
Effects of Cocrystallization on the Structure and Properties of Melt-Cast Explosive 2,4-Dinitroanisole: A Computational Study



Abstract
:1. Introduction
2. Results
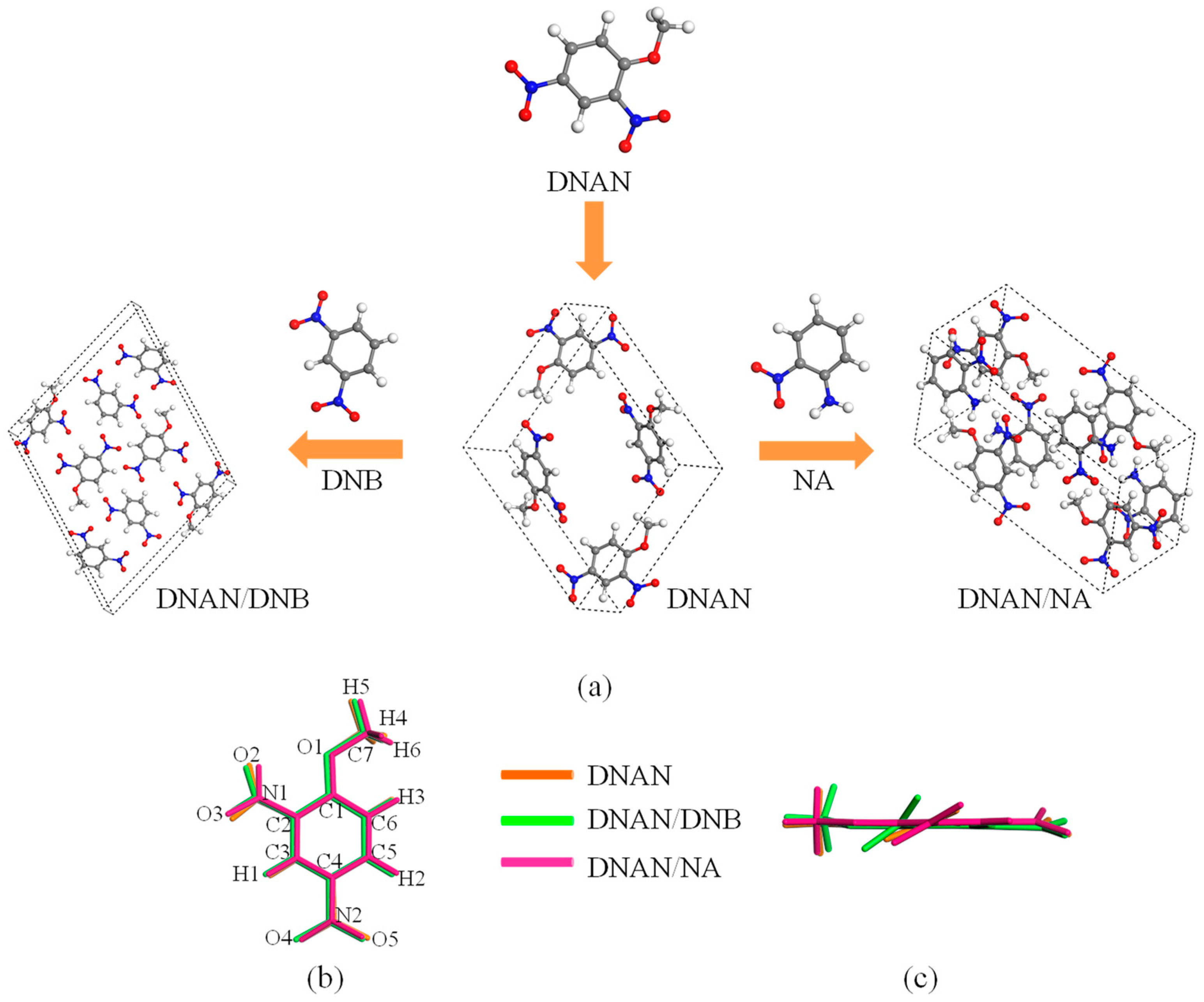
2.1. Crystal Structure and Stacking
2.1.1. Crystal Structure
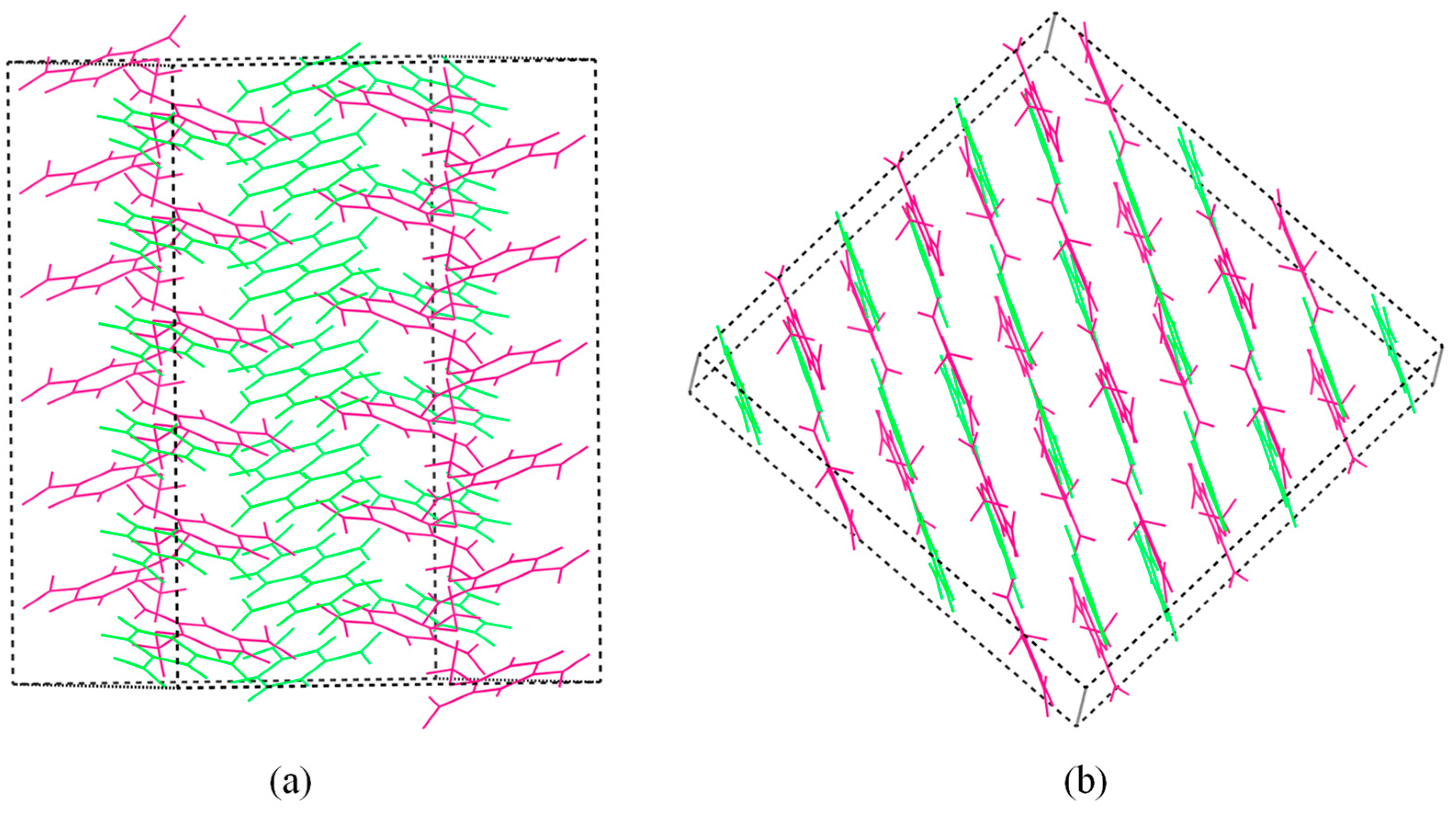
2.1.2. Crystal Stacking
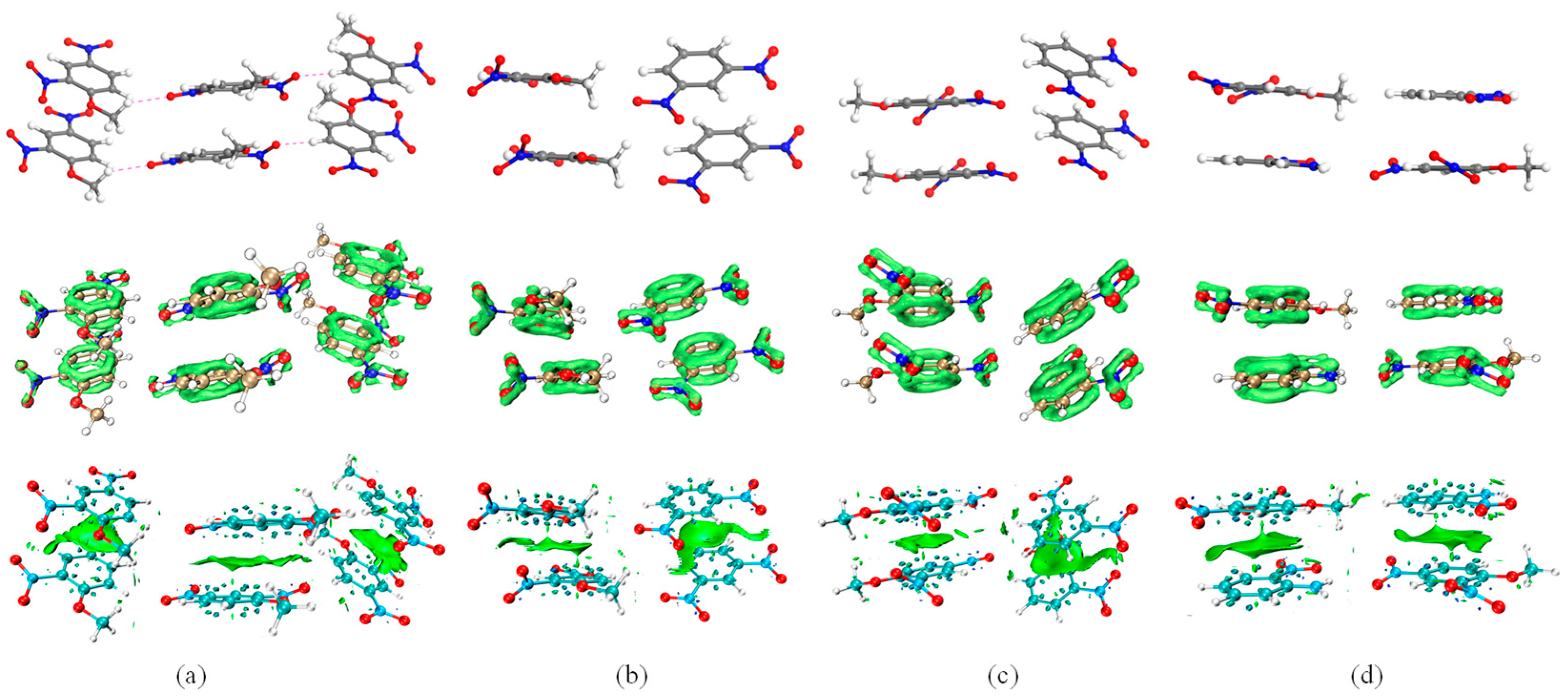
2.2. Non-Covalent Interactions
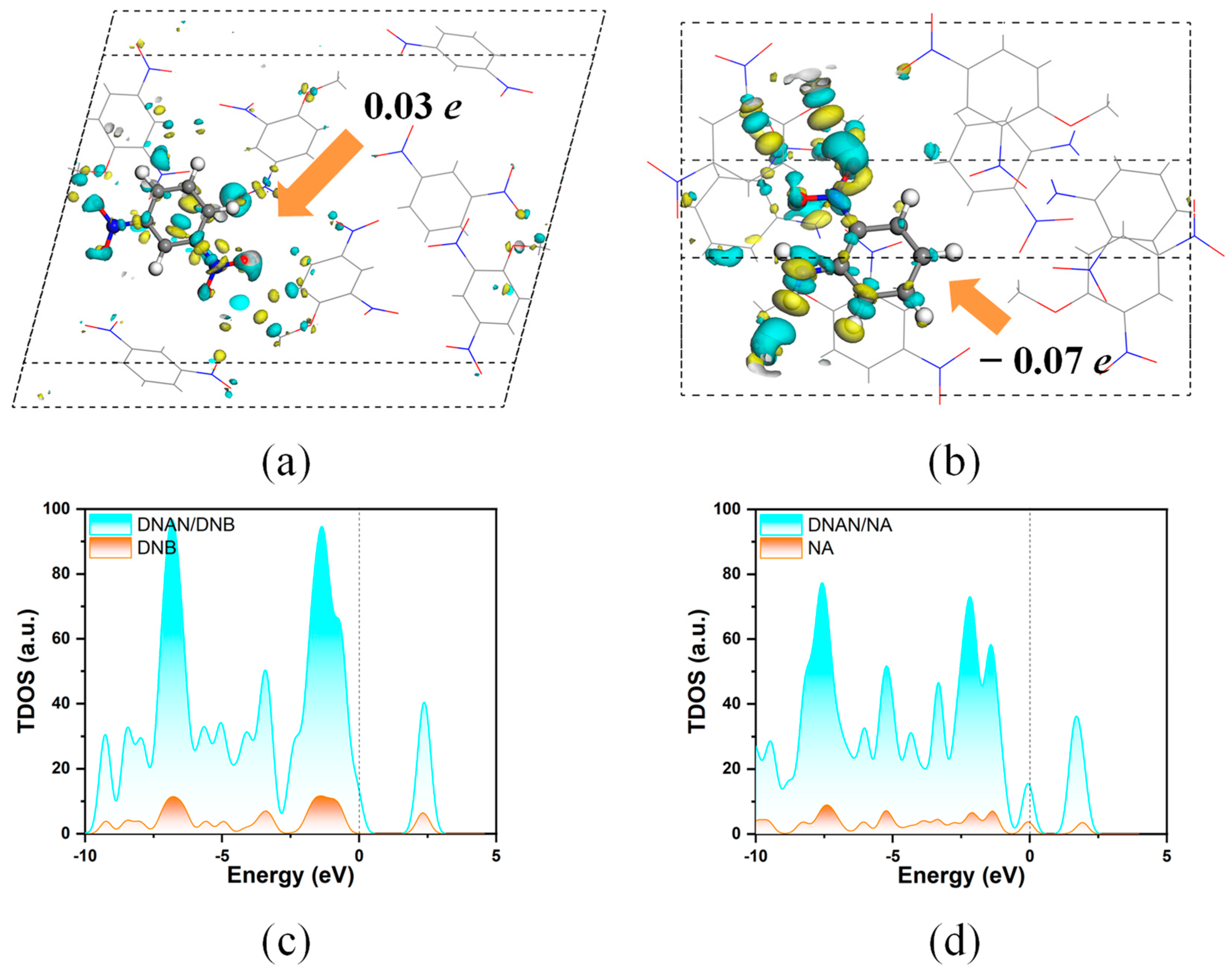
2.2.1. Interlayer Interactions
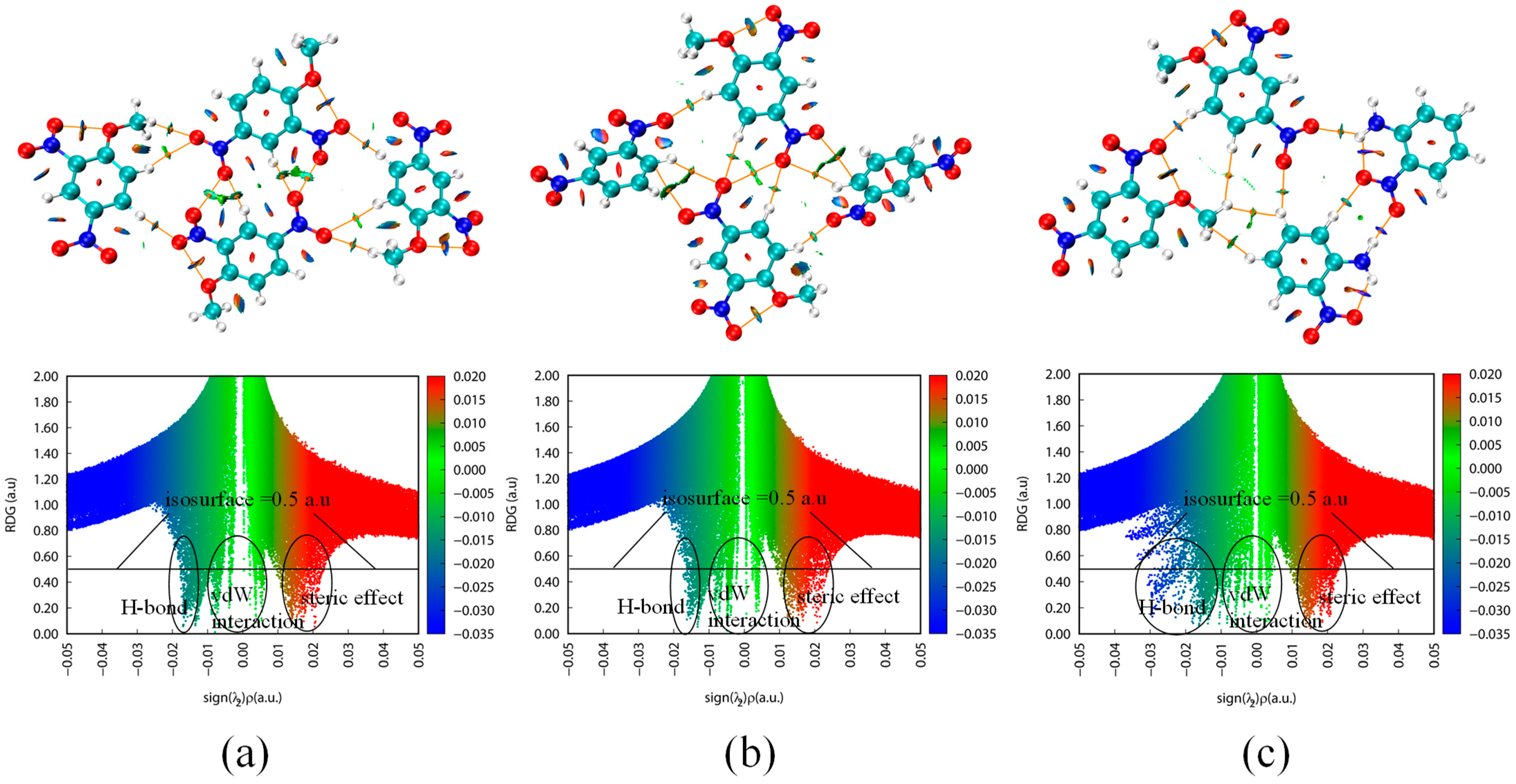
2.2.2. Intralayer Interactions
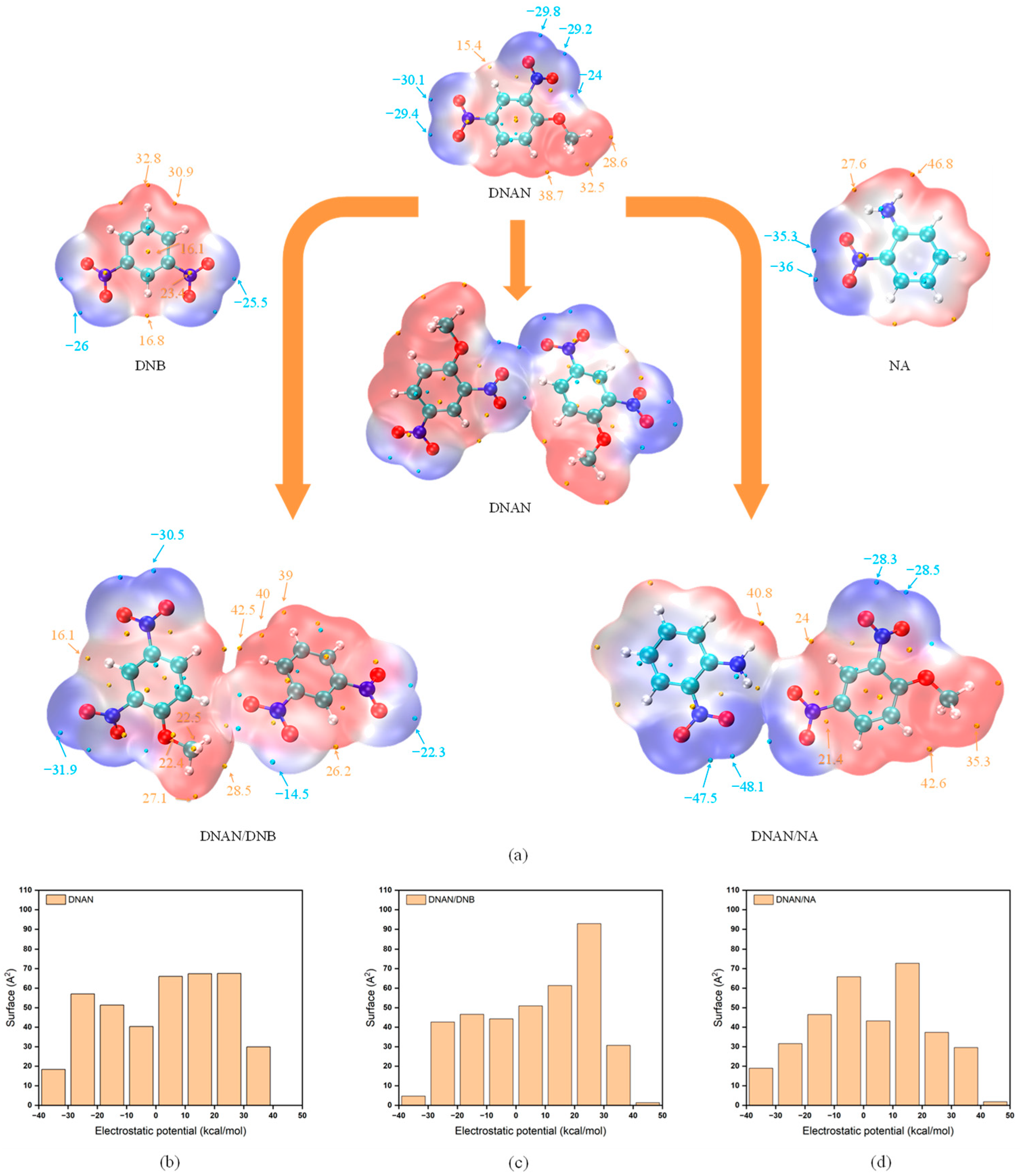
2.2.3. ESP Analysis
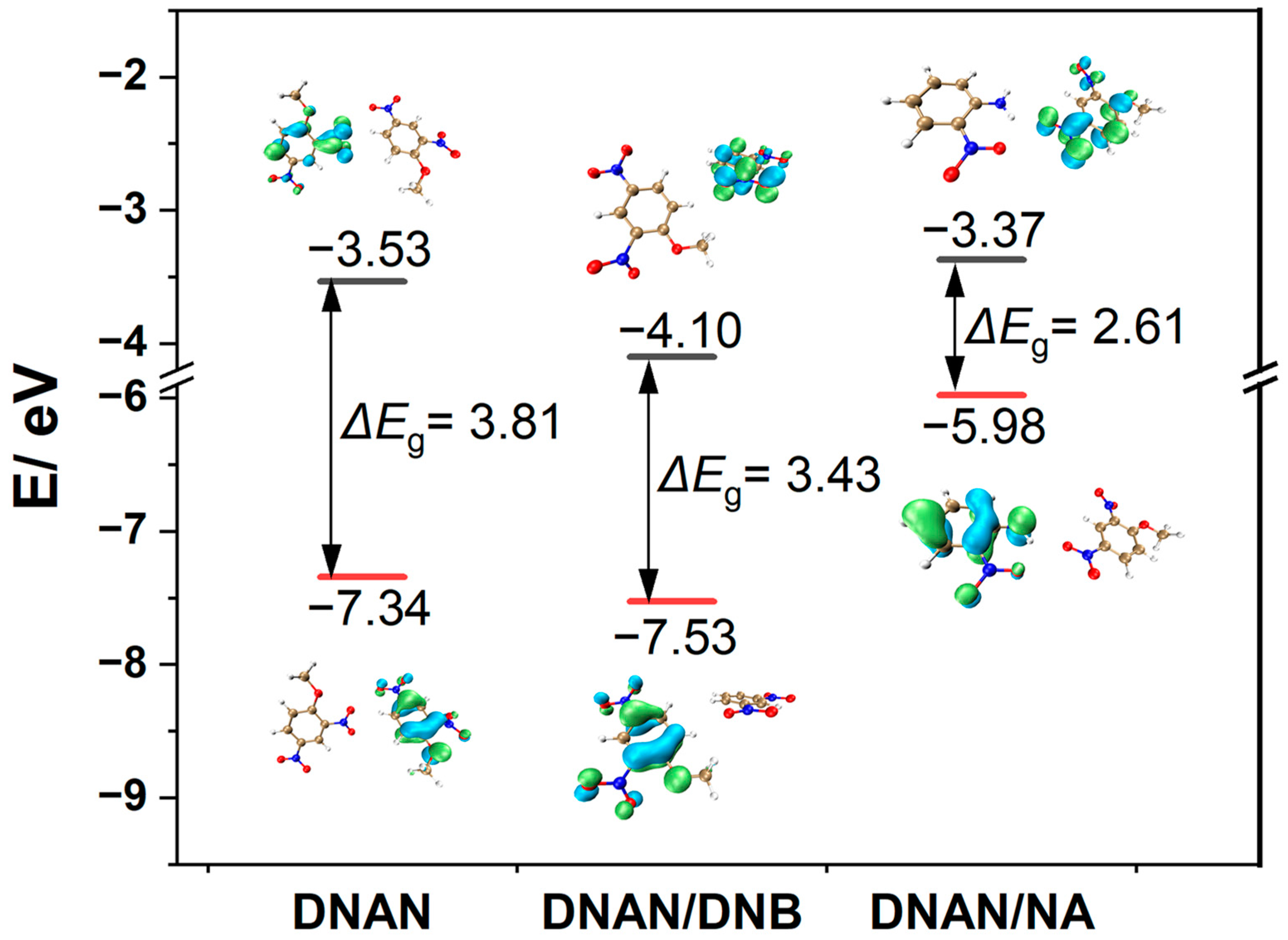
2.3. Electronic Properties
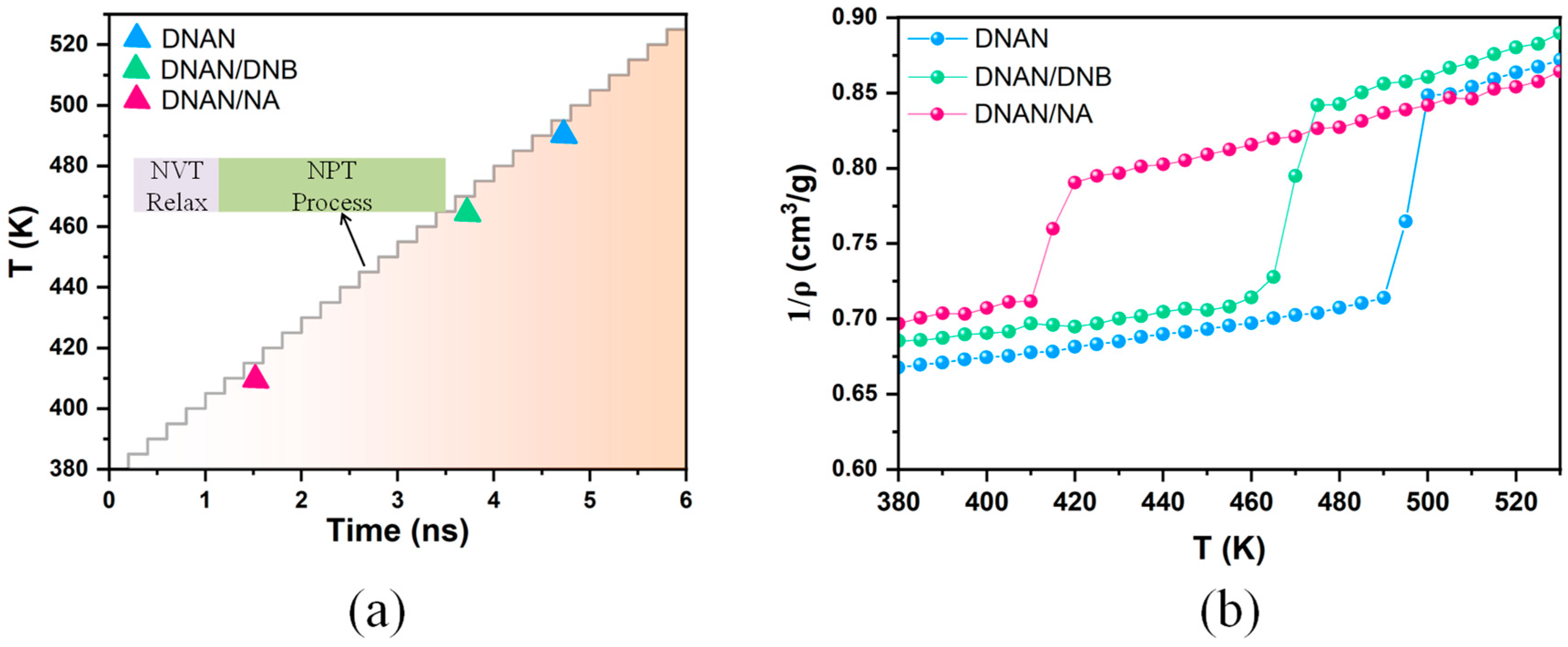
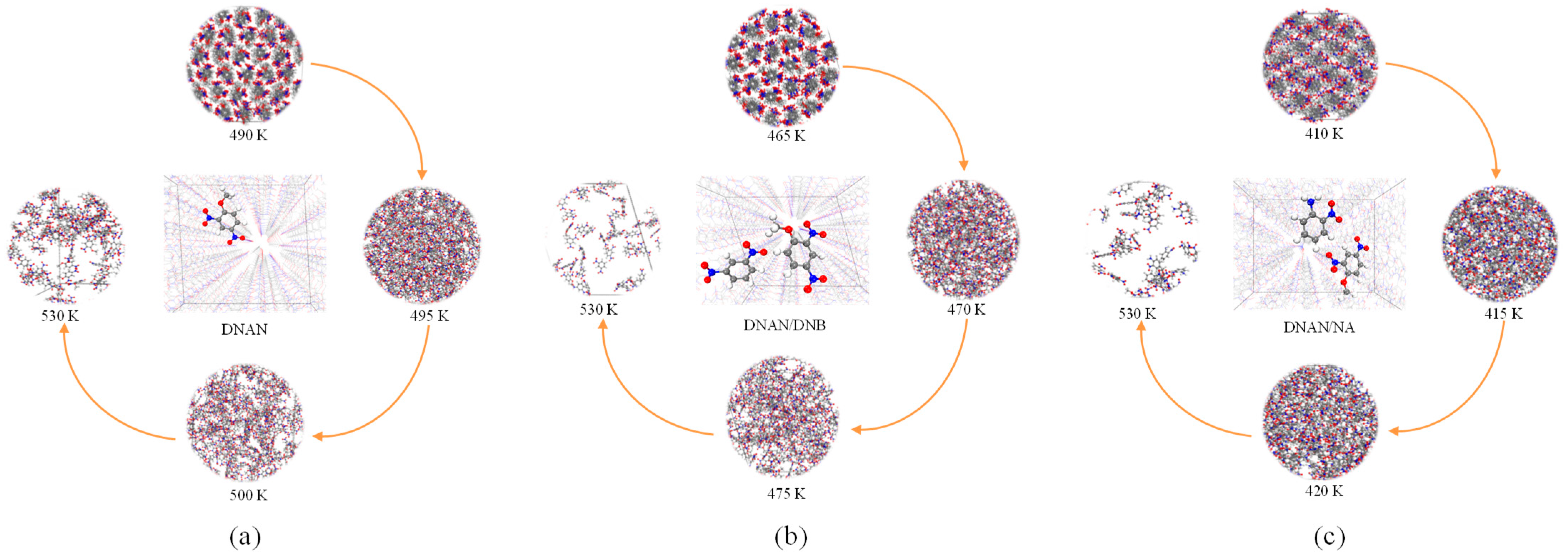
2.4. Melting Process Simulations
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bu, R.; Jiao, F.; Liu, G.; Zhao, J.; Zhang, C. Categorizing and Understanding Energetic Crystals. Cryst. Growth Des. 2020, 21, 3–15. [Google Scholar] [CrossRef]
- Jiao, F.; Xiong, Y.; Li, H.; Zhang, C. Alleviating the energy & safety contradiction to construct new low sensitivity and highly energetic materials through crystal engineering. CrystEngComm 2018, 20, 1757–1768. [Google Scholar]
- Ravi, P.; Badgujar, D.M.; Gore, G.M.; Tewari, S.P.; Sikder, A.K. Review on Melt Cast Explosives. Propellants Explos. Pyrotech. 2011, 36, 393–403. [Google Scholar] [CrossRef]
- Bennion, J.C.; Siddiqi, Z.R.; Matzger, A.J. A melt castable energetic cocrystal. Chem. Commun. 2017, 53, 6065–6068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acree, W.E. thermodynamic properties of organic compounds. Thermochim. Acta 1993, 219, 97–104. [Google Scholar] [CrossRef]
- Provatas, A.; Wall, C. Ageing of Australian DNAN Based Melt-Cast Insensitive Explosives. Propellants Explos. Pyrotech. 2016, 41, 555–561. [Google Scholar] [CrossRef]
- Sikder, N.; Sikder, A.K.; Bulakh, N.R.; Gandhe, B.R. 1,3,3-Trinitroazetidine (TNAZ), a melt-cast explosive: Synthesis, characterization and thermal behaviour. J. Hazard Mater. 2004, 113, 35–43. [Google Scholar] [CrossRef]
- Kamenar, B.P.C.K. Molecular Complexes. Part 1. The crystal and molecular structure of the 1: 1 adduct of benzotrifuroxan and 13,14-Dithiatricyclo trtradeca-4,6,10,12-tetraene. J. Chem. Soc. 1965, 82, 4838–4851. [Google Scholar] [CrossRef]
- Muravyev, N.V.; Fershtat, L.L.; Dalinger, I.L.; Suponitsky, K.Y.; Ananyev, I.V.; Melnikov, I.N. Rational Screening of Cocrystals using Thermal Analysis: Benchmarking on Energetic Materials. Cryst. Growth Des. 2022, 22, 7349–7362. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, H.; Xu, J.; Wang, S.; Zhu, C.; Wang, H.; Ding, R.; Yu, Z.; Sun, J. Two Novel Melt-Cast Cocrystal Explosives Based on 2,4-Dinitroanisole with Significantly Decreased Melting Point. Cryst. Growth Des. 2019, 19, 6826–6830. [Google Scholar] [CrossRef]
- Li, X.; Ju, X. Molecular Dynamic Simulation of Melting Points of Trans-1,4,5,8-tetranitro-1,4,5,8-tetraazadacalin (TNAD) with Some Propellants. Chin. J. Chem. Phys. 2014, 27, 412–418. [Google Scholar] [CrossRef]
- Liu, Y.; Lai, W.; Yu, T.; Ma, Y.; Guo, W.; Ge, Z. Melting Point Prediction of Energetic Materials via Continuous Heating Simulation on Solid-to-Liquid Phase Transition. ACS Omega 2019, 4, 4320–4324. [Google Scholar] [CrossRef]
- Takahashi, H.; Tamura, R. Low temperature phase transition induced biaxial negative thermal expansion of 2,4-dinitroanisole. CrystEngComm 2015, 17, 8888–8896. [Google Scholar] [CrossRef]
- Bu, R.; Xiong, Y.; Zhang, C. π–π Stacking Contributing to the Low or Reduced Impact Sensitivity of Energetic Materials. Cryst. Growth Des. 2020, 20, 2824–2841. [Google Scholar] [CrossRef]
- Tian, B.; Xiong, Y.; Chen, L.; Zhang, C. Relationship between the crystal packing and impact sensitivity of energetic materials. CrystEngComm 2018, 20, 837–848. [Google Scholar] [CrossRef]
- Zhang, C.; Jiao, F.; Li, H. Crystal Engineering for Creating Low Sensitivity and Highly Energetic Materials. Cryst. Growth Des. 2018, 18, 5713–5726. [Google Scholar] [CrossRef]
- Kretic, D.S.; Radovanovic, J.I.; Veljkovic, D.Z. Can the sensitivity of energetic materials be tuned by using hydrogen bonds? Another look at the role of hydrogen bonding in the design of high energetic compounds. Phys. Chem. Chem. Phys. 2021, 23, 7472–7479. [Google Scholar] [CrossRef]
- Liu, G.; Wei, S.-H.; Zhang, C. Review of the Intermolecular Interactions in Energetic Molecular Cocrystals. Cryst. Growth Des. 2020, 20, 7065–7079. [Google Scholar] [CrossRef]
- Zhao, X.; Huang, S.; Liu, Y.; Li, J.; Zhu, W. Effects of Noncovalent Interactions on the Impact Sensitivity of HNS-Based Cocrystals: A DFT Study. Cryst. Growth Des. 2018, 19, 756–767. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Interaction Region Indicator: A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions. Chem.–Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Johnson, E.; KEINAN, S.; Paula, M.; Julia, C.; Cohen, A.; Yang, W. Reavealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Garcia, J.; Yang, W.; Johnson, E.R. Analysis of hydrogen-bond interaction potentials from the electron density: Integration of noncovalent interaction regions. J. Phys. Chem. A 2011, 115, 12983–12990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. WIREs Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Fukui, K. Role of frontier orbitals in chemical reaction. Science 1982, 218, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Segall, M.D.; Lindan, P.J.D.; Lindan, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Fischer, T.H.; Almlof, J. General methods for geometry and wave function optimization. J. Phys. Chem. 1992, 96, 9768–9774. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Schmider, H.L.; Becke, A.D. Chemical content of the kinetic energy density. J. Mol. Struct. 2000, 527, 51–61. [Google Scholar] [CrossRef]
- Gonthier, J.F.; Steinmann, S.N.; Roch, L.; Ruggi, A.; Luisier, N.; Severin, K.; Corminboeuf, C. pi-Depletion as a criterion to predict pi-stacking ability. Chem. Commun. 2012, 48, 9239–9241. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, Q. A simple method of identifying π orbitals for non-planar systems and a protocol of studying π electronic structure. Theor. Chem. Acc. 2020, 139, 25. [Google Scholar] [CrossRef]
- Becke, A. The Quantum Theory of Atoms in Molecules-From Solid State to DNA and Drug Design; WILEY-VCH: New York, NY, USA, 2007. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Clark, S.J.S.M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Fuer Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Sun, H. COMPASS: An ab Initio Force-Field Optimized for Condensed-Phase Applicationss- Overview with Details on Alkane and Benzene Compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNAN | DNAN/DNB | DNAN/NA | ||
|---|---|---|---|---|
| Space group | P21/n | P21/n | P21/n | |
| This work | 3.88 | 18.86 | 7.58 | |
| a (Å) | Expt. [10] | 3.91 | 18.81 | 7.43 |
| Error (%) | 0.77% | −0.26% | −2.02% | |
| This work | 13.54 | 3.79 | 17.55 | |
| b (Å) | Expt. [10] | 13.78 | 3.87 | 17.57 |
| Error (%) | 1.74% | 2.07% | 0.11% | |
| This work | 15.32 | 21.82 | 11.49 | |
| c (Å) | Expt. [10] | 15.42 | 21.91 | 11.49 |
| Error (%) | 0.65% | 0.41% | 0% | |
| This work | 90.00 | 90.00 | 90.00 | |
| α (°) | Expt. [10] | 90.00 | 90.00 | 90.00 |
| Error (%) | 0% | 0% | 0% | |
| This work | 95.30 | 106.70 | 95.22 | |
| β (°) | Expt. [10] | 95.31 | 106.70 | 95.21 |
| Error (%) | 0.01% | 0% | −0.01% | |
| This work | 90.00 | 90.00 | 90.00 | |
| γ (°) | Expt. [10] | 90.00 | 90.00 | 90.00 |
| Error (%) | 0% | 0% | 0% |
| DNAN | DNAN/DNB | DNAN/NA | |
|---|---|---|---|
| Bond length (Å) | |||
| C2-NO2 | 1.459 | 1.456 | 1.451 |
| C4-NO2 | 1.45 | 1.45 | 1.448 |
| C1-OCH3 | 1.337 | 1.335 | 1.337 |
| O1-CH3 | 1.449 | 1.458 | 1.446 |
| Torsion angles (°) | |||
| C3-C4-N2-O5 | −169.7 | −169.1 | −162.3 |
| C3-C2-N1-O2 | −156.9 | −139.2 | −150.2 |
| C2-C1-O1-C7 | 179.1 | 171.2 | 177.7 |
| Type | Crystal | Interactions | Electron Number × 10 | qbind × 103 |
|---|---|---|---|---|
| Interlayer | DNAN | DNAN-DNAN | 0.21 | 0.05 |
| DNAN/DNB | DNAN-DNAN | 0.21 | 0.32 | |
| DNB-DNB | 0.26 | 0.02 | ||
| DNAN/NA | DNAN-NA | 0.33 | 0.45 | |
| Intralayer | DNAN | C10-H3⋯O40 | 0.02 | −0.43 |
| C6-H7⋯O57 | 0.03 | −0.67 | ||
| C68-H69⋯O60 | 0.01 | 0 | ||
| C66-H67⋯O37 | 0.03 | −0.68 | ||
| C70-H73⋯O60 | 0.02 | −0.43 | ||
| C8-H9⋯O40 | 0.01 | 0 | ||
| DNAN/DNB | C8-H9⋯O40 | 0.02 | −0.36 | |
| C48-H49⋯O54 | 0.03 | −0.33 | ||
| C32-H33⋯O2 | 0.03 | −0.33 | ||
| C10-H11⋯O53 | 0.01 | −0.26 | ||
| C62-H63⋯O1 | 0.01 | −0.27 | ||
| C69-H70⋯O24 | 0.02 | −0.36 | ||
| DNAN/NA | N24-H26⋯O3 | 0.02 | −0.43 | |
| C14-H15⋯O53 | 0.03 | −0.86 | ||
| C48-H49⋯O4 | 0.02 | −0.36 | ||
| C50-H51⋯O22 | 0.02 | −0.53 | ||
| N40-H41⋯O21 | 0.06 | −1.56 | ||
| C48-H49⋯H70 | 0.01 | −0.01 | ||
| C46-H47⋯H72 | 0.03 | −0.42 |
| Crystal | Type | Interaction | ρ | ▽2ρ | GBCP | VBCP | EBCP/HBCP | Eint1 | Eint2 |
|---|---|---|---|---|---|---|---|---|---|
| DNAN | Intra- molecular | O17-O16 | 0.017 | 0.070 | 0.016 | −0.014 | 0.002 | 17.59 | 17.95 |
| O56-O57 | 0.017 | 0.070 | 0.016 | −0.014 | 0.002 | 17.49 | 17.74 | ||
| O76-O77 | 0.017 | 0.070 | 0.016 | −0.014 | 0.002 | 17.59 | 17.95 | ||
| O36-O37 | 0.017 | 0.070 | 0.016 | −0.014 | 0.002 | 17.49 | 17.74 | ||
| Inter- molecular | C10-H3⋯O40 | 0.009 | 0.034 | 0.007 | −0.006 | 0.001 | 7.95 | 7.44 | |
| C8-H9⋯O40 | 0.004 | 0.013 | 0.003 | −0.002 | 0.001 | 3.16 | 2.95 | ||
| C6-H7⋯O57 | 0.013 | 0.042 | 0.009 | −0.007 | 0.002 | 9.97 | 9.57 | ||
| O58-O39 | 0.007 | 0.027 | 0.006 | −0.005 | 0.001 | 6.83 | 7.09 | ||
| C43-H44⋯O39 | 0.008 | 0.029 | 0.006 | −0.005 | 0.001 | 6.89 | 6.46 | ||
| C23-H24⋯O59 | 0.008 | 0.029 | 0.006 | −0.005 | 0.001 | 6.89 | 6.46 | ||
| O38-O59 | 0.007 | 0.027 | 0.006 | −0.005 | 0.001 | 6.83 | 7.09 | ||
| C68-H69⋯O60 | 0.004 | 0.013 | 0.003 | −0.002 | 0.001 | 3.16 | 2.95 | ||
| C70-H73⋯O60 | 0.009 | 0.034 | 0.007 | −0.006 | 0.001 | 7.96 | 7.45 | ||
| C66-H67⋯O37 | 0.013 | 0.042 | 0.009 | −0.007 | 0.002 | 9.96 | 9.57 | ||
| DNAN/ DNB | Intra- molecular | O1-O5 | 0.013 | 0.052 | 0.012 | −0.010 | 0.001 | 13.27 | 13.78 |
| O57-O53 | 0.013 | 0.052 | 0.012 | −0.010 | 0.001 | 13.27 | 13.78 | ||
| Inter- molecular | C8-H9⋯O40 | 0.008 | 0.031 | 0.006 | −0.005 | 0.001 | 7.31 | 6.85 | |
| O2-C32 | 0.005 | 0.017 | 0.003 | −0.003 | 0.001 | 3.91 | 3.49 | ||
| C10-H11⋯O53 | 0.007 | 0.027 | 0.006 | −0.004 | 0.001 | 6.28 | 5.78 | ||
| C32-H33⋯O2 | 0.004 | 0.013 | 0.003 | −0.002 | 0.001 | 3.17 | 2.96 | ||
| O1-O53 | 0.005 | 0.019 | 0.004 | −0.003 | 0.001 | 4.52 | 4.33 | ||
| C48-H49⋯O54 | 0.004 | 0.013 | 0.003 | −0.002 | 0.001 | 3.17 | 2.96 | ||
| C62-H63⋯O1 | 0.007 | 0.027 | 0.006 | −0.004 | 0.001 | 6.28 | 5.78 | ||
| C48-O54 | 0.005 | 0.017 | 0.003 | −0.003 | 0.001 | 3.91 | 3.49 | ||
| C69-H70⋯O24 | 0.008 | 0.031 | 0.006 | −0.005 | 0.001 | 7.31 | 6.85 | ||
| DNAN/ NA | Intra- molecular | N40-H42⋯O38 | 0.030 | 0.116 | 0.026 | −0.024 | 0.003 | 29.74 | 31.36 |
| O53-O57 | 0.016 | 0.066 | 0.015 | −0.013 | 0.002 | 16.64 | 17.04 | ||
| N24-H26⋯O22 | 0.029 | 0.115 | 0.026 | −0.023 | 0.003 | 29.44 | 30.75 | ||
| O1-O5 | 0.016 | 0.067 | 0.015 | −0.013 | 0.002 | 16.72 | 17.09 | ||
| Inter- molecular | C46-H47⋯H72 | 0.006 | 0.021 | 0.004 | −0.003 | 0.001 | 4.83 | 4.26 | |
| N40-H41⋯O21 | 0.023 | 0.085 | 0.019 | −0.016 | 0.002 | 21.23 | 21.49 | ||
| C48-H49⋯H70 | 0.004 | 0.012 | 0.002 | −0.002 | 0.001 | 2.66 | 2.40 | ||
| C50-H51⋯O22 | 0.011 | 0.033 | 0.007 | −0.006 | 0.001 | 8.01 | 7.97 | ||
| C48-H49⋯O4 | 0.008 | 0.031 | 0.006 | −0.005 | 0.001 | 7.30 | 6.88 | ||
| C12-H13⋯H70 | 0.002 | 0.007 | 0.001 | −0.001 | 0.000 | 1.50 | 1.25 | ||
| C14-H15⋯O53 | 0.015 | 0.061 | 0.012 | −0.010 | 0.003 | 14.04 | 12.79 | ||
| N24-H26⋯O3 | 0.010 | 0.037 | 0.008 | −0.006 | 0.002 | 8.80 | 8.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Yang, L.; Zhu, W. Effects of Cocrystallization on the Structure and Properties of Melt-Cast Explosive 2,4-Dinitroanisole: A Computational Study. Molecules 2022, 27, 9010. https://doi.org/10.3390/molecules27249010
Wang D, Yang L, Zhu W. Effects of Cocrystallization on the Structure and Properties of Melt-Cast Explosive 2,4-Dinitroanisole: A Computational Study. Molecules. 2022; 27(24):9010. https://doi.org/10.3390/molecules27249010
Chicago/Turabian StyleWang, Defu, Lei Yang, and Weihua Zhu. 2022. "Effects of Cocrystallization on the Structure and Properties of Melt-Cast Explosive 2,4-Dinitroanisole: A Computational Study" Molecules 27, no. 24: 9010. https://doi.org/10.3390/molecules27249010
APA StyleWang, D., Yang, L., & Zhu, W. (2022). Effects of Cocrystallization on the Structure and Properties of Melt-Cast Explosive 2,4-Dinitroanisole: A Computational Study. Molecules, 27(24), 9010. https://doi.org/10.3390/molecules27249010