40) |
| 840/839 (m) | 843.5 | 46.8 | γaCH (44), γbCH (15), γCO (15), γCF (14), γcCH (12) |
| 774 (w) | 771.6 | 23.1 | γaCH (34), γdCH (25), γcCH (20), γbCH (14) |
| 681/680/678 (w) | 681.8 | 23.7 | τaR (89) |
| n.o. | 608.7 | <0.1 | γCF (40), γCO (37), τbR (15) |
| 456 (vw) | 457.2 | 9.4 | τcR (59), τbR (17), γCF (12), γCO (11) |
| n.i. | 348.8 | 97.0 | τOH (99) |
| n.i. | 236.3 | 1.9 | τbR (58), τcR (33), γCF (12) |
| n.i. | 221.6 | <0.1 | τaR (23), τbR (23), γCO (22), τcR (18), γCF (16) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes Jesus, A.J.; de Lucena Júnior, J.R.; Fausto, R.; Reva, I. Infrared Spectra and Phototransformations of meta-Fluorophenol Isolated in Argon and Nitrogen Matrices. Molecules 2022, 27, 8248. https://doi.org/10.3390/molecules27238248
Lopes Jesus AJ, de Lucena Júnior JR, Fausto R, Reva I. Infrared Spectra and Phototransformations of meta-Fluorophenol Isolated in Argon and Nitrogen Matrices. Molecules. 2022; 27(23):8248. https://doi.org/10.3390/molecules27238248
Chicago/Turabian StyleLopes Jesus, A. J., Juracy Regis de Lucena Júnior, Rui Fausto, and Igor Reva. 2022. "Infrared Spectra and Phototransformations of meta-Fluorophenol Isolated in Argon and Nitrogen Matrices" Molecules 27, no. 23: 8248. https://doi.org/10.3390/molecules27238248
APA StyleLopes Jesus, A. J., de Lucena Júnior, J. R., Fausto, R., & Reva, I. (2022). Infrared Spectra and Phototransformations of meta-Fluorophenol Isolated in Argon and Nitrogen Matrices. Molecules, 27(23), 8248. https://doi.org/10.3390/molecules27238248