
Determination of Organophosphate Ester Metabolites in Seafood Species by QuEChERS-SPE Followed by LC-HRMS



Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Standards
2.2. Sampling
2.3. QuEChERS Extraction and Clean-Up
2.4. Liquid Chromatography Coupled to High-Resolution Mass Spectrometry
3. Results and Discussion
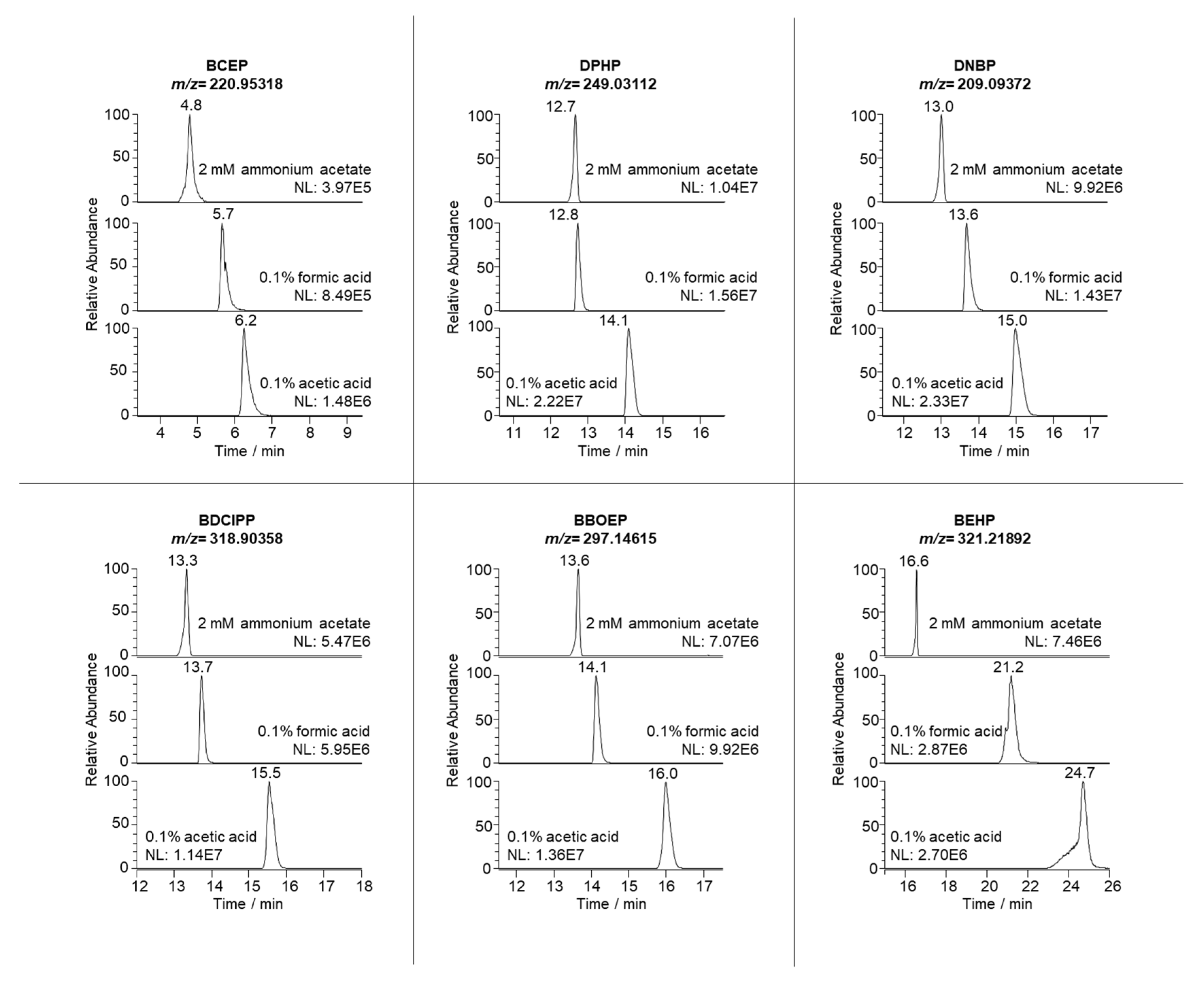
3.1. Liquid Chromatography Coupled to High-Resolution Mass Spectrometry
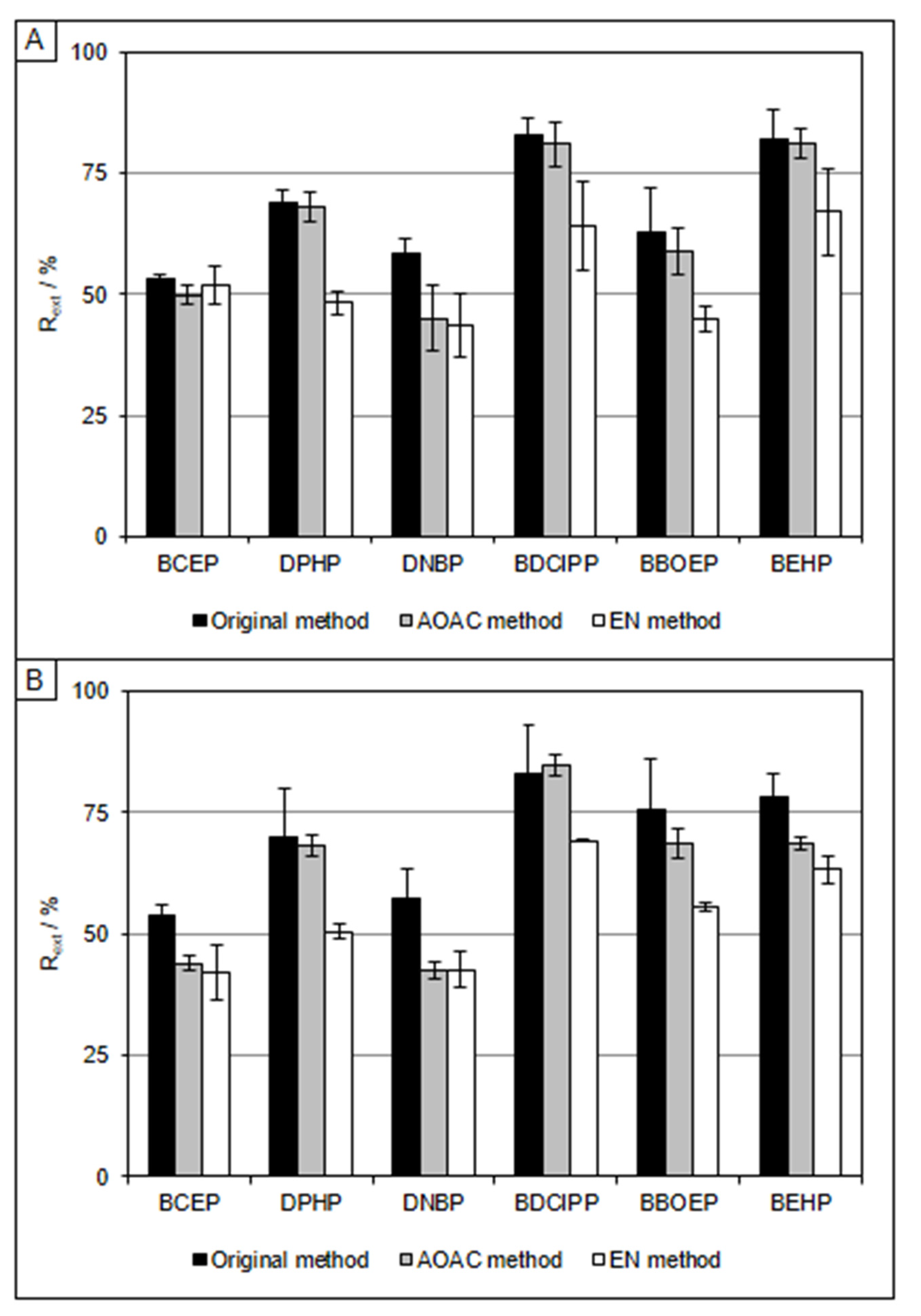
3.2. QuEChERS Extraction
3.3. Clean-Up Strategies
3.4. Method Validation
3.5. Application to Seafood Samples
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Han, L.; Sapozhnikova, Y.; Nuñez, A. Analysis and Occurrence of Organophosphate Esters in Meats and Fish Consumed in the United States. J. Agric. Food Chem. 2019, 67, 12652–12662. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhong, W.; Xiao, B.; Liu, Q.; Yang, L.; Covaci, A.; Zhu, L. Bioavailability and Biomagnification of Organophosphate Esters in the Food Web of Taihu Lake, China: Impacts of Chemical Properties and Metabolism. Environ. Int. 2019, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Yang, L.; Yu, Y.; Guan, Q.; Liu, X.; Li, L.; Chen, D. Co-Existence of Organophosphate Di- and Tri-Esters in House Dust from South China and Midwestern United States: Implications for Human Exposure. Environ. Sci. Technol. 2019, 53, 4784–4793. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, X.; Thai, P.; Mueller, J.F.; Gallen, C.; Li, Y.; Baduel, C. Development and Validation of a Multi-Residue Method for the Analysis of Brominated and Organophosphate Flame Retardants in Indoor Dust. Talanta 2017, 164, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, W.; Martínez-Moral, M.P.; Sun, H.; Kannan, K. Metabolites of Organophosphate Esters in Urine from the United States: Concentrations, Temporal Variability, and Exposure Assessment. Environ. Int. 2019, 122, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Guo, J.; Wang, Y.; Li, Z.; Liang, K.; Corcoran, M.B.; Hosseini, S.; Bonina, S.M.C.; Rockne, K.J.; Sturchio, N.C.; et al. Organophosphate Esters in Sediment of the Great Lakes. Environ. Sci. Technol. 2017, 51, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Bin, L.; Cui, J.; Nyobe, D.; Li, P.; Huang, S.; Fu, F.; Tang, B. Tracing the Occurrence of Organophosphate Ester along the River Flow Path and Textile Wastewater Treatment Processes by Using Dissolved Organic Matters as an Indicator. Sci. Total Environ. 2020, 722, 137895. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shao, H.; Wu, M.; Zhang, J.; Li, D.; Li, J.; Wang, H.; Shi, W.; Xu, G. Occurrence, Distribution, and Potential Sources of Organophosphate Esters in Urban and Rural Surface Water in Shanghai, China. Arch. Environ. Contam. Toxicol. 2019, 77, 115–126. [Google Scholar] [CrossRef]
- Strobel, A.; Willmore, W.G.; Sonne, C.; Dietz, R.; Letcher, R.J. Organophosphate Esters in East Greenland Polar Bears and Ringed Seals: Adipose Tissue Concentrations and In Vitro Depletion and Metabolite Formation. Chemosphere 2018, 196, 240–250. [Google Scholar] [CrossRef]
- Greaves, A.K.; Letcher, R.J. A Review of Organophosphate Esters in the Environment from Biological Effects to Distribution and Fate. Bull. Environ. Contam. Toxicol. 2017, 98, 2–7. [Google Scholar] [CrossRef]
- Wang, G.; Du, Z.; Chen, H.; Su, Y.; Gao, S.; Mao, L. Tissue-Specific Accumulation, Depuration, and Transformation of Triphenyl Phosphate (TPHP) in Adult Zebrafish (Danio Rerio). Environ. Sci. Technol. 2016, 50, 13555–13564. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, H.; Du, Z.; Li, J.; Wang, Z.; Gao, S. In Vivo Metabolism of Organophosphate Flame Retardants and Distribution of Their Main Metabolites in Adult Zebrafish. Sci. Total Environ. 2017, 590–591, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Huang, C.; Rao, K.; Xu, Y.; Wang, Z. Characterized In Vitro Metabolism Kinetics of Alkyl Organophosphate Esters in Fish Liver and Intestinal Microsomes. Environ. Sci. Technol. 2018, 52, 3202–3210. [Google Scholar] [CrossRef] [PubMed]
- Greaves, A.K.; Su, G.; Letcher, R.J. Environmentally Relevant Organophosphate Triesters in Herring Gulls: In Vitro Biotransformation and Kinetics and Diester Metabolite Formation Using a Hepatic Microsomal Assay. Toxicol. Appl. Pharmacol. 2016, 308, 59–65. [Google Scholar] [CrossRef]
- Xu, L.; Hu, Q.; Liu, J.; Liu, S.; Liu, C.; Deng, Q.; Zeng, X.; Yu, Z. Occurrence of Organophosphate Esters and Their Diesters Degradation Products in Industrial Wastewater Treatment Plants in China: Implication for the Usage and Potential Degradation during Production Processing. Environ. Pollut. 2019, 250, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.A.; Reddam, A.; Dasgupta, S.; Zhang, S.; Stapleton, H.M.; Volz, D.C. Diphenyl Phosphate-Induced Toxicity during Embryonic Development. Environ. Sci. Technol. 2019, 53, 3908–3916. [Google Scholar] [CrossRef]
- Lee, J.S.; Morita, Y.; Kawai, Y.K.; Covaci, A.; Kubota, A. Developmental Circulatory Failure Caused by Metabolites of Organophosphorus Flame Retardants in Zebrafish, Danio Rerio. Chemosphere 2020, 246, 125738. [Google Scholar] [CrossRef]
- Noyes, P.D.; Haggard, D.E.; Gonnerman, G.D.; Tanguay, R.L. Advanced Morphological—Behavioral Test Platform Reveals Neurodevelopmental Defects in Embryonic Zebrafish Exposed to Comprehensive Suite of Halogenated and Organophosphate Flame Retardants. Toxicol. Sci. 2015, 145, 177–195. [Google Scholar] [CrossRef] [Green Version]
- Su, G.; Crump, D.; Letcher, R.J.; Kennedy, S.W. Rapid In Vitro Metabolism of the Flame Retardant Triphenyl Phosphate and Effects on Cytotoxicity and MRNA Expression in Chicken Embryonic Hepatocytes. Environ. Sci. Technol. 2014, 48, 13511–13519. [Google Scholar] [CrossRef]
- Yang, F.W.; Zhao, G.P.; Ren, F.Z.; Pang, G.F.; Li, Y.X. Assessment of the endocrine-disrupting effects of diethyl phosphate, a nonspecific metabolite of organophosphorus pesticides, by in vivo and in silico approaches. Environ. Int. 2020, 135, 105383. [Google Scholar] [CrossRef]
- ECHA Annex III Inventory. Available online: https://echa.europa.eu/information-on-chemicals/annex-iii-inventory (accessed on 10 October 2022).
- Auta, H.S.; Emenike, C.U.; Fauziah, S.H. Distribution and Importance of Microplastics in the Marine Environment: A Review of the Sources, Fate, Effects, and Potential Solutions. Environ. Int. 2017, 102, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Castro-Jiménez, J.; Ratola, N. An Innovative Approach for the Simultaneous Quantitative Screening of Organic Plastic Additives in Complex Matrices in Marine Coastal Areas. Environ. Sci. Pollut. Res. 2020, 27, 11450–11457. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Miller, P.; von Hippel, F.A.; Buck, C.L.; Carpenter, D.O.; Salamova, A. Legacy and Emerging Semi-Volatile Organic Compounds in Sentinel Fish from an Arctic Formerly Used Defense Site in Alaska. Environ. Pollut. 2020, 259, 113872. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Shi, H.; Du, Z.; Chen, H.; Peng, J.; Gao, S. Bioaccumulation Mechanism of Organophosphate Esters in Adult Zebrafish (Danio Rerio). Environ. Pollut. 2017, 229, 177–187. [Google Scholar] [CrossRef]
- Hou, R.; Liu, C.; Gao, X.; Xu, Y.; Zha, J.; Wang, Z. Accumulation and Distribution of Organophosphate Flame Retardants (PFRs) and Their Di-Alkyl Phosphates (DAPs) Metabolites in Different Freshwater Fish from Locations around Beijing, China. Environ. Pollut. 2017, 229, 548–556. [Google Scholar] [CrossRef]
- Cequier, E.; Marcé, R.M.; Becher, G.; Thomsen, C. A High-Throughput Method for Determination of Metabolites of Organophosphate Flame Retardants in Urine by Ultra Performance Liquid Chromatography-High Resolution Mass Spectrometry. Anal. Chim. Acta 2014, 845, 98–104. [Google Scholar] [CrossRef]
- Wang, Y.; Kannan, P.; Halden, R.U.; Kannan, K. A Nationwide Survey of 31 Organophosphate Esters in Sewage Sludge from the United States. Sci. Total Environ. 2019, 655, 446–453. [Google Scholar] [CrossRef]
- Alves, A.; Covaci, A.; Voorspoels, S. Method Development for Assessing the Human Exposure to Organophosphate Flame Retardants in Hair and Nails. Chemosphere 2017, 168, 692–698. [Google Scholar] [CrossRef]
- Choi, Y.; Jeon, J.; Choi, Y.; Kim, S.D. Characterizing Biotransformation Products and Pathways of the Flame Retardant Triphenyl Phosphate in Daphnia Magna Using Non-Target Screening. Sci. Total Environ. 2020, 708, 135106. [Google Scholar] [CrossRef]
- Fu, L.; Du, B.; Wang, F.; Lam, J.C.W.; Zeng, L.; Zeng, E.Y. Organophosphate Triesters and Diester Degradation Products in Municipal Sludge from Wastewater Treatment Plants in China: Spatial Patterns and Ecological Implications. Environ. Sci. Technol. 2017, 51, 13614–13623. [Google Scholar] [CrossRef]
- Hidalgo-Serrano, M.; Borrull, F.; Marcé, R.M.; Pocurull, E. Simple Method for Determining Phthalate Diesters and Their Metabolites in Seafood Species Using QuEChERS Extraction and Liquid Chromatography-High Resolution Mass Spectrometry. Food Chem. 2021, 336, 127722. [Google Scholar] [CrossRef] [PubMed]
- Castro, Ó.; Pocurull, E.; Borrull, F. Determination of Organophosphate Ester Flame Retardants and Plasticisers in Fish Samples by QuEChERs Followed by Gas Chromatography-Tandem Mass Spectrometry. Exposure and Risk Assessment through Fish Consumption. J. Chromatogr. A 2020, 1626, 461356. [Google Scholar] [CrossRef]
- Perestrelo, R.; Silva, P.; Porto-Figueira, P.; Pereira, J.A.M.; Silva, C.; Medina, S.; Câmara, J.S. QuEChERS—Fundamentals, Relevant Improvements, Applications and Future Trends. Anal. Chim. Acta 2019, 1070, 1–28. [Google Scholar] [CrossRef]
- Schindler, B.K.; Förster, K.; Angerer, J. Determination of Human Urinary Organophosphate Flame Retardant Metabolites by Solid-Phase Extraction and Gas Chromatography-Tandem Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 375–381. [Google Scholar] [CrossRef]
- Kim, J.W.; Isobe, T.; Chang, K.H.; Amano, A.; Maneja, R.H.; Zamora, P.B.; Siringan, F.P.; Tanabe, S. Levels and Distribution of Organophosphorus Flame Retardants and Plasticizers in Fishes from Manila Bay, the Philippines. Environ. Pollut. 2011, 159, 3653–3659. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound and Structure | Quantifier Ion (m/z) | Qualifier Ions (m/z) |
|---|---|---|
Bis(2-chloroethyl) phosphate (BCEP) | [C4H8PO4Cl2]− 220.95318 | [PO3]− 78.95796 |
Diphenyl phosphate (DPHP) | [C12H10PO4]− 249.03112 | [C6H5O]− 93.03349 [C6H4PO3]− 154.98926 |
Dibutyl phosphate (DNBP) | [C8H18PO4]− 209.09372 | [PO3]− 78.95796 [C4H10PO4]− 153.03112 |
Bis(1,3-dichloro-2-propyl) phosphate (BDCIPP) | [C6H11PO4Cl337Cl]− 318.90358 | [PO3]− 78.95796 |
Bis(2-butoxyethyl) phosphate (BBOEP) | [C12H26PO6]− 297.14615 | [PO3]− 78.95796 [C6H14PO5]− 197.05734 |
Bis(2-ethylhexyl) phosphate (BEHP) | [C16H34PO4]− 321.21892 | [PO3]− 78.95796 [C8H18PO4]− 209.09372 |
| Compound | tR (min) | Hake (Merluccius merluccius) | Mackerel (Scomber scombrus) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Rrel (%) | ME (%) | MLOD (ng g−1) | MLOQ (ng g−1) | Rrel (%) | ME (%) | MLOD (ng g−1) | MLOQ (ng g−1) | ||
| BCEP | 4.8 | 113 | −2 | 25 | 50 | 107 | −1 | 50 | 75 |
| DPHP | 12.6 | 108 | −40 | 1.0 | 7.5 | 99 | −45 | 1.0 | 2.5 |
| DNBP | 13.0 | 138 | −39 | 2.5 | 5.0 | 122 | −35 | 1.0 | 2.5 |
| BDCIPP | 13.3 | 104 | −24 | 2.5 | 5.0 | 100 | −11 | 7.5 | 10 |
| BBOEP | 13.6 | 108 | −28 | 25 | 50 | 89 | −21 | 25 | 50 |
| BEHP | 16.6 | 38 | −70 | 50 | 75 | 22 | −56 | 50 | 75 |
| Compound | Seafood with Low Lipid Content (<10%) | Seafood with High Lipid Content (>10%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Hake (Merluccius merluccius) | Sole (Solea solea) | Cod (Gadus morhua) | Squid (Loligo vulgaris) | Sardine (Sardina pilchardus) | Tuna (Thunnus thynnus) | Mackerel (Scomber scombrus) | Salmon (Salmo salar) | |
| DPHP | 59 (7) | 54 (4) | 68 (4) | 48 (8) | 42 (1) | 75 (4) | 100 (1) | 55 (10) |
| DNBP | 67 (2) | 36 (13) | 54 (0.3) | 32 (3) | 33 (7) | 50 (9) | 79 (1) | 30 (2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hidalgo-Serrano, M.; Borrull, F.; Pocurull, E.; Marcé, R.M. Determination of Organophosphate Ester Metabolites in Seafood Species by QuEChERS-SPE Followed by LC-HRMS. Molecules 2022, 27, 8635. https://doi.org/10.3390/molecules27238635
Hidalgo-Serrano M, Borrull F, Pocurull E, Marcé RM. Determination of Organophosphate Ester Metabolites in Seafood Species by QuEChERS-SPE Followed by LC-HRMS. Molecules. 2022; 27(23):8635. https://doi.org/10.3390/molecules27238635
Chicago/Turabian StyleHidalgo-Serrano, Míriam, Francesc Borrull, Eva Pocurull, and Rosa Maria Marcé. 2022. "Determination of Organophosphate Ester Metabolites in Seafood Species by QuEChERS-SPE Followed by LC-HRMS" Molecules 27, no. 23: 8635. https://doi.org/10.3390/molecules27238635
APA StyleHidalgo-Serrano, M., Borrull, F., Pocurull, E., & Marcé, R. M. (2022). Determination of Organophosphate Ester Metabolites in Seafood Species by QuEChERS-SPE Followed by LC-HRMS. Molecules, 27(23), 8635. https://doi.org/10.3390/molecules27238635