
Spotlight on Nociceptin/Orphanin FQ Receptor in the Treatment of Pain


Abstract
:1. Introduction
2. Structure of NOP Receptor
3. The Distribution and Signaling Pathway of NOP Receptor
4. Ligands of NOP Receptor
4.1. Peptide Ligands Related to N/OFQ Targeting Pain
4.1.1. [Nphe1]N/OFQ(1-13)NH2
4.1.2. [Nphe1, Arg14, Lys15]N/OFQ-NH2 (UFP-101)
4.1.3. [(pF)Phe4Aib7Arg14Lys15]N/OFQ-NH2 (UFP-112)
4.1.4. [Phe1Ψ(CH2-NH)Gly2(pF)Phe4Aib7Arg14Lys15]N/OFQ-NH2 (UFP-113)
4.1.5. PWT2-N/OFQ
4.2. Non-Peptide NOP Receptor Ligands Targeting Pain
4.2.1. Ro 65-6570
4.2.2. Ro 64-6198
4.2.3. SCH221510
4.3. Bifunctional and Mixed NOP Receptor Compounds
4.3.1. SR 16435
4.3.2. AT-121
4.3.3. Buprenorphine and Its Analog BU08028
4.3.4. BPR1M97
4.3.5. BU10038
4.3.6. JTC-801
4.3.7. Cebranopadol
5. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gaskin, D.J.; Richard, P. The economic costs of pain in the United States. J. Pain 2012, 13, 715–724. [Google Scholar] [CrossRef]
- Brownstein, M.J. A brief history of opiates, opioid peptides, and opioid receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 5391–5393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkow, N.D.; Collins, F.S. The Role of Science in Addressing the Opioid Crisis. N. Engl. J. Med. 2017, 377, 391–394. [Google Scholar] [CrossRef]
- Ballantyne, J.C.; Kalso, E.; Stannard, C. WHO analgesic ladder: A good concept gone astray. BMJ 2016, 352, i20. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Roth, B.L. New Technologies for Elucidating Opioid Receptor Function. Trends Pharmacol. Sci. 2016, 37, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Khademi, H.; Kamangar, F.; Brennan, P.; Malekzadeh, R. Opioid Therapy and its Side Effects: A Review. Arch. Iran. Med. 2016, 19, 870–876. [Google Scholar] [PubMed]
- Iwanicki, J.L.; Severtson, S.G.; Margolin, Z.; Dasgupta, N.; Green, J.L.; Dart, R.C. Consistency between Opioid-Related Mortality Trends Derived From Poison Center and National Vital Statistics System, United States, 2006–2016. Am. J. Public Health 2018, 108, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Kirson, N.Y.; Scarpati, L.M.; Enloe, C.J.; Dincer, A.P.; Birnbaum, H.G.; Mayne, T.J. The Economic Burden of Opioid Abuse: Updated Findings. J. Manag. Care Spec. Pharm. 2017, 23, 427–445. [Google Scholar] [CrossRef]
- Available online: https://www.cdc.gov/nchs/nvss/vsrr/drug-overdose-data.htm (accessed on 12 January 2022).
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Anton, B.; Fein, J.; To, T.; Li, X.; Silberstein, L.; Evans, C.J. Immunohistochemical localization of ORL-1 in the central nervous system of the rat. J. Comp. Neurol. 1996, 368, 229–251. [Google Scholar] [CrossRef]
- Mollereau, C.; Mouledous, L. Tissue distribution of the opioid receptor-like (ORL1) receptor. Peptides 2000, 21, 907–917. [Google Scholar] [CrossRef]
- Neal, C.R., Jr.; Mansour, A.; Reinscheid, R.; Nothacker, H.P.; Civelli, O.; Akil, H.; Watson, S.J., Jr. Opioid receptor-like (ORL1) receptor distribution in the rat central nervous system: Comparison of ORL1 receptor mRNA expression with (125)I-[(14)Tyr]-orphanin FQ binding. J. Comp. Neurol. 1999, 412, 563–605. [Google Scholar] [CrossRef]
- Meunier, J.C.; Mollereau, C.; Toll, L.; Suaudeau, C.; Moisand, C.; Alvinerie, P.; Butour, J.L.; Guillemot, J.C.; Ferrara, P.; Monsarrat, B.; et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature 1995, 377, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Reinscheid, R.K.; Nothacker, H.P.; Bourson, A.; Ardati, A.; Henningsen, R.A.; Bunzow, J.R.; Grandy, D.K.; Langen, H.; Monsma, F.J., Jr.; Civelli, O. Orphanin FQ: A neuropeptide that activates an opioidlike G protein-coupled receptor. Science 1995, 270, 792–794. [Google Scholar] [CrossRef]
- Xu, X.J.; Hao, J.X.; Wiesenfeld-Hallin, Z. Nociceptin or antinociceptin: Potent spinal antinociceptive effect of orphanin FQ/nociceptin in the rat. Neuroreport 1996, 7, 2092–2094. [Google Scholar]
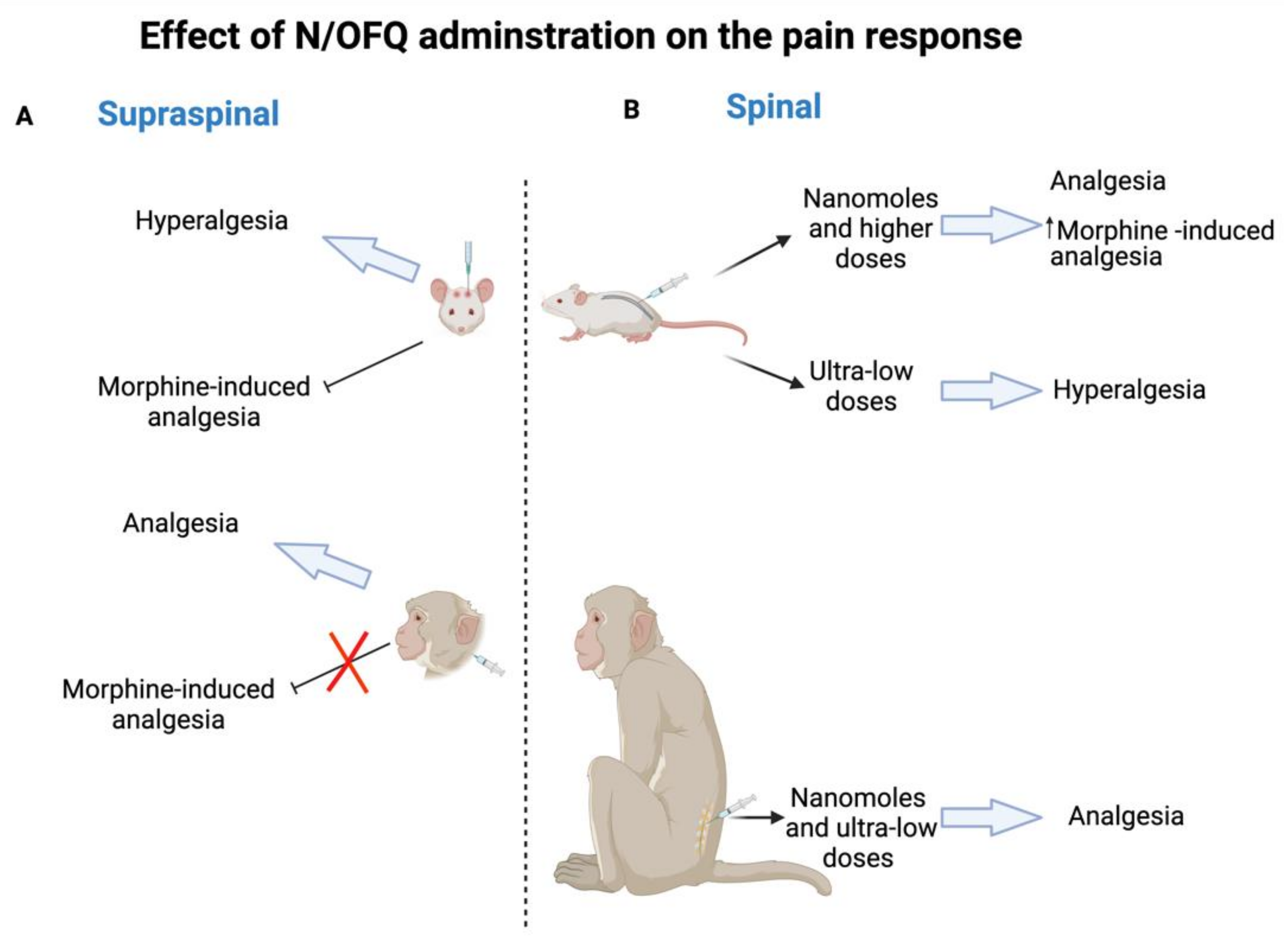
- Ko, M.C.; Naughton, N.N. Antinociceptive effects of nociceptin/orphanin FQ administered intrathecally in monkeys. J. Pain 2009, 10, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.C.; Wei, H.; Woods, J.H.; Kennedy, R.T. Effects of intrathecally administered nociceptin/orphanin FQ in monkeys: Behavioral and mass spectrometric studies. J. Pharmacol. Exp. Ther. 2006, 318, 1257–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Nozaki-Taguchi, N.; Kimura, S. Analgesic effect of intrathecally administered nociceptin, an opioid receptor-like1 receptor agonist, in the rat formalin test. Neuroscience 1997, 81, 249–254. [Google Scholar] [CrossRef]
- Bunzow, J.R.; Saez, C.; Mortrud, M.; Bouvier, C.; Williams, J.T.; Low, M.; Grandy, D.K. Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a mu, delta or kappa opioid receptor type. FEBS Lett. 1994, 347, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K.; Kato, S.; Mori, K.; Nishi, M.; Takeshima, H.; Iwabe, N.; Miyata, T.; Houtani, T.; Sugimoto, T. cDNA cloning and regional distribution of a novel member of the opioid receptor family. FEBS Lett. 1994, 343, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Mollereau, C.; Parmentier, M.; Mailleux, P.; Butour, J.L.; Moisand, C.; Chalon, P.; Caput, D.; Vassart, G.; Meunier, J.C. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994, 341, 33–38. [Google Scholar] [CrossRef]
- Nishi, M.; Takeshima, H.; Mori, M.; Nakagawara, K.; Takeuchi, T. Structure and chromosomal mapping of genes for the mouse kappa-opioid receptor and an opioid receptor homologue (MOR-C). Biochem. Biophys. Res. Commun. 1994, 205, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Osinski, M.A.; Pampusch, M.S.; Murtaugh, M.P.; Brown, D.R. Cloning, expression and functional role of a nociceptin/orphanin FQ receptor in the porcine gastrointestinal tract. Eur. J. Pharmacol. 1999, 365, 281–289. [Google Scholar] [CrossRef]
- Wick, M.J.; Minnerath, S.R.; Lin, X.; Elde, R.; Law, P.Y.; Loh, H.H. Isolation of a novel cDNA encoding a putative membrane receptor with high homology to the cloned mu, delta, and kappa opioid receptors. Brain Res. Mol. 1994, 27, 37–44. [Google Scholar] [CrossRef]
- Mollereau, C.; Moisand, C.; Butour, J.L.; Parmentier, M.; Meunier, J.C. Replacement of Gln280 by His in TM6 of the human ORL1 receptor increases affinity but reduces intrinsic activity of opioids. FEBS Lett. 1996, 395, 17–21. [Google Scholar] [CrossRef]
- Mollereau, C.; Simons, M.J.; Soularue, P.; Liners, F.; Vassart, G.; Meunier, J.C.; Parmentier, M. Structure, tissue distribution, and chromosomal localization of the prepronociceptin gene. Proc. Natl. Acad. Sci. USA 1996, 93, 8666–8670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothacker, H.P.; Reinscheid, R.K.; Mansour, A.; Henningsen, R.A.; Ardati, A.; Monsma, F.J., Jr.; Watson, S.J.; Civelli, O. Primary structure and tissue distribution of the orphanin FQ precursor. Proc. Natl. Acad. Sci. USA 1996, 93, 8677–8682. [Google Scholar] [CrossRef] [Green Version]
- Toll, L.; Bruchas, M.R.; Calo, G.; Cox, B.M.; Zaveri, N.T. Nociceptin/Orphanin FQ Receptor Structure, Signaling, Ligands, Functions, and Interactions with Opioid Systems. Pharmacol. Rev. 2016, 68, 419–457. [Google Scholar] [CrossRef]
- Cox, B.M.; Christie, M.J.; Devi, L.; Toll, L.; Traynor, J.R. Challenges for opioid receptor nomenclature: IUPHAR Review 9. Br. J. Pharmacol. 2015, 172, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.L.; Thompson, A.A.; Trapella, C.; Guerrini, R.; Malfacini, D.; Patel, N.; Han, G.W.; Cherezov, V.; Caló, G.; Katritch, V.; et al. The Importance of Ligand-Receptor Conformational Pairs in Stabilization: Spotlight on the N/OFQ G Protein-Coupled Receptor. Structure 2015, 23, 2291–2299. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H.; Vardy, E.; Huang, X.P.; Trapella, C.; Guerrini, R.; Calo, G.; et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 2012, 485, 395–399. [Google Scholar] [CrossRef]
- Akuzawa, N.; Takeda, S.; Ishiguro, M. Structural modelling and mutation analysis of a nociceptin receptor and its ligand complexes. J. Biochem. 2007, 141, 907–916. [Google Scholar] [CrossRef]
- Daga, P.R.; Zaveri, N.T. Homology modeling and molecular dynamics simulations of the active state of the nociceptin receptor reveal new insights into agonist binding and activation. Proteins 2012, 80, 1948–1961. [Google Scholar] [CrossRef] [Green Version]
- Topham, C.M.; Moulédous, L.; Poda, G.; Maigret, B.; Meunier, J.C. Molecular modelling of the ORL1 receptor and its complex with nociceptin. Protein Eng. 1998, 11, 1163–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, A.; Brunori, G.; Mercatelli, D.; Wu, J.; Cippitelli, A.; Zou, B.; Xie, X.S.; Williams, M.; Zaveri, N.T.; Low, S.; et al. Knock-In Mice with NOP-eGFP Receptors Identify Receptor Cellular and Regional Localization. J. Neurosci. 2015, 35, 11682–11693. [Google Scholar] [CrossRef] [Green Version]
- Peluso, J.; LaForge, K.S.; Matthes, H.W.; Kreek, M.J.; Kieffer, B.L.; Gavériaux-Ruff, C. Distribution of nociceptin/orphanin FQ receptor transcript in human central nervous system and immune cells. J. Neuroimmunol. 1998, 81, 184–192. [Google Scholar] [CrossRef]
- Neal, C.R., Jr.; Akil, H.; Watson, S.J., Jr. Expression of orphanin FQ and the opioid receptor-like (ORL1) receptor in the developing human and rat brain. J. Chem. Neuroanat. 2001, 22, 219–249. [Google Scholar] [CrossRef]
- Lambert, D.G. The nociceptin/orphanin FQ receptor: A target with broad therapeutic potential. Nat. Rev. Drug Discov. 2008, 7, 694–710. [Google Scholar] [CrossRef]
- Childers, S.R.; Creese, I.; Snowman, A.M.; Synder, S.H. Opiate receptor binding affected differentially by opiates and opioid peptides. Eur. J. Pharmacol. 1979, 55, 11–18. [Google Scholar] [CrossRef]
- Childers, S.R.; Snyder, S.H. Guanine nucleotides differentiate agonist and antagonist interactions with opiate receptors. Life Sci. 1978, 23, 759–761. [Google Scholar] [CrossRef]
- Connor, M.; Christie, M.J. Modulation of Ca2+ channel currents of acutely dissociated rat periaqueductal grey neurons. J. Physiol. 1998, 509, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Darlison, M.G.; Greten, F.R.; Harvey, R.J.; Kreienkamp, H.J.; Stühmer, T.; Zwiers, H.; Lederis, K.; Richter, D. Opioid receptors from a lower vertebrate (Catostomus commersoni): Sequence, pharmacology, coupling to a G-protein-gated inward-rectifying potassium channel (GIRK1), and evolution. Proc. Natl. Acad. Sci. USA 1997, 94, 8214–8219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.R.; Planer, W.; Siuda, E.R.; Zhao, H.C.; Stickler, L.; Chang, S.D.; Baird, M.A.; Cao, Y.Q.; Bruchas, M.R. Serine 363 is required for nociceptin/orphanin FQ opioid receptor (NOPR) desensitization, internalization, and arrestin signaling. J. Biol. Chem. 2012, 287, 42019–42030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, N.; Roberts, K.; Pal, K.; Bentolila, L.A.; Fultz, E.; Minasyan, A.; Cahill, C.; Pradhan, A.; Conner, D.; DeFea, K.; et al. Select G-protein-coupled receptors modulate agonist-induced signaling via a ROCK, LIMK, and beta-arrestin 1 pathway. Cell Rep. 2013, 5, 1010–1021. [Google Scholar] [CrossRef] [Green Version]
- Stein, C. Opioid Receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, K.E.; Bruchas, M.R. NOP Receptor Signaling Cascades. Handb. Exp. Pharmacol. 2019, 254, 131–139. [Google Scholar]
- Armstead, W.M. Differential activation of ERK, p38, and JNK MAPK by nociceptin/orphanin FQ in the potentiation of prostaglandin cerebrovasoconstriction after brain injury. Eur. J. Pharmacol. 2006, 529, 129–135. [Google Scholar] [CrossRef]
- New, D.C.; Wong, Y.H. The ORL1 receptor: Molecular pharmacology and signalling mechanisms. Neurosignals 2002, 11, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Marti, M.; Stocchi, S.; Paganini, F.; Mela, F.; De Risi, C.; Calo, G.; Guerrini, R.; Barnes, T.A.; Lambert, D.G.; Beani, L.; et al. Pharmacological profiles of presynaptic nociceptin/orphanin FQ receptors modulating 5-hydroxytryptamine and noradrenaline release in the rat neocortex. Br. J. Pharmacol. 2003, 138, 91–98. [Google Scholar] [CrossRef]
- Nicol, B.; Lambert, D.G.; Rowbotham, D.J.; Smart, D.; McKnight, A.T. Nociceptin induced inhibition of K+ evoked glutamate release from rat cerebrocortical slices. Br. J. Pharmacol. 1996, 119, 1081–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, S.S. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol. Rev. 2001, 53, 1–24. [Google Scholar]
- Krupnick, J.G.; Benovic, J.L. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev. Pharmacol. Toxicol. 1998, 38, 289–319. [Google Scholar] [CrossRef]
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef] [Green Version]
- Mogil, J.S.; Grisel, J.E.; Reinscheid, R.K.; Civelli, O.; Belknap, J.K.; Grandy, D.K. Orphanin FQ is a functional anti-opioid peptide. Neuroscience 1996, 75, 333–337. [Google Scholar] [CrossRef]
- Mogil, J.S.; Grisel, J.E.; Zhangs, G.; Belknap, J.K.; Grandy, D.K. Functional antagonism of mu-, delta- and kappa-opioid antinociception by orphanin FQ. Neurosci. Lett. 1996, 214, 131–134. [Google Scholar] [CrossRef]
- Morgan, M.M.; Grisel, J.E.; Robbins, C.S.; Grandy, D.K. Antinociception mediated by the periaqueductal gray is attenuated by orphanin FQ. Neuroreport 1997, 8, 3431–3434. [Google Scholar] [CrossRef] [PubMed]
- Toll, L.; Ozawa, A.; Cippitelli, A. NOP-Related Mechanisms in Pain and Analgesia. Handb. Exp. Pharmacol. 2019, 254, 165–186. [Google Scholar] [PubMed]
- Guerrini, R.; Calo, G.; Rizzi, A.; Bianchi, C.; Lazarus, L.H.; Salvadori, S.; Temussi, P.A.; Regoli, D. Address and message sequences for the nociceptin receptor: A structure-activity study of nociceptin-(1-13)-peptide amide. J. Med. Chem. 1997, 40, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Calo, G.; Guerrini, R.; Bigoni, R.; Rizzi, A.; Marzola, G.; Okawa, H.; Bianchi, C.; Lambert, D.G.; Salvadori, S.; Regoli, D. Characterization of [Nphe(1)]nociceptin(1-13)NH(2), a new selective nociceptin receptor antagonist. Br. J. Pharmacol. 2000, 129, 1183–1193. [Google Scholar]
- Calo, G.; Rizzi, A.; Rizzi, D.; Bigoni, R.; Guerrini, R.; Marzola, G.; Marti, M.; McDonald, J.; Morari, M.; Lambert, D.G.; et al. [Nphe1,Arg14,Lys15]nociceptin-NH2, a novel potent and selective antagonist of the nociceptin/orphanin FQ receptor. Br. J. Pharmacol. 2002, 136, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrini, R.; Calo, G.; Rizzi, A.; Bigoni, R.; Bianchi, C.; Salvadori, S.; Regoli, D. A new selective antagonist of the nociceptin receptor. Br. J. Pharmacol. 1998, 123, 163–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzi, A.; Salis, M.B.; Ciccocioppo, R.; Marzola, G.; Bigoni, R.; Guerrini, R.; Massi, M.; Madeddu, P.; Salvadori, S.; Regoli, D.; et al. Pharmacological characterisation of [(pX)Phe4]nociceptin(1-13)NH2 analogues. 2. In vivo studies. Naunyn Schmiedebergs Arch. Pharmacol. 2002, 365, 450–456. [Google Scholar] [CrossRef]
- Bigoni, R.; Rizzi, D.; Rizzi, A.; Camarda, V.; Guerrini, R.; Lambert, D.G.; Hashiba, E.; Berger, H.; Salvadori, S.; Regoli, D.; et al. Pharmacological characterisation of [(pX)Phe4]nociceptin(1-13)amide analogues. 1. In vitro studies. Naunyn Schmiedebergs Arch. Pharmacol. 2002, 365, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Nazzaro, C.; Rizzi, A.; Salvadori, S.; Guerrini, R.; Regoli, D.; Zeilhofer, H.U.; Calo, G. UFP-101 antagonizes the spinal antinociceptive effects of nociceptin/orphanin FQ: Behavioral and electrophysiological studies in mice. Peptides 2007, 28, 663–669. [Google Scholar] [CrossRef]
- Rizzi, A.; Spagnolo, B.; Wainford, R.D.; Fischetti, C.; Guerrini, R.; Marzola, G.; Baldisserotto, A.; Salvadori, S.; Regoli, D.; Kapusta, D.R.; et al. In vitro and in vivo studies on UFP-112, a novel potent and long lasting agonist selective for the nociceptin/orphanin FQ receptor. Peptides 2007, 28, 1240–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, E.; Calò, G.; Guerrini, R.; Ko, M.C. Long-lasting antinociceptive spinal effects in primates of the novel nociceptin/orphanin FQ receptor agonist UFP-112. Pain 2010, 148, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Micheli, L.; Di Cesare Mannelli, L.; Guerrini, R.; Trapella, C.; Zanardelli, M.; Ciccocioppo, R.; Rizzi, A.; Ghelardini, C.; Calò, G. Acute and subchronic antinociceptive effects of nociceptin/orphanin FQ receptor agonists infused by intrathecal route in rats. Eur. J. Pharmacol. 2015, 754, 73–81. [Google Scholar] [CrossRef]
- Rizzi, A.; Malfacini, D.; Cerlesi, M.C.; Ruzza, C.; Marzola, E.; Bird, M.F.; Rowbotham, D.J.; Salvadori, S.; Guerrini, R.; Lambert, D.G.; et al. In vitro and in vivo pharmacological characterization of nociceptin/orphanin FQ tetrabranched derivatives. Br. J. Pharmacol. 2014, 171, 4138–4153. [Google Scholar] [CrossRef] [Green Version]
- Rizzi, A.; Sukhtankar, D.D.; Ding, H.; Hayashida, K.; Ruzza, C.; Guerrini, R.; Calò, G.; Ko, M.C. Spinal antinociceptive effects of the novel NOP receptor agonist PWT2-nociceptin/orphanin FQ in mice and monkeys. Br. J. Pharmacol. 2015, 172, 3661–3670. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, R.; Caló, G.; Bigoni, R.; Rizzi, A.; Varani, K.; Toth, G.; Gessi, S.; Hashiba, E.; Hashimoto, Y.; Lambert, D.G.; et al. Further studies on nociceptin-related peptides: Discovery of a new chemical template with antagonist activity on the nociceptin receptor. J. Med. Chem. 2000, 43, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Sujaku, T.; Chuman, Y.; Nakashima, R.; Nose, T.; Costa, T.; Yamada, Y.; Yokoyama, M.; Nagahisa, A.; Shimohigashi, Y. Highly potent nociceptin analog containing the Arg-Lys triple repeat. Biochem. Biophys. Res. Commun. 2000, 278, 493–498. [Google Scholar] [CrossRef]
- Dooley, C.T.; Houghten, R.A. Orphanin FQ: Receptor binding and analog structure activity relationships in rat brain. Life Sci. 1996, 59, PL23-9. [Google Scholar] [CrossRef]
- Reinscheid, R.K.; Ardati, A.; Monsma, F.J., Jr.; Civelli, O. Structure-activity relationship studies on the novel neuropeptide orphanin FQ. J. Biol. Chem. 1996, 271, 14163–14168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Miller, W.; Valenzano, K.J.; Kyle, D.J. Novel, potent ORL-1 receptor agonist peptides containing alpha-Helix-promoting conformational constraints. J. Med. Chem. 2002, 45, 5280–5286. [Google Scholar] [CrossRef]
- Arduin, M.; Spagnolo, B.; Calò, G.; Guerrini, R.; Carrà, G.; Fischetti, C.; Trapella, C.; Marzola, E.; McDonald, J.; Lambert, D.G.; et al. Synthesis and biological activity of nociceptin/orphanin FQ analogues substituted in position 7 or 11 with Calpha, alpha-dialkylated amino acids. Bioorg. Med. Chem. 2007, 15, 4434–4443. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Marzola, E.; Trapella, C.; Pela’, M.; Molinari, S.; Cerlesi, M.C.; Malfacini, D.; Rizzi, A.; Salvadori, S.; Calo’, G. A novel and facile synthesis of tetra branched derivatives of nociceptin/orphanin FQ. Bioorg. Med. Chem. 2014, 22, 3703–3712. [Google Scholar] [CrossRef] [PubMed]
- Byford, A.J.; Anderson, A.; Jones, P.S.; Palin, R.; Houghton, A.K. The hypnotic, electroencephalographic, and antinociceptive properties of nonpeptide ORL1 receptor agonists after intravenous injection in rodents. Anesth. Analg. 2007, 104, 174–179. [Google Scholar] [CrossRef]
- Rizzi, A.; Ruzza, C.; Bianco, S.; Trapella, C.; Calo’, G. Antinociceptive action of NOP and opioid receptor agonists in the mouse orofacial formalin test. Peptides 2017, 94, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Jenck, F.; Wichmann, J.; Dautzenberg, F.M.; Moreau, J.L.; Ouagazzal, A.M.; Martin, J.R.; Lundstrom, K.; Cesura, A.M.; Poli, S.M.; Roever, S.; et al. A synthetic agonist at the orphanin FQ/nociceptin receptor ORL1: Anxiolytic profile in the rat. Proc. Natl. Acad. Sci. USA 2000, 97, 4938–4943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.D.; Brieaddy, L.E.; Harvey, J.D.; Lewin, A.H.; Mascarella, S.W.; Seltzman, H.H.; Reddy, P.A.; Decker, A.M.; McElhinny, C.J., Jr.; Zhong, D.; et al. Novel Synthesis and Pharmacological Characterization of NOP Receptor Agonist 8-[(1S,3aS)-2,3,3a,4,5,6-Hexahydro-1H-phenalen-1-yl]-1-phenyl-1,3,8-triazaspiro[4.5]decan-4-one (Ro 64-6198). ACS Chem. Neurosci. 2015, 6, 1956–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, D.; Wichmann, J.; Tekeshima, H.; Kieffer, B.L.; Ouagazzal, A.M. Effects of nociceptin/orphanin FQ receptor (NOP) agonist, Ro64-6198, on reactivity to acute pain in mice: Comparison to morphine. Eur. J. Pharmacol. 2008, 579, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Varty, G.B.; Lu, S.X.; Morgan, C.A.; Cohen-Williams, M.E.; Hodgson, R.A.; Smith-Torhan, A.; Zhang, H.; Fawzi, A.B.; Graziano, M.P.; Ho, G.D.; et al. The anxiolytic-like effects of the novel, orally active nociceptin opioid receptor agonist 8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol (SCH 221510). J. Pharmacol. Exp. Ther. 2008, 326, 672–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobczak, M.; Mokrowiecka, A.; Cygankiewicz, A.I.; Zakrzewski, P.K.; Sałaga, M.; Storr, M.; Kordek, R.; Małecka-Panas, E.; Krajewska, W.M.; Fichna, J. Anti-inflammatory and antinociceptive action of an orally available nociceptin receptor agonist SCH 221510 in a mouse model of inflammatory bowel diseases. J. Pharmacol. Exp. Ther. 2014, 348, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Röver, S.; Adam, G.; Cesura, A.M.; Galley, G.; Jenck, F.; Monsma, F.J., Jr.; Wichmann, J.; Dautzenberg, F.M. High-affinity, non-peptide agonists for the ORL1 (orphanin FQ/nociceptin) receptor. J. Med. Chem. 2000, 43, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Hashiba, E.; Harrison, C.; Galo’, G.; Guerrini, R.; Rowbotham, D.J.; Smith, G.; Lambert, D.G. Characterisation and comparison of novel ligands for the nociceptin/orphanin FQ receptor. Naunyn Schmiedebergs Arch. Pharmacol. 2001, 363, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Azevedo Neto, J.; Ruzza, C.; Sturaro, C.; Malfacini, D.; Pacifico, S.; Zaveri, N.T.; Calò, G. Functional Selectivity Does Not Predict Antinociceptive/Locomotor Impairing Potencies of NOP Receptor Agonists. Front. Neurosci. 2021, 15, 657153. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.; Malfacini, D.; Journigan, B.V.; Bird, M.F.; Trapella, C.; Guerrini, R.; Lambert, D.G.; Calo’, G.; Zaveri, N.T. In vitro pharmacological characterization of a novel unbiased NOP receptor-selective nonpeptide agonist AT-403. Pharmacol. Res. Perspect. 2017, 5, e00333. [Google Scholar] [CrossRef]
- Wichmann, J.; Adam, G.; Röver, S.; Cesura, A.M.; Dautzenberg, F.M.; Jenck, F. 8-acenaphthen-1-yl-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one derivatives as orphanin FQ receptor agonists. Bioorg. Med. Chem. Lett. 1999, 9, 2343–2348. [Google Scholar] [CrossRef]
- Wichmann, J.; Adam, G.; Röver, S.; Hennig, M.; Scalone, M.; Cesura, A.M.; Dautzenberg, F.M.; Jenck, F. Synthesis of (1S,3aS)-8-(2,3,3a,4,5, 6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4. 5]decan-4-one, a potent and selective orphanin FQ (OFQ) receptor agonist with anxiolytic-like properties. Eur. J. Med. Chem. 2000, 35, 839–851. [Google Scholar] [CrossRef]
- Shoblock, J.R. The pharmacology of Ro 64-6198, a systemically active, nonpeptide NOP receptor (opiate receptor-like 1, ORL-1) agonist with diverse preclinical therapeutic activity. CNS Drug Rev. 2007, 13, 107–136. [Google Scholar] [CrossRef]
- Ding, H.; Hayashida, K.; Suto, T.; Sukhtankar, D.D.; Kimura, M.; Mendenhall, V.; Ko, M.C. Supraspinal actions of nociceptin/orphanin FQ, morphine and substance P in regulating pain and itch in non-human primates. Br. J. Pharmacol. 2015, 172, 3302–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obara, I.; Przewlocki, R.; Przewlocka, B. Spinal and local peripheral antiallodynic activity of Ro64-6198 in neuropathic pain in the rat. Pain 2005, 116, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Varty, G.B.; Hyde, L.A.; Hodgson, R.A.; Lu, S.X.; McCool, M.F.; Kazdoba, T.M.; Del Vecchio, R.A.; Guthrie, D.H.; Pond, A.J.; Grzelak, M.E.; et al. Characterization of the nociceptin receptor (ORL-1) agonist, Ro64-6198, in tests of anxiety across multiple species. Psychopharmacology 2005, 182, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Sukhtankar, D.D.; Zaveri, N.T.; Husbands, S.M.; Ko, M.C. Effects of spinally administered bifunctional nociceptin/orphanin FQ peptide receptor/μ-opioid receptor ligands in mouse models of neuropathic and inflammatory pain. J. Pharmacol. Exp. Ther. 2013, 346, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Sukhtankar, D.D.; Lagorio, C.H.; Ko, M.C. Effects of the NOP agonist SCH221510 on producing and attenuating reinforcing effects as measured by drug self-administration in rats. Eur. J. Pharmacol. 2014, 745, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Liu, L. ORL1 Activation Mediates a Novel ORL1 Receptor Agonist SCH221510 Analgesia in Neuropathic Pain in Rats. J. Mol. Neurosci. 2018, 66, 10–16. [Google Scholar] [CrossRef]
- Cremeans, C.M.; Gruley, E.; Kyle, D.J.; Ko, M.C. Roles of mu-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. J. Pharmacol. Exp. Ther. 2012, 343, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.Q.; Wang, Z.Y.; Chen, J.M.; Wu, N.; Li, J. Involvement of the nociceptin opioid peptide receptor in morphine-induced antinociception, tolerance and physical dependence in female mice. Metab. Brain Dis. 2021, 36, 2243–2253. [Google Scholar] [CrossRef]
- Kiguchi, N.; Ko, M.C. Effects of NOP-Related Ligands in Nonhuman Primates. Handb. Exp. Pharmacol. 2019, 254, 323–343. [Google Scholar] [PubMed]
- Lutfy, K.; Hossain, S.M.; Khaliq, I.; Maidment, N.T. Orphanin FQ/nociceptin attenuates the development of morphine tolerance in rats. Br. J. Pharmacol. 2001, 134, 529–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, N.P.; Lee, Y.; Maidment, N.T. Orphanin FQ/nociceptin blocks acquisition of morphine place preference. Brain Res. 1999, 832, 168–170. [Google Scholar] [CrossRef]
- Murphy, N.P.; Ly, H.T.; Maidment, N.T. Intracerebroventricular orphanin FQ/nociceptin suppresses dopamine release in the nucleus accumbens of anaesthetized rats. Neuroscience 1996, 75, 1–4. [Google Scholar] [CrossRef]
- Rutten, K.; De Vry, J.; Bruckmann, W.; Tzschentke, T.M. Effects of the NOP receptor agonist Ro65-6570 on the acquisition of opiate- and psychostimulant-induced conditioned place preference in rats. Eur. J. Pharmacol. 2010, 645, 119–126. [Google Scholar] [CrossRef]
- Raffa, R.B.; Pergolizzi, J.V., Jr.; Tallarida, R.J. The determination and application of fixed-dose analgesic combinations for treating multimodal pain. J. Pain 2010, 11, 701–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiguchi, N.; Ding, H.; Kishioka, S.; Ko, M.C. Nociceptin/Orphanin FQ Peptide Receptor-Related Ligands as Novel Analgesics. Curr. Top. Med. Chem. 2020, 20, 2878–2888. [Google Scholar] [CrossRef] [PubMed]
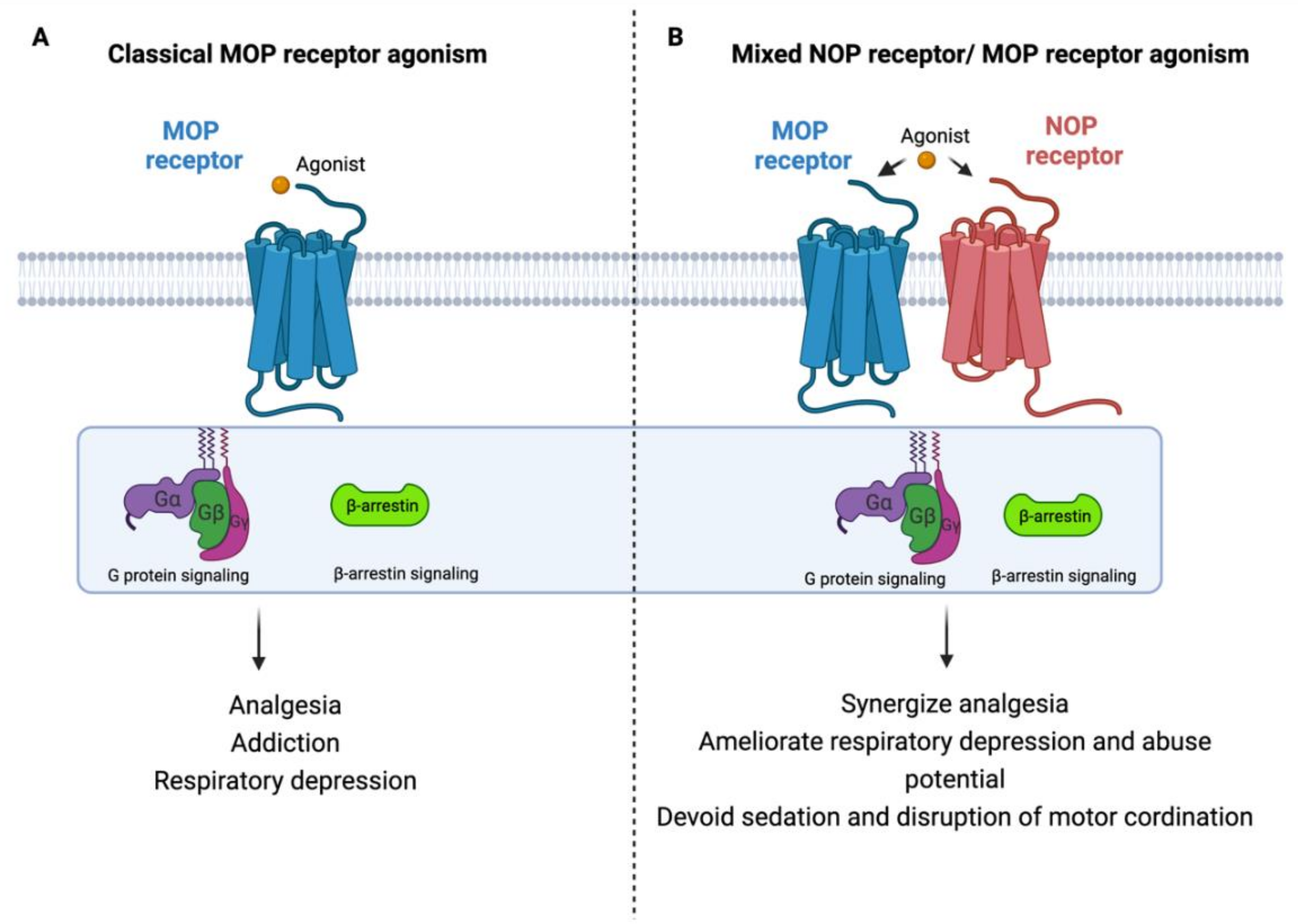
- Kiguchi, N.; Ding, H.; Ko, M.C. Therapeutic potentials of NOP and MOP receptor coactivation for the treatment of pain and opioid abuse. J. Neurosci. Res. 2020, 100, 191–202. [Google Scholar] [CrossRef]
- Mustazza, C.; Pieretti, S.; Marzoli, F. Nociceptin/Orphanin FQ Peptide (NOP) Receptor Modulators: An Update in Structure-Activity Relationships. Curr. Med. Chem. 2018, 25, 2353–2384. [Google Scholar] [CrossRef] [PubMed]
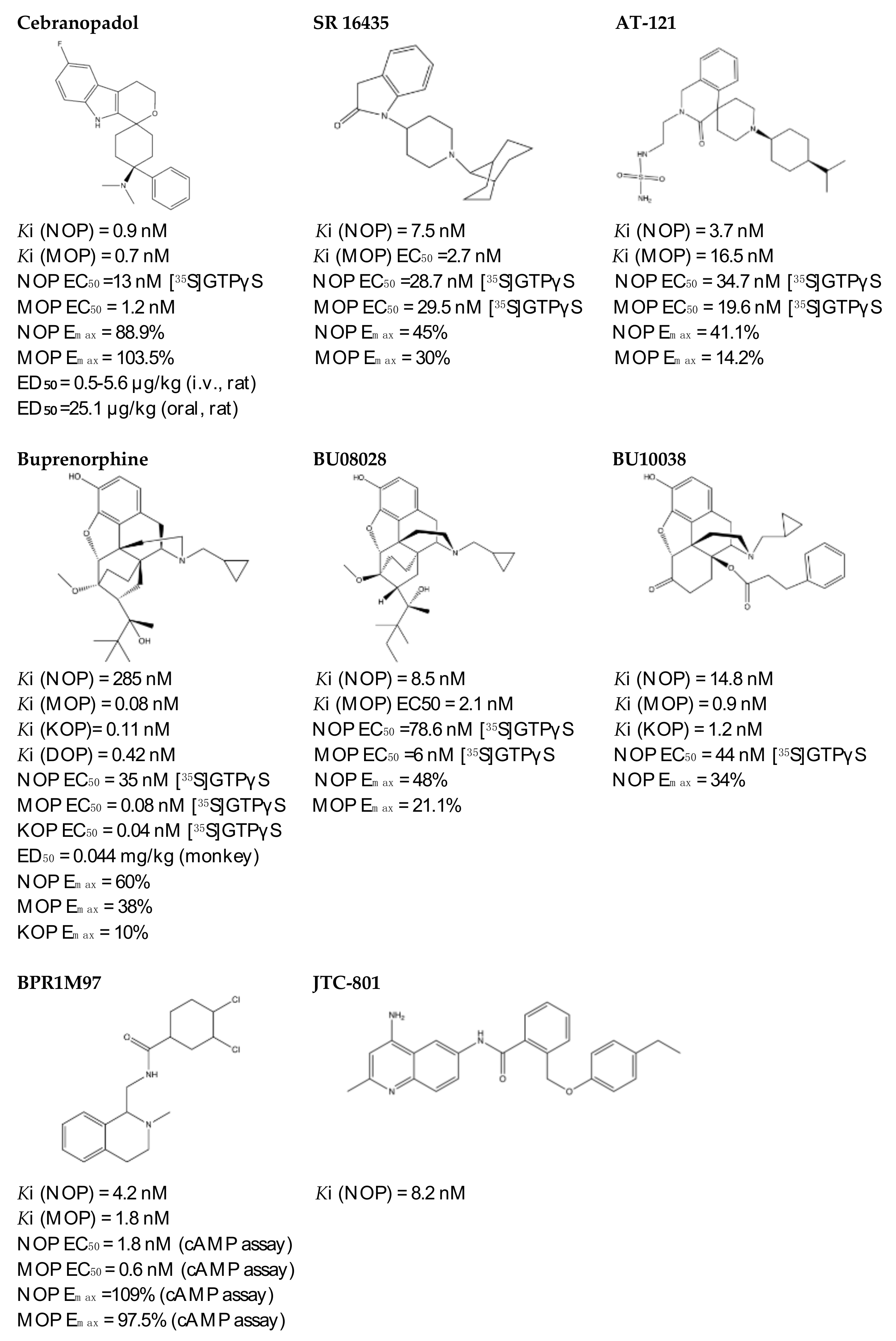
- Khroyan, T.V.; Zaveri, N.T.; Polgar, W.E.; Orduna, J.; Olsen, C.; Jiang, F.; Toll, L. SR 16435 [1-(1-(bicyclo[3.3.1]nonan-9-yl)piperidin-4-yl)indolin-2-one], a novel mixed nociceptin/orphanin FQ/mu-opioid receptor partial agonist: Analgesic and rewarding properties in mice. J. Pharmacol. Exp. Ther. 2007, 320, 934–943. [Google Scholar] [CrossRef]
- Linz, K.; Christoph, T.; Tzschentke, T.M.; Koch, T.; Schiene, K.; Gautrois, M.; Schröder, W.; Kögel, B.Y.; Beier, H.; Englberger, W.; et al. Cebranopadol: A novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J. Pharmacol. Exp. Ther. 2014, 349, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Schunk, S.; Linz, K.; Hinze, C.; Frormann, S.; Oberbörsch, S.; Sundermann, B.; Zemolka, S.; Englberger, W.; Germann, T.; Christoph, T.; et al. Discovery of a Potent Analgesic NOP and Opioid Receptor Agonist: Cebranopadol. ACS Med. Chem. Lett. 2014, 5, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Zaveri, N.T.; Jiang, F.; Olsen, C.M.; Deschamps, J.R.; Parrish, D.; Polgar, W.; Toll, L. A novel series of piperidin-4-yl-1,3-dihydroindol-2-ones as agonist and antagonist ligands at the nociceptin receptor. J. Med. Chem. 2004, 47, 2973–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Kiguchi, N.; Yasuda, D.; Daga, P.R.; Polgar, W.E.; Lu, J.J.; Czoty, P.W.; Kishioka, S.; Zaveri, N.T.; Ko, M.C. A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Sci. Transl. Med. 2018, 10, eaar3483. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Kehner, G.B.; Cowan, A.; Liu-Chen, L.Y. Comparison of pharmacological activities of buprenorphine and norbuprenorphine: Norbuprenorphine is a potent opioid agonist. J. Pharmacol. Exp. Ther. 2001, 297, 688–695. [Google Scholar] [PubMed]
- Khroyan, T.V.; Polgar, W.E.; Cami-Kobeci, G.; Husbands, S.M.; Zaveri, N.T.; Toll, L. The first universal opioid ligand, (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028): Characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. J. Pharmacol. Exp. Ther. 2011, 336, 952–961. [Google Scholar]
- Kiguchi, N.; Ding, H.; Cami-Kobeci, G.; Sukhtankar, D.D.; Czoty, P.W.; DeLoid, H.B.; Hsu, F.C.; Toll, L.; Husbands, S.M.; Ko, M.C. BU10038 as a safe opioid analgesic with fewer side-effects after systemic and intrathecal administration in primates. Br. J. Anaesth. 2019, 122, e146–e156. [Google Scholar] [CrossRef] [Green Version]
- Chao, P.K.; Chang, H.F.; Chang, W.T.; Yeh, T.K.; Ou, L.C.; Chuang, J.Y.; Tsu-An Hsu, J.; Tao, P.L.; Loh, H.H.; Shih, C.; et al. BPR1M97, a dual mu opioid receptor/nociceptin-orphanin FQ peptide receptor agonist, produces potent antinociceptive effects with safer properties than morphine. Neuropharmacology 2020, 166, 107678. [Google Scholar] [CrossRef]
- Shinkai, H.; Ito, T.; Iida, T.; Kitao, Y.; Yamada, H.; Uchida, I. 4-Aminoquinolines: Novel nociceptin antagonists with analgesic activity. J. Med. Chem. 2000, 43, 4667–4677. [Google Scholar] [CrossRef] [PubMed]
- Christoph, T.; Kögel, B.; Schiene, K.; Méen, M.; De Vry, J.; Friderichs, E. Broad analgesic profile of buprenorphine in rodent models of acute and chronic pain. Eur. J. Pharmacol. 2005, 507, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Czoty, P.W.; Kiguchi, N.; Cami-Kobeci, G.; Sukhtankar, D.D.; Nader, M.A.; Husbands, S.M.; Ko, M.C. A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proc. Natl. Acad. Sci. USA 2016, 113, E5511–E5518. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.R.; Ke, Y.Y.; Yeh, T.K.; Lin, S.Y.; Ou, L.C.; Chen, S.C.; Chang, W.T.; Chang, H.F.; Wu, Z.H.; Hsieh, C.C.; et al. Discovery, structure-activity relationship studies, and anti-nociceptive effects of N-(1,2,3,4-tetrahydro-1-isoquinolinylmethyl)benzamides as novel opioid receptor agonists. Eur. J. Med. Chem. 2017, 126, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Zaveri, N.T. Nociceptin Opioid Receptor (NOP) as a Therapeutic Target: Progress in Translation from Preclinical Research to Clinical Utility. J. Med. Chem. 2016, 59, 7011–7028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, D.G.; Bird, M.F.; Rowbotham, D.J. Cebranopadol: A first in-class example of a nociceptin/orphanin FQ receptor and opioid receptor agonist. Br. J. Anaesth. 2015, 114, 364–366. [Google Scholar] [CrossRef] [Green Version]
- Rizzi, A.; Cerlesi, M.C.; Ruzza, C.; Malfacini, D.; Ferrari, F.; Bianco, S.; Costa, T.; Guerrini, R.; Trapella, C.; Calo’, G. Pharmacological characterization of cebranopadol a novel analgesic acting as mixed nociceptin/orphanin FQ and opioid receptor agonist. Pharmacol. Res. Perspect. 2016, 4, e00247. [Google Scholar] [CrossRef] [PubMed]
- Tzschentke, T.M.; Linz, K.; Koch, T.; Christoph, T. Cebranopadol: A Novel First-in-Class Potent Analgesic Acting via NOP and Opioid Receptors. Handb. Exp. Pharmacol. 2019, 254, 367–398. [Google Scholar]
- Mann, A.; Moulédous, L.; Froment, C.; O’Neill, P.R.; Dasgupta, P.; Günther, T.; Brunori, G.; Kieffer, B.L.; Toll, L.; Bruchas, M.R.; et al. Agonist-selective NOP receptor phosphorylation correlates in vitro and in vivo and reveals differential post-activation signaling by chemically diverse agonists. Sci. Signal. 2019, 12, eaau8072. [Google Scholar] [CrossRef] [PubMed]
- Kleideiter, E.; Piana, C.; Wang, S.; Nemeth, R.; Gautrois, M. Clinical Pharmacokinetic Characteristics of Cebranopadol, a Novel First-in-Class Analgesic. Clin. Pharmacokinet. 2018, 57, 31–50. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/NCT00872885 (accessed on 12 January 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT01939366 (accessed on 12 January 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT01357837 (accessed on 12 January 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT01709214 (accessed on 12 January 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT01725087 (accessed on 12 January 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT01347671 (accessed on 12 January 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT00878293 (accessed on 12 January 2022).
- Eerdekens, M.H.; Kapanadze, S.; Koch, E.D.; Kralidis, G.; Volkers, G.; Ahmedzai, S.H.; Meissner, W. Cancer-related chronic pain: Investigation of the novel analgesic drug candidate cebranopadol in a randomized, double-blind, noninferiority trial. Eur. J. Pain 2019, 23, 577–588. [Google Scholar] [CrossRef]
- Koch, E.D.; Kapanadze, S.; Eerdekens, M.H.; Kralidis, G.; Létal, J.; Sabatschus, I.; Ahmedzai, S.H. Cebranopadol, a Novel First-in-Class Analgesic Drug Candidate: First Experience With Cancer-Related Pain for up to 26 Weeks. J. Pain Symptom. Manag. 2019, 58, 390–399. [Google Scholar] [CrossRef]
- Che, T.; Roth, B.L. Structural Insights Accelerate the Discovery of Opioid Alternatives. Annu. Rev. Biochem. 2021, 90, 739–761. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Name/Structure | Category | In Vitro Human NOP Receptor | In Vivo | Ref | |||
|---|---|---|---|---|---|---|---|
| Receptor Binding pKi | [35S]GTPγS pKB/pA2 | Ca+2 Mobilization pKB/pA2 | Administration Route/Dose/Species | Effect | |||
| [Nphe1]N/OFQ(113)NH2 | Selective NOP receptor antagonist | 8.39 | 7.33 | 6.29 | (30 nmol) i.c.v. mice | Analgesia Promote morphine-induced analgesia. | [61] |
| [Nphe1, Arg14, Lys15]N/OFQ-NH2 (UFP-101) | Selective NOP receptor antagonist | 10.24 | 8.85 | 7.66 | (10 nmol) i.c.v. mice | Long lasting analgesia Block N/OFQ effect on locomotor activity | [62] |
| (10 nmol) i.t. mice | Block N/OFQ (i.t.1 nmol) analgesic effect | [66] | |||||
| [(pF)Phe4Aib7Arg14Lys15]N/OFQ-NH2 (UFP-112) | Selective NOP receptor agonist | 10.55 | 10.55 | 9.05 | (1–100 pmol) i.c.v. mice | Hyperalgesia Decrease locomotor activity | [67] |
| (1–100 pmol) i.t. mice | Long lasting dose dependent analgesia | [67] | |||||
| (0.1 and 10 nmol/kg) Intravenous (i.v.) rats | Decrease heart rate Decrease blood pressure Decrease urinary sodium excretion Increase urine flow | [67] | |||||
| (1–10 nmol) i.t. monkey | Dose-dependent analgesia without inducing itch Promotes morphine-induced analgesia without increasing itch response | [68] | |||||
| [Phe1Ψ(CH2-NH)Gly2(pF)Phe4Aib7Arg14Lys15]N/OFQ-NH2 (UFP-113) | Selective NOP receptor partial agonist | 10.26 | 9.72 | 7.97 | (0.001–1 nmol) i.t. rats | Analgesia | [69] |
PWT2-N/OFQ | Selective NOP receptor agonist | 10.3 | 10.12 | 8.83 | (250 pmol) i.c.v. mice | Decease locomotor activity | [70] |
| (2.5–250 pmol) i.t. mice | Dose-dependent analgesia | [71] | |||||
| (0.3, 1, and 3 nmol) i.t. monkey | Analgesia No itching No sedation No impairment in motor activity | [71] | |||||
| Name/Structure | Category | In Vitro Human NOP Receptor | In Vivo | Ref | |||
|---|---|---|---|---|---|---|---|
| Receptor Binding pKi | [35S]GTPγS pEC50 | Ca+2 MobilizationpEC 50 | Administration Route/Dose/Species | Effect | |||
Ro 65-6570 | NOP receptor non peptide agonist | 8.6 | (0.1–1 mg/kg) (0.03 to 1 μmol/kg) i.v. mice | Analgesia | [79,80] | ||
Ro 64-6198 | NOP receptor nonpeptide agonist | 9.41 | 8.09 | 7.98 | (3 mg/kg) (1 mg/kg) intraperitoneal (i.p) mice (0.3 to 3 mg/kg) i.p. mice | Analgesia Additive analgesia anxiolytic-like effects | [81,82,83] |
| (0.001–0.06 mg/kg), subcutaneous (s.c.) monkey | Analgesia No depression No itching No reinforcing | [17] | |||||
SCH221510 | Selective NOP receptor nonpeptide agonist | 0.3 | 12 | 1–30 mg/kg) peroral (p.o.) rat | anxiolytic-like effects | [84] | |
| (0.1–3.0 mg/kg) i.p., p.o., intracolonic mice | potent anti-inflammatory and analgesic effect | [85] | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Daibani, A.; Che, T. Spotlight on Nociceptin/Orphanin FQ Receptor in the Treatment of Pain. Molecules 2022, 27, 595. https://doi.org/10.3390/molecules27030595
El Daibani A, Che T. Spotlight on Nociceptin/Orphanin FQ Receptor in the Treatment of Pain. Molecules. 2022; 27(3):595. https://doi.org/10.3390/molecules27030595
Chicago/Turabian StyleEl Daibani, Amal, and Tao Che. 2022. "Spotlight on Nociceptin/Orphanin FQ Receptor in the Treatment of Pain" Molecules 27, no. 3: 595. https://doi.org/10.3390/molecules27030595
APA StyleEl Daibani, A., & Che, T. (2022). Spotlight on Nociceptin/Orphanin FQ Receptor in the Treatment of Pain. Molecules, 27(3), 595. https://doi.org/10.3390/molecules27030595