
Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors



Abstract
:1. Introduction
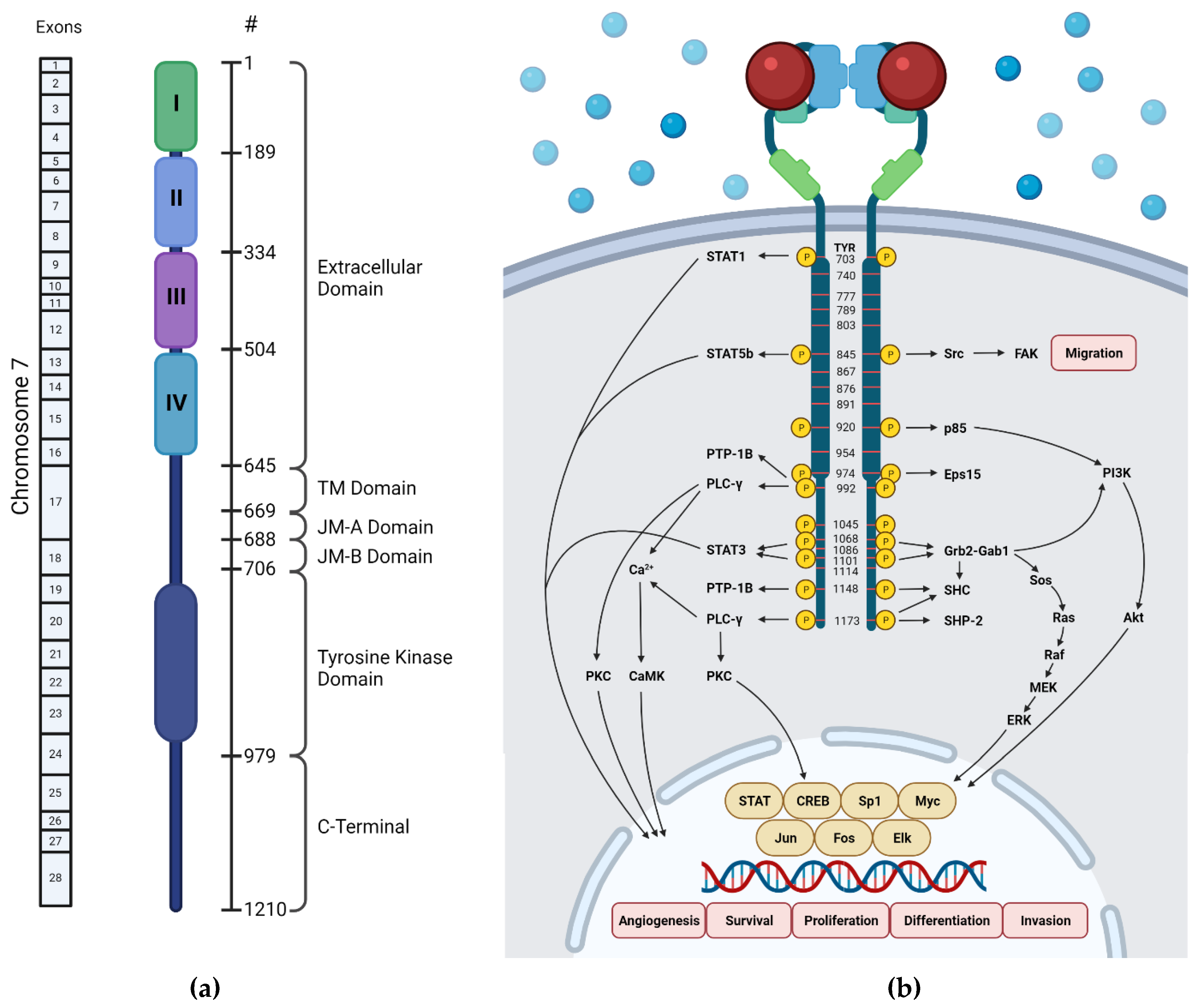
2. Structure of EGFR
2.1. Extracellular Domain
2.2. Transmembrane Domain
2.3. Juxtamembrane Domain
2.4. Kinase Domain
2.5. C-Terminal
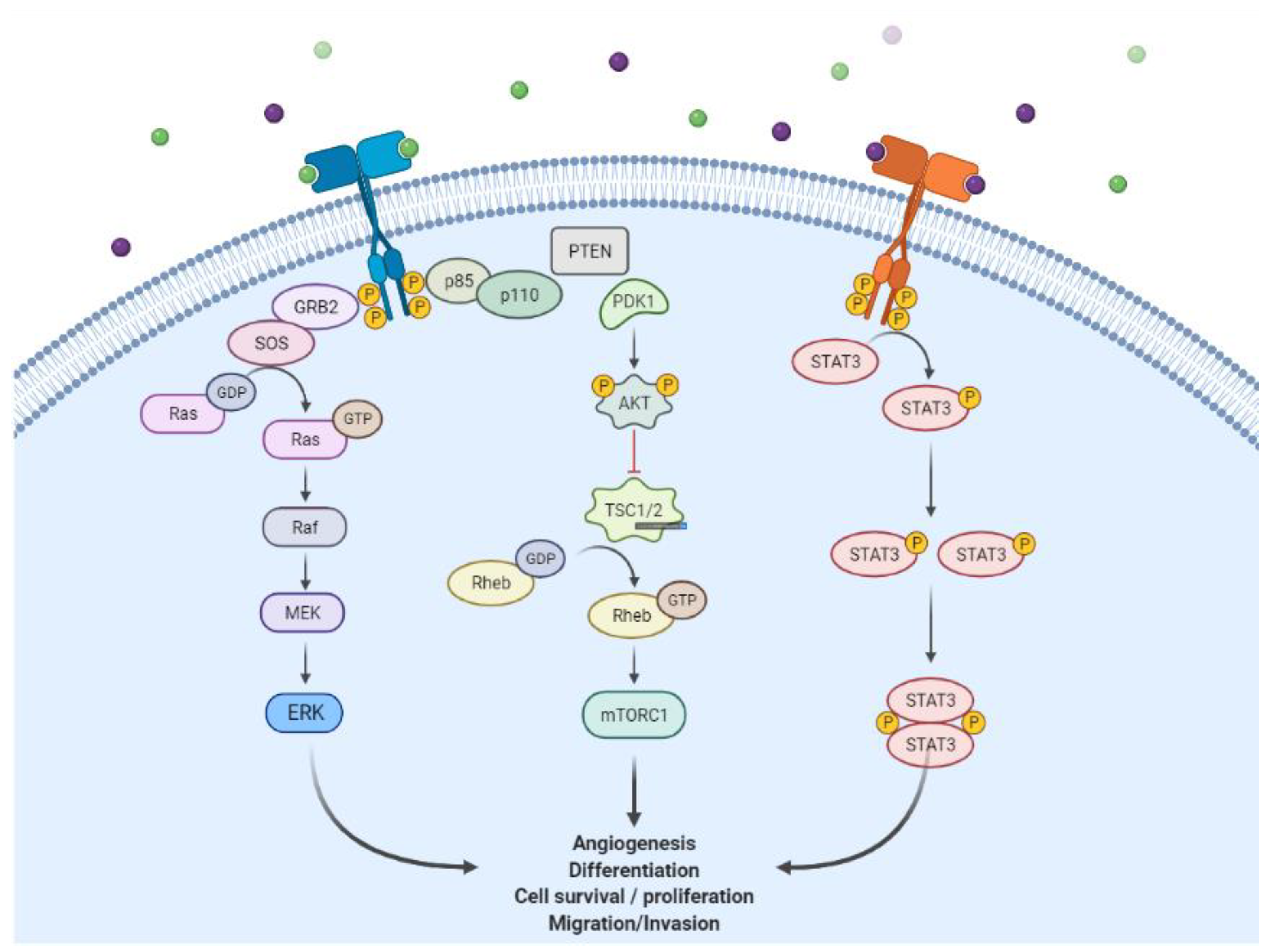
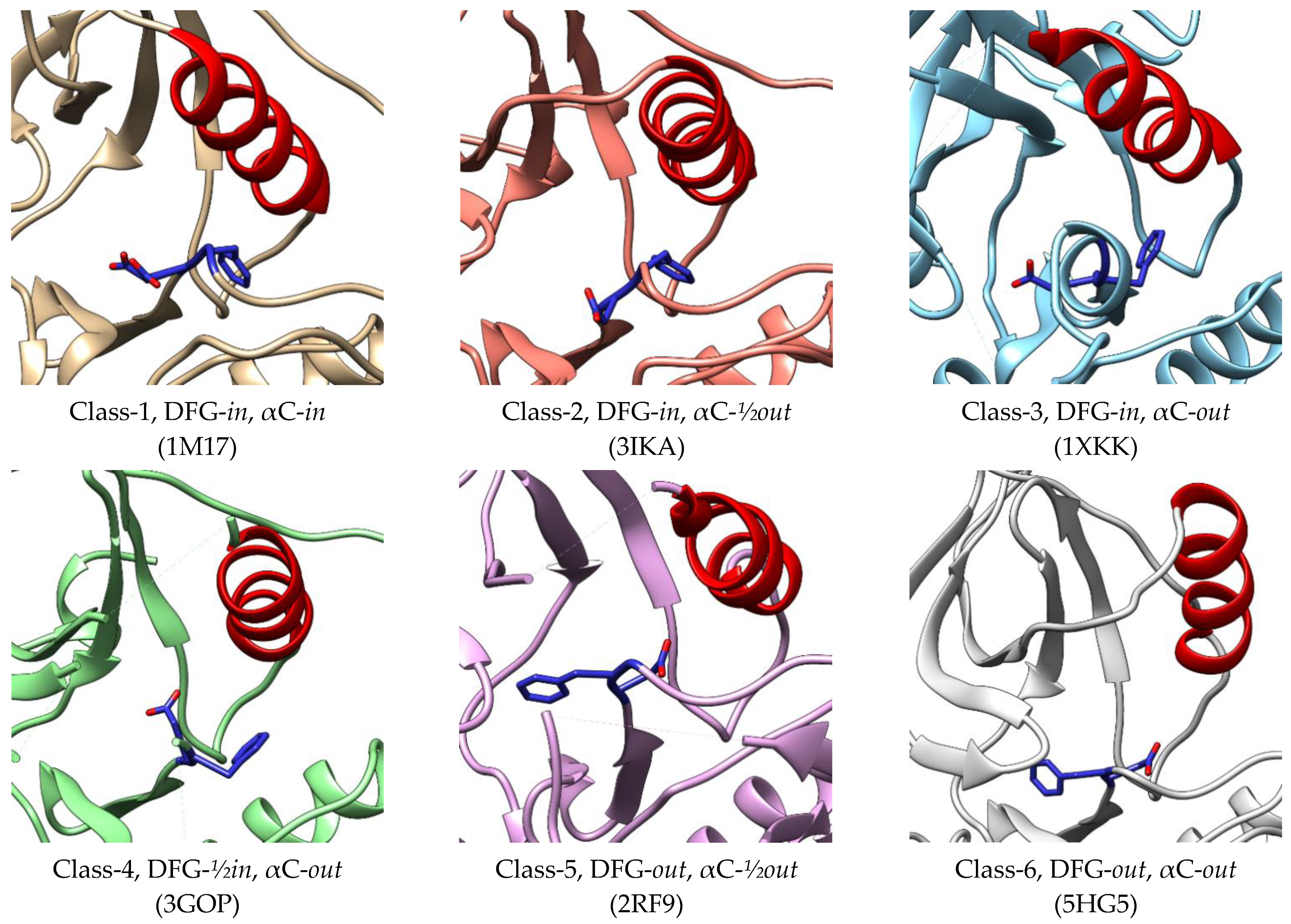
3. Active–Inactive Conformation
4. Mutation of EGFR and Resistance Mechanism
5. ATP-Binding Site
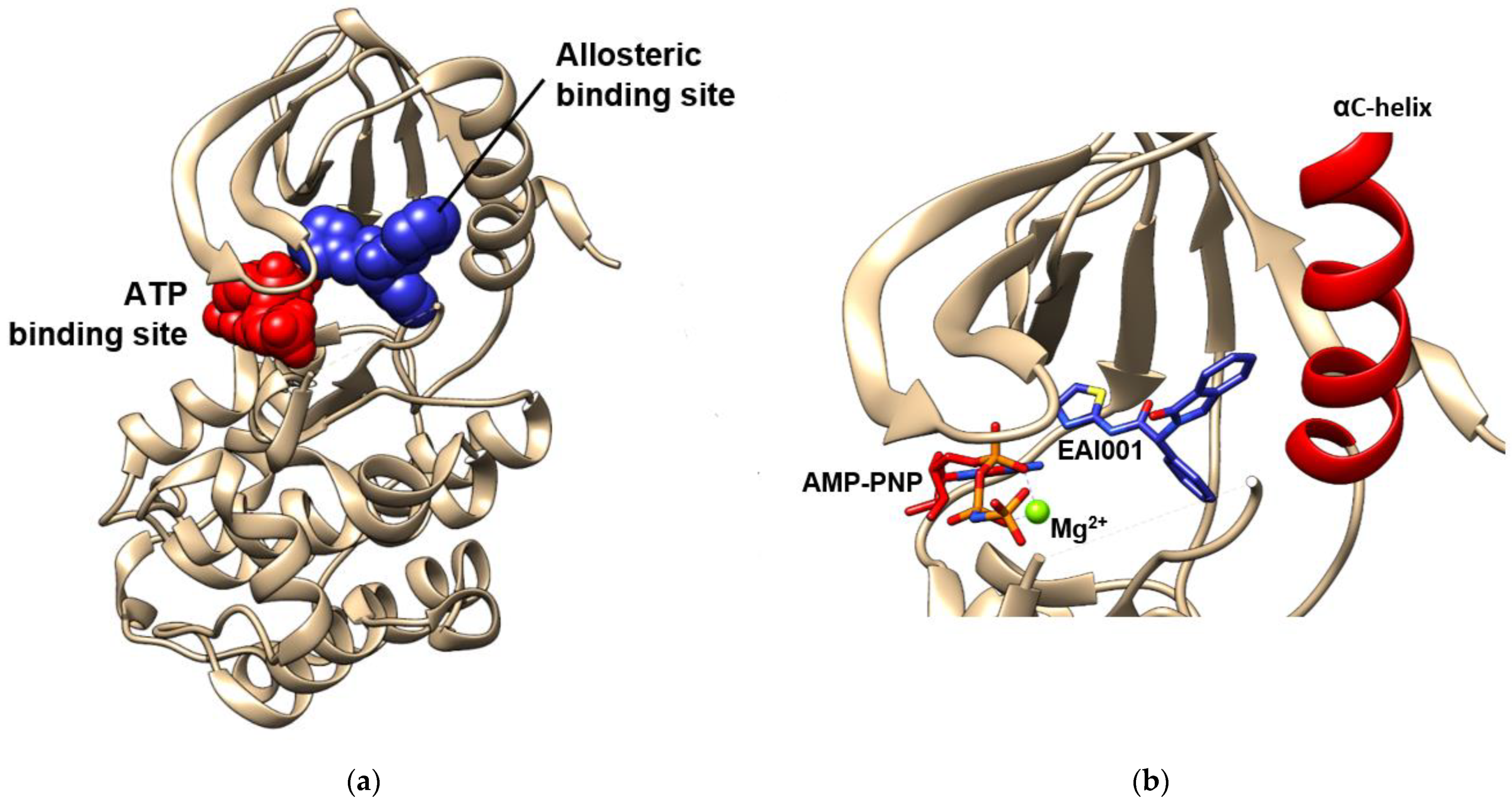
6. Allosteric Site
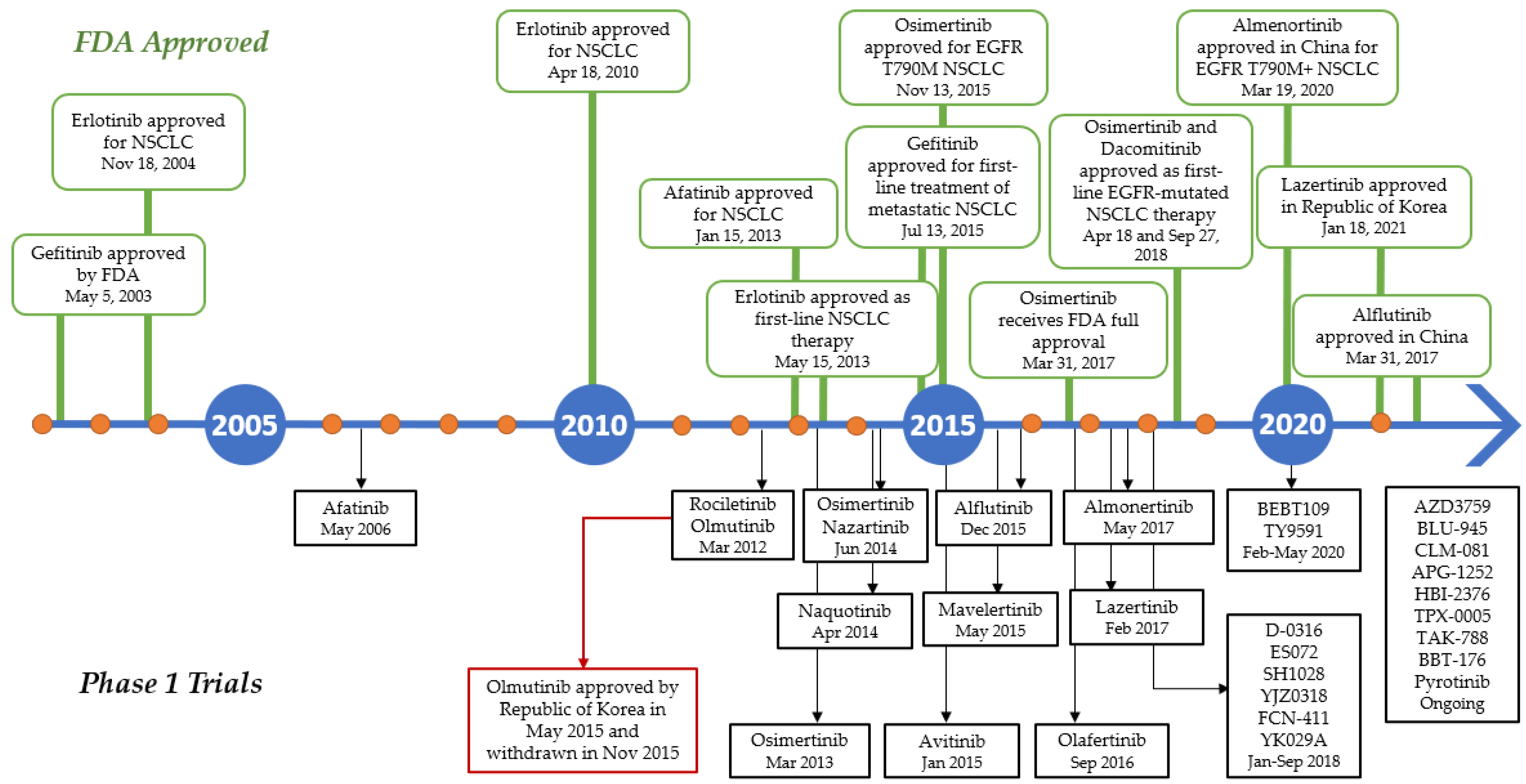
7. EGFR Tyrosine Kinase Inhibitors
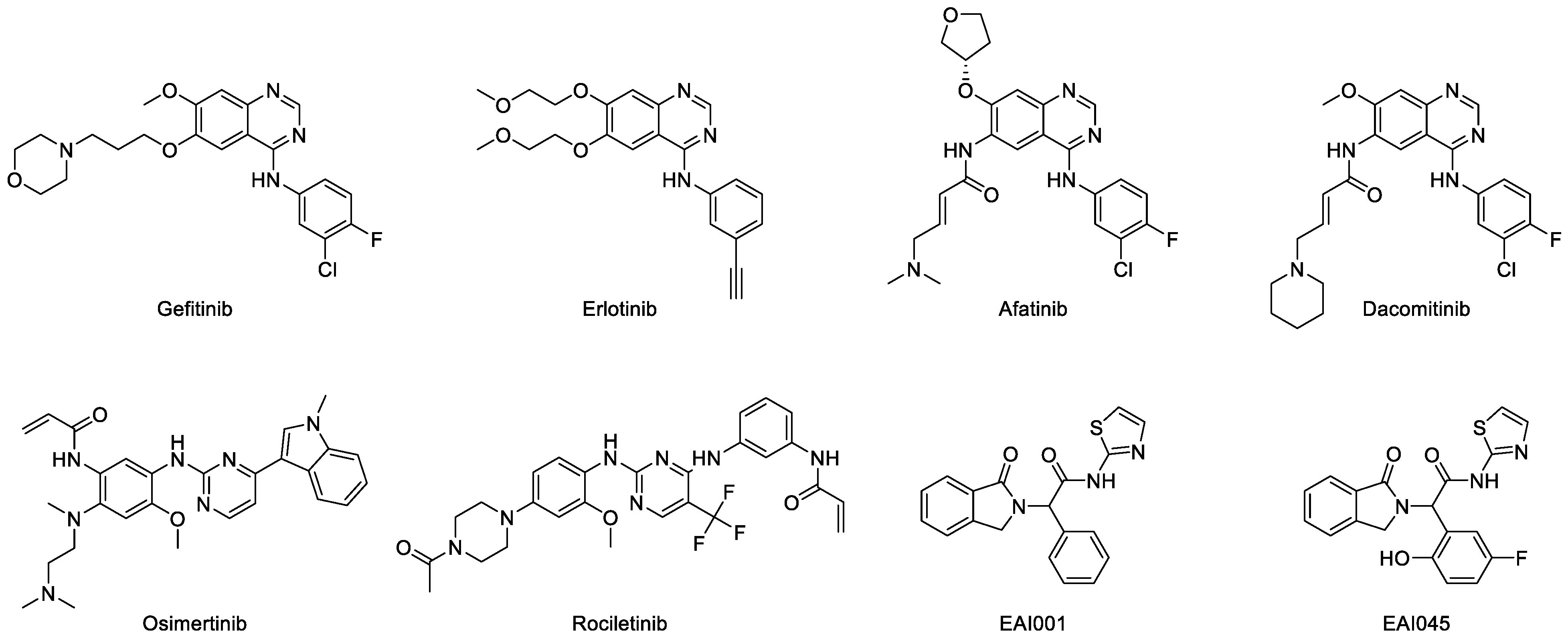
7.1. First-Generation Inhibitors
7.2. Second-Generation Inhibitors
7.3. Third-Generation Inhibitors
7.4. Fourth-Generation Inhibitors
7.5. Other Allosteric Inhibitors
7.6. Small Molecules in the Clinical Trials
8. Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB Signalling Network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Zhang, G.; Haura, E.B. Targeting Epidermal Growth Factor Receptor: Central Signaling Kinase in Lung Cancer. Biochem. Pharmacol. 2010, 80, 613–623. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Carotenuto, A.; Rachiglio, A.; Gallo, M.; Maiello, M.R.; Aldinucci, D.; Pinto, A.; Normanno, N. The Role of the EGFR Signaling in Tumor Microenvironment. J. Cell. Physiol. 2008, 214, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Hidalgo, M. Pharmacogenomics of Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors. Biochim. Biophys. Acta (BBA) Rev. Cancer 2006, 1766, 217–229. [Google Scholar] [CrossRef]
- Mosesson, Y.; Yarden, Y. Oncogenic Growth Factor Receptors: Implications for Signal Transduction Therapy. Semin. Cancer Biol. 2004, 14, 262–270. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Al Olayan, A.; al Hussaini, H.; Rahman Jazieh, A. The Roles of Epidermal Growth Factor Receptor (EGFR) Inhibitors in the Management of Lung Cancer. J. Infect. Public Health 2012, 5, S50–S60. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Engelman, J.A.; Johnson, B.E. Epidermal Growth Factor Receptor Mutations in Non–Small-Cell Lung Cancer: Implications for Treatment and Tumor Biology. J. Clin. Oncol. 2005, 23, 3227–3234. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [Green Version]
- Woodburn, J.R. The Epidermal Growth Factor Receptor and Its Inhibition in Cancer Therapy. Pharmacol. Ther. 1999, 82, 241–250. [Google Scholar] [CrossRef]
- Huang, H.-J.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.-D.; Huang, C.-M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The Enhanced Tumorigenic Activity of a Mutant Epidermal Growth Factor Receptor Common in Human Cancers Is Mediated by Threshold Levels of Constitutive Tyrosine Phosphorylation and Unattenuated Signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, J.; Hsuan, J.; Waterfield, M. Analysis of Mammalian Fibroblast Transformation by Normal and Mutated Human EGF Receptors. Oncogene 1989, 4, 273–283. [Google Scholar]
- Hazan, R.B.; Norton, L. The Epidermal Growth Factor Receptor Modulates the Interaction of E-Cadherin with the Actin Cytoskeleton. J. Biol. Chem. 1998, 273, 9078–9084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Bunn, P.A. Targeting the Epidermal Growth Factor Receptor in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2003, 9, 5813–5824. [Google Scholar] [PubMed]
- Ellerbroek, S.M.; Halbleib, J.M.; Benavidez, M.; Warmka, J.K.; Wattenberg, E.V.; Stack, M.S.; Hudson, L.G. Phosphatidylinositol 3-Kinase Activity in Epidermal Growth Factor-Stimulated Matrix Metalloproteinase-9 Production and Cell Surface Association. Cancer Res. 2001, 61, 1855–1861. [Google Scholar] [PubMed]
- Roskoski, R. Small Molecule Inhibitors Targeting the EGFR/ErbB Family of Protein-Tyrosine Kinases in Human Cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Fu, L. Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Chang, Y. Epidermal Growth Factor Receptor (EGFR) Phosphorylation, Signaling and Trafficking in Prostate Cancer. In Prostate Cancer—From Bench to Bedside; InTech: London, UK, 2011. [Google Scholar]
- Ullrich, A.; Schlessinger, J. Signal Transduction by Receptors with Tyrosine Kinase Activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J.; et al. Human Epidermal Growth Factor Receptor CDNA Sequence and Aberrant Expression of the Amplified Gene in A431 Epidermoid Carcinoma Cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Lelimousin, M.; Limongelli, V.; Sansom, M.S.P. Conformational Changes in the Epidermal Growth Factor Receptor: Role of the Transmembrane Domain Investigated by Coarse-Grained MetaDynamics Free Energy Calculations. J. Am. Chem. Soc. 2016, 138, 10611–10622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocharov, E.V.; Bragin, P.E.; Pavlov, K.V.; Bocharova, O.V.; Mineev, K.S.; Polyansky, A.A.; Volynsky, P.E.; Efremov, R.G.; Arseniev, A.S. The Conformation of the Epidermal Growth Factor Receptor Transmembrane Domain Dimer Dynamically Adapts to the Local Membrane Environment. Biochemistry 2017, 56, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Endres, N.F.; Das, R.; Smith, A.W.; Arkhipov, A.; Kovacs, E.; Huang, Y.; Pelton, J.G.; Shan, Y.; Shaw, D.E.; Wemmer, D.E.; et al. Conformational Coupling across the Plasma Membrane in Activation of the EGF Receptor. Cell 2013, 152, 543–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janosi, L.; Prakash, A.; Doxastakis, M. Lipid-Modulated Sequence-Specific Association of Glycophorin A in Membranes. Biophys. J. 2010, 99, 284–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, D.; Marrink, S.J. Lipid-Mediated Interactions Tune the Association of Glycophorin A Helix and Its Disruptive Mutants in Membranes. Phys. Chem. Chem. Phys. 2010, 12, 12987–12996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, A.S.; Polyansky, A.A.; Fleck, M.; Volynsky, P.E.; Efremov, R.G. Adaptable Lipid Matrix Promotes Protein–Protein Association in Membranes. J. Chem. Theory Comput. 2015, 11, 4415–4426. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Mayzel, M.L.; Volynsky, P.E.; Mineev, K.S.; Tkach, E.N.; Ermolyuk, Y.S.; Schulga, A.A.; Efremov, R.G.; Arseniev, A.S. Left-Handed Dimer of EphA2 Transmembrane Domain: Helix Packing Diversity among Receptor Tyrosine Kinases. Biophys. J. 2010, 98, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Muhle-Goll, C.; Hoffmann, S.; Afonin, S.; Grage, S.L.; Polyansky, A.A.; Windisch, D.; Zeitler, M.; Bürck, J.; Ulrich, A.S. Hydrophobic Matching Controls the Tilt and Stability of the Dimeric Platelet-Derived Growth Factor Receptor (PDGFR) β Transmembrane Segment. J. Biol. Chem. 2012, 287, 26178–26186. [Google Scholar] [CrossRef] [Green Version]
- Brewer, M.R.; Choi, S.H.; Alvarado, D.; Moravcevic, K.; Pozzi, A.; Lemmon, M.A.; Carpenter, G. The Juxtamembrane Region of the EGF Receptor Functions as an Activation Domain. Mol. Cell 2009, 34, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Aifa, S.; Aydin, J.; Nordvall, G.; Lundström, I.; Svensson, S.P.S.; Hermanson, O. A Basic Peptide within the Juxtamembrane Region Is Required for EGF Receptor Dimerization. Exp. Cell Res. 2005, 302, 108–114. [Google Scholar] [CrossRef]
- Thiel, K.W.; Carpenter, G. Epidermal Growth Factor Receptor Juxtamembrane Region Regulates Allosteric Tyrosine Kinase Activation. Proc. Natl. Acad. Sci. USA 2007, 104, 19238–19243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkhipov, A.; Shan, Y.; Kim, E.T.; Shaw, D.E. Membrane Interaction of Bound Ligands Contributes to the Negative Binding Cooperativity of the EGF Receptor. PLoS Comput. Biol. 2014, 10, e1003742. [Google Scholar] [CrossRef] [Green Version]
- Hedger, G.; Sansom, M.S.P.; Koldsø, H. The Juxtamembrane Regions of Human Receptor Tyrosine Kinases Exhibit Conserved Interaction Sites with Anionic Lipids. Sci. Rep. 2015, 5, 9198. [Google Scholar] [CrossRef] [Green Version]
- Kil, S.J.; Carlin, C. EGF Receptor Residues Leu679, Leu680 Mediate Selective Sorting of Ligand-Receptor Complexes in Early Endosomal Compartments. J. Cell. Physiol. 2000, 185, 47–60. [Google Scholar] [CrossRef]
- He, C.; Hobert, M.; Friend, L.; Carlin, C. The Epidermal Growth Factor Receptor Juxtamembrane Domain Has Multiple Basolateral Plasma Membrane Localization Determinants, Including a Dominant Signal with a Polyproline Core. J. Biol. Chem. 2002, 277, 38284–38293. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.-C. Nuclear Localization of EGF Receptor and Its Potential New Role as a Transcription Factor. Nat. Cell Biol. 2001, 3, 802–808. [Google Scholar] [CrossRef]
- Martín-Nieto, J.; Villalobo, A. The Human Epidermal Growth Factor Receptor Contains a Juxtamembrane Calmodulin-Binding Site. Biochemistry 1998, 37, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.; Ling, N.; Cooper, J.A. Protein Kinase C Phosphorylation of the EGF Receptor at a Threonine Residue Close to the Cytoplasmic Face of the Plasma Membrane. Nature 1984, 311, 480–483. [Google Scholar] [CrossRef]
- Davis, R.J.; Czech, M.P. Tumor-Promoting Phorbol Diesters Cause the Phosphorylation of Epidermal Growth Factor Receptors in Normal Human Fibroblasts at Threonine-654. Proc. Natl. Acad. Sci. USA 1985, 82, 1974–1978. [Google Scholar] [CrossRef] [Green Version]
- Takishima, K.; Griswold-Prenner, I.; Ingebritsen, T.; Rosner, M.R. Epidermal Growth Factor (EGF) Receptor T669 Peptide Kinase from 3T3-L1 Cells Is an EGF-Stimulated “MAP” Kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 2520–2524. [Google Scholar] [CrossRef] [Green Version]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jura, N.; Zhang, X.; Endres, N.F.; Seeliger, M.A.; Schindler, T.; Kuriyan, J. Catalytic Control in the EGF Receptor and Its Connection to General Kinase Regulatory Mechanisms. Mol. Cell 2011, 42, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Fernandez, M.L.; Clarke, D.T.; Roberts, S.K.; Zanetti-Domingues, L.C.; Gervasio, F.L. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer. Cells 2019, 8, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertics, P.J.; Gill, G.N. Self-Phosphorylation Enhances the Protein-Tyrosine Kinase Activity of the Epidermal Growth Factor Receptor. J. Biol. Chem. 1985, 260, 14642–14647. [Google Scholar] [CrossRef]
- Bertics, P.J.; Chen, W.S.; Hubler, L.; Lazar, C.S.; Rosenfeld, M.G.; Gill, G.N. Alteration of Epidermal Growth Factor Receptor Activity by Mutation of Its Primary Carboxyl-Terminal Site of Tyrosine Self-Phosphorylation. J. Biol. Chem. 1988, 263, 3610–3617. [Google Scholar] [CrossRef]
- Mustafa, M.; Mirza, A.; Kannan, N. Conformational Regulation of the EGFR Kinase Core by the Juxtamembrane and C-Terminal Tail: A Molecular Dynamics Study. Proteins Struct. Funct. Bioinform. 2011, 79, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.; Das, R.; Wang, Q.; Collier, T.S.; Cantor, A.; Huang, Y.; Wong, K.; Mirza, A.; Barros, T.; Grob, P.; et al. Analysis of the Role of the C-Terminal Tail in the Regulation of the Epidermal Growth Factor Receptor. Mol. Cell. Biol. 2015, 35, 3083–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, M.; Fleishman, S.J.; Ben-Tal, N. A Putative Mechanism for Downregulation of the Catalytic Activity of the EGF Receptor via Direct Contact between Its Kinase and C-Terminal Domains. Structure 2004, 12, 2265–2275. [Google Scholar] [CrossRef] [Green Version]
- Sorkin, A.; Mazzotti, M.; Sorkina, T.; Scotto, L.; Beguinot, L. Epidermal Growth Factor Receptor Interaction with Clathrin Adaptors Is Mediated by the Tyr974-Containing Internalization Motif. J. Biol. Chem. 1996, 271, 13377–13384. [Google Scholar] [CrossRef] [Green Version]
- Bublil, E.M.; Pines, G.; Patel, G.; Fruhwirth, G.; Ng, T.; Yarden, Y. Kinase-Mediated Quasi-Dimers of EGFR. FASEB J. 2010, 24, 4744–4755. [Google Scholar] [CrossRef]
- Jura, N.; Endres, N.F.; Engel, K.; Deindl, S.; Das, R.; Lamers, M.H.; Wemmer, D.E.; Zhang, X.; Kuriyan, J. Mechanism for Activation of the EGF Receptor Catalytic Domain by the Juxtamembrane Segment. Cell 2009, 137, 1293–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canagarajah, B.J.; Khokhlatchev, A.; Cobb, M.H.; Goldsmith, E.J. Activation Mechanism of the MAP Kinase ERK2 by Dual Phosphorylation. Cell 1997, 90, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Hasenahuer, M.A.; Barletta, G.P.; Fernandez-Alberti, S.; Parisi, G.; Fornasari, M.S. Pockets as Structural Descriptors of EGFR Kinase Conformations. PLoS ONE 2017, 12, e0189147. [Google Scholar] [CrossRef] [Green Version]
- Nagar, B. C-Abl Tyrosine Kinase and Inhibition by the Cancer Drug Imatinib (Gleevec/STI-571). J. Nutr. 2007, 137, 1518S–1523S. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.R. Crystal Structure of the Activated Insulin Receptor Tyrosine Kinase in Complex with Peptide Substrate and ATP Analog. EMBO J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Wei, L.; Hendrickson, W.A. Crystal Structure of the Tyrosine Kinase Domain of the Human Insulin Receptor. Nature 1994, 372, 746–754. [Google Scholar] [CrossRef]
- Hari, S.B.; Merritt, E.A.; Maly, D.J. Sequence Determinants of a Specific Inactive Protein Kinase Conformation. Chem. Biol. 2013, 20, 806–815. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M Mutation in EGFR Kinase Causes Drug Resistance by Increasing the Affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Xie, L.; Bourne, P.E. Structural Insights into Characterizing Binding Sites in Epidermal Growth Factor Receptor Kinase Mutants. J. Chem. Inf. Model. 2019, 59, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel Mutant-Selective EGFR Kinase Inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib). Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Pickin, K.A.; Bose, R.; Jura, N.; Cole, P.A.; Kuriyan, J. Inhibition of the EGF Receptor by Binding of MIG6 to an Activating Kinase Domain Interface. Nature 2007, 450, 741–744. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Nair, S.K.; Murray, B.W.; Almaden, C.; Bailey, S.; Baxi, S.; Behenna, D.; Cho-Schultz, S.; Dalvie, D.; Dinh, D.M.; et al. Discovery of 1-{(3 R,4 R)-3-[({5-Chloro-2-[(1-Methyl-1 H -Pyrazol-4-Yl)Amino]-7 H -Pyrrolo[2,3-d]Pyrimidin-4-Yl}oxy)Methyl]-4-Methoxypyrrolidin-1-Yl}prop-2-En-1-One (PF-06459988), a Potent, WT Sparing, Irreversible Inhibitor of T790M-Containing EGFR Mutants. J. Med. Chem. 2016, 59, 2005–2024. [Google Scholar] [CrossRef]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Ishii, G.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (MTOR) Axis Is Responsible for Aerobic Glycolysis Mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-Mutated Lung Adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.A.; Luwor, R.B.; Burgess, A.W. Epidermal Growth Factor Receptor: Structure-Function Informing the Design of Anticancer Therapeutics. Exp. Cell Res. 2018, 371, 1–9. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, H.; Gazdar, A.F. Somatic Mutations of Epidermal Growth Factor Receptor Signaling Pathway in Lung Cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and Biological Features Associated with Epidermal Growth Factor Receptor Gene Mutations in Lung Cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Choong, N.W.; Dietrich, S.; Seiwert, T.Y.; Tretiakova, M.S.; Nallasura, V.; Davies, G.C.; Lipkowitz, S.; Husain, A.N.; Salgia, R.; Ma, P.C. Gefitinib Response of Erlotinib-Refractory Lung Cancer Involving Meninges—Role of EGFR Mutation. Nat. Clin. Pract. Oncol. 2006, 3, 50–57. [Google Scholar] [CrossRef]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and Clinical Relevance of the Epidermal Growth Factor Receptor in Human Cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible Inhibitors of the EGF Receptor May Circumvent Acquired Resistance to Gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Yoshida, K.; Hida, T.; Tsuboi, M.; Tada, H.; Kuwano, H.; Mitsudomi, T. Analysis of Epidermal Growth Factor Receptor Gene Mutation in Patients with Non–Small Cell Lung Cancer and Acquired Resistance to Gefitinib. Clin. Cancer Res. 2006, 12, 5764–5769. [Google Scholar] [CrossRef] [Green Version]
- Balak, M.N.; Gong, Y.; Riely, G.J.; Somwar, R.; Li, A.R.; Zakowski, M.F.; Chiang, A.; Yang, G.; Ouerfelli, O.; Kris, M.G.; et al. Novel D761Y and Common Secondary T790M Mutations in Epidermal Growth Factor Receptor–Mutant Lung Adenocarcinomas with Acquired Resistance to Kinase Inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [Google Scholar] [CrossRef] [Green Version]
- Carter, T.A.; Wodicka, L.M.; Shah, N.P.; Velasco, A.M.; Fabian, M.A.; Treiber, D.K.; Milanov, Z.V.; Atteridge, C.E.; Biggs, W.H.; Edeen, P.T.; et al. Inhibition of Drug-Resistant Mutants of ABL, KIT, and EGF Receptor Kinases. Proc. Natl. Acad. Sci. USA 2005, 102, 11011–11016. [Google Scholar] [CrossRef] [Green Version]
- Greulich, H.; Chen, T.-H.; Feng, W.; Jänne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic Transformation by Inhibitor-Sensitive and -Resistant EGFR Mutants. PLoS Med. 2005, 2, e313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, Y.; Suda, K.; Kimura, H.; Matsumoto, K.; Arao, T.; Nagai, T.; Saijo, N.; Yatabe, Y.; Mitsudomi, T.; Nishio, K. Highly Sensitive Detection of EGFR T790M Mutation Using Colony Hybridization Predicts Favorable Prognosis of Patients with Lung Cancer Harboring Activating EGFR Mutation. J. Thorac. Oncol. 2012, 7, 1640–1644. [Google Scholar] [CrossRef] [Green Version]
- Su, K.-Y.; Chen, H.-Y.; Li, K.-C.; Kuo, M.-L.; Yang, J.C.-H.; Chan, W.-K.; Ho, B.-C.; Chang, G.-C.; Shih, J.-Y.; Yu, S.-L.; et al. Pretreatment Epidermal Growth Factor Receptor (EGFR) T790M Mutation Predicts Shorter EGFR Tyrosine Kinase Inhibitor Response Duration in Patients with Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2012, 30, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Ji, H.; Yuza, Y.; Meyerson, M.; Wong, K.-K.; Tenen, D.G.; Halmos, B. An Alternative Inhibitor Overcomes Resistance Caused by a Mutation of the Epidermal Growth Factor Receptor. Cancer Res. 2005, 65, 7096–7101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Mukohara, T.; Zejnullahu, K.; Lifshits, E.; Borras, A.M.; Gale, C.-M.; Naumov, G.N.; Yeap, B.Y.; Jarrell, E.; Sun, J.; et al. Allelic Dilution Obscures Detection of a Biologically Significant Resistance Mutation in EGFR-Amplified Lung Cancer. J. Clin. Investig. 2006, 116, 2695–2706. [Google Scholar] [CrossRef]
- Costa, D.B.; Halmos, B.; Kumar, A.; Schumer, S.T.; Huberman, M.S.; Boggon, T.J.; Tenen, D.G.; Kobayashi, S. BIM Mediates EGFR Tyrosine Kinase Inhibitor-Induced Apoptosis in Lung Cancers with Oncogenic EGFR Mutations. PLoS Med. 2007, 4, e315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bean, J.; Riely, G.J.; Balak, M.; Marks, J.L.; Ladanyi, M.; Miller, V.A.; Pao, W. Acquired Resistance to Epidermal Growth Factor Receptor Kinase Inhibitors Associated with a Novel T854A Mutation in a Patient with EGFR -Mutant Lung Adenocarcinoma. Clin. Cancer Res. 2008, 14, 7519–7525. [Google Scholar] [CrossRef] [Green Version]
- Avizienyte, E.; Ward, R.A.; Garner, A.P. Comparison of the EGFR Resistance Mutation Profiles Generated by EGFR-Targeted Tyrosine Kinase Inhibitors and the Impact of Drug Combinations. Biochem. J. 2008, 415, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Sousa, A.C.; Silveira, C.; Janeiro, A.; Malveiro, S.; Oliveira, A.R.; Felizardo, M.; Nogueira, F.; Teixeira, E.; Martins, J.; Carmo-Fonseca, M. Detection of Rare and Novel EGFR Mutations in NSCLC Patients: Implications for Treatment-Decision. Lung Cancer 2020, 139, 35–40. [Google Scholar] [CrossRef]
- Naidoo, J.; Sima, C.S.; Rodriguez, K.; Busby, N.; Nafa, K.; Ladanyi, M.; Riely, G.J.; Kris, M.G.; Arcila, M.E.; Yu, H.A. Epidermal Growth Factor Receptor Exon 20 Insertions in Advanced Lung Adenocarcinomas: Clinical Outcomes and Response to Erlotinib. Cancer 2015, 121, 3212–3220. [Google Scholar] [CrossRef] [Green Version]
- Weber, F.; Fukino, K.; Sawada, T.; Williams, N.; Sweet, K.; Brena, R.M.; Plass, C.; Caldes, T.; Mutter, G.L.; Villalona-Calero, M.A.; et al. Variability in Organ-Specific EGFR Mutational Spectra in Tumour Epithelium and Stroma May Be the Biological Basis for Differential Responses to Tyrosine Kinase Inhibitors. Br. J. Cancer 2005, 92, 1922–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preusser, M.; Berghoff, A.S.; Koller, R.; Zielinski, C.C.; Hainfellner, J.A.; Liebmann-Reindl, S.; Popitsch, N.; Geier, C.B.; Streubel, B.; Birner, P. Spectrum of Gene Mutations Detected by next Generation Exome Sequencing in Brain Metastases of Lung Adenocarcinoma. Eur. J. Cancer 2015, 51, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Gajiwala, K.S. EGFR: Tale of the C-Terminal Tail. Protein Sci. 2013, 22, 995–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Deng, G.; Liu, X.; Cho, W.C. Monoclonal Antibodies in Lung Cancer. Expert Opin. Biol. Ther. 2013, 13, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Riely, G.J.; Lovly, C.M. Therapeutic Strategies Utilized in the Setting of Acquired Resistance to EGFR Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2014, 20, 5898–5907. [Google Scholar] [CrossRef] [Green Version]
- Remon, J.; Morán, T.; Majem, M.; Reguart, N.; Dalmau, E.; Márquez-Medina, D.; Lianes, P. Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer: A New Era Begins. Cancer Treat. Rev. 2014, 40, 93–101. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, X.; Jin, H. EGFR-TKI Resistance in NSCLC Patients: Mechanisms and Strategies. Am. J. Cancer Res. 2014, 4, 411–435. [Google Scholar]
- Zhao, Z.; Xie, L.; Xie, L.; Bourne, P.E. Delineation of Polypharmacology across the Human Structural Kinome Using a Functional Site Interaction Fingerprint Approach. J. Med. Chem. 2016, 59, 4326–4341. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) Resistance with Mutant-Selective Allosteric Inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, P.A.; Moody, C.L.; Murali, R. Allosteric Modulation of Ras and the PI3K/AKT/MTOR Pathway: Emerging Therapeutic Opportunities. Front. Physiol. 2014, 5, 478. [Google Scholar] [CrossRef] [Green Version]
- Ling, Y.; Jing, M.; Wang, X. Allosteric Therapies for Lung Cancer. Cancer Metastasis Rev. 2015, 34, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.-J.; Jang, H. Dynamic Protein Allosteric Regulation and Disease. Adv. Exp. Med. Biol. 2019, 1163, 25–43. [Google Scholar] [PubMed]
- Tsai, C.-J.; Nussinov, R. Emerging Allosteric Mechanism of EGFR Activation in Physiological and Pathological Contexts. Biophys. J. 2019, 117, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Tsai, C.-J. Allostery in Disease and in Drug Discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, L.; Rastelli, G. AC Helix Displacement as a General Approach for Allosteric Modulation of Protein Kinases. Drug Discov. Today 2013, 18, 407–414. [Google Scholar] [CrossRef]
- Purba, E.; Saita, E.; Maruyama, I. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.L.; Vellanki, P.J.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Shen, Y.L.; Xia, H.; Li, Y.; Liu, J.; Zirkelbach, J.F.; et al. FDA Approval Summary: Osimertinib for Adjuvant Treatment of Surgically Resected Non–Small Cell Lung Cancer, a Collaborative Project Orbis Review. Clin. Cancer Res. 2021, 27, 6638–6643. [Google Scholar] [CrossRef]
- Fan, W.-L.; Shiao, M.-S.; Hui, R.C.-Y.; Su, S.-C.; Wang, C.-W.; Chang, Y.-C.; Chung, W.-H. HLA Association with Drug-Induced Adverse Reactions. J. Immunol. Res. 2017, 2017, 3186328. [Google Scholar] [CrossRef]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; McGuinn, W.D.; Morse, D.; Abraham, S.; Rahman, A.; Liang, C.; Lostritto, R.; et al. United States Food and Drug Administration Drug Approval Summary. Clin. Cancer Res. 2004, 10, 1212–1218. [Google Scholar] [CrossRef] [Green Version]
- Barker, A.J.; Gibson, K.H.; Grundy, W.; Godfrey, A.A.; Barlow, J.J.; Healy, M.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Scarlett, L.; et al. Studies Leading to the Identification of ZD1839 (IressaTM): An Orally Active, Selective Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Targeted to the Treatment of Cancer. Bioorg. Med. Chem. Lett. 2001, 11, 1911–1914. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. A Novel Approach in the Treatment of Cancer: Targeting the Epidermal Growth Factor Receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar] [PubMed]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.-Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-Institutional Randomized Phase II Trial of Gefitinib for Previously Treated Patients with Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Schiller, J.; Fukuoka, M.; Natale, R.; Lynch, T.; Averbuch, S.; Kay, A. Results from Two Phase II Trials (IDEAL 1 and IDEAL 2) of ZD1839 in Patients with Locally Advanced or Matastatic Non-Small Cell Lung Cancer. (Thoracic Oncology: 12:00 p.m.–1:45 p.m.). Chest 2002, 122, 168S. [Google Scholar]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, J.T.J.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; et al. Efficacy of Gefitinib, an Inhibitor of the Epidermal Growth Factor Receptor Tyrosine Kinase, in Symptomatic Patients with Non–Small Cell Lung Cancer. JAMA 2003, 290, 2149–2158. [Google Scholar] [CrossRef] [Green Version]
- Cella, D.; Herbst, R.S.; Lynch, T.J.; Prager, D.; Belani, C.P.; Schiller, J.H.; Heyes, A.; Ochs, J.S.; Wolf, M.K.; Kay, A.C.; et al. Clinically Meaningful Improvement in Symptoms and Quality of Life for Patients With Non-Small-Cell Lung Cancer Receiving Gefitinib in a Randomized Controlled Trial. J. Clin. Oncol. 2005, 23, 2946–2954. [Google Scholar] [CrossRef]
- Nishiwaki, Y.; Yano, S.; Tamura, T.; Nakagawa, K.; Kudoh, S.; Horai, T.; Noda, K.; Takata, I.; Watanabe, K.; Saka, H.; et al. Subset Analysis of Data in the Japanese Patients with NSCLC from IDEAL 1 Study on Gefitinib. Gan To Kagaku Ryoho 2004, 31, 567–573. [Google Scholar]
- Thatcher, N.; Chang, A.; Parikh, P.; Rodrigues Pereira, J.; Ciuleanu, T.; von Pawel, J.; Thongprasert, S.; Tan, E.H.; Pemberton, K.; Archer, V.; et al. Gefitinib plus Best Supportive Care in Previously Treated Patients with Refractory Advanced Non-Small-Cell Lung Cancer: Results from a Randomised, Placebo-Controlled, Multicentre Study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005, 366, 1527–1537. [Google Scholar] [CrossRef]
- Chang, A.; Parikh, P.; Thongprasert, S.; Huat Tan, E.; Perng, R.P.; Ganzon, D.; Yang, C.-H.; Tsao, C.-J.; Watkins, C.; Botwood, N.; et al. Gefitinib (IRESSA) in Patients of Asian Origin with Refractory Advanced Non-Small Cell Lung Cancer: Subset Analysis from the ISEL Study. J. Thorac. Oncol. 2006, 1, 847–855. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Bunn, P.A.; Franklin, W.A.; Dziadziuszko, R.; Thatcher, N.; Chang, A.; Parikh, P.; Pereira, J.R.; Ciuleanu, T.; et al. Molecular Predictors of Outcome with Gefitinib in a Phase III Placebo-Controlled Study in Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2006, 24, 5034–5042. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, F.R.; Dziadziuszko, R.; Thatcher, N.; Mann, H.; Watkins, C.; Parums, D.V.; Speake, G.; Holloway, B.; Bunn, P.A.; Franklin, W.A. Epidermal Growth Factor Receptor Immunohistochemistry. Cancer 2008, 112, 1114–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib Binds Both Inactive and Active Conformations of the EGFR Tyrosine Kinase Domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänne, P.A.; Wang, X.; Socinski, M.A.; Crawford, J.; Stinchcombe, T.E.; Gu, L.; Capelletti, M.; Edelman, M.J.; Villalona-Calero, M.A.; Kratzke, R.; et al. Randomized Phase II Trial of Erlotinib Alone or With Carboplatin and Paclitaxel in Patients Who Were Never or Light Former Smokers with Advanced Lung Adenocarcinoma: CALGB 30406 Trial. J. Clin. Oncol. 2012, 30, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Ansari, R.; Bustin, F.; Flynn, P.; Hart, L.; Otterson, G.A.; Vlahovic, G.; Soh, C.-H.; O’Connor, P.; Hainsworth, J. Efficacy of Bevacizumab plus Erlotinib versus Erlotinib Alone in Advanced Non-Small-Cell Lung Cancer after Failure of Standard First-Line Chemotherapy (BeTa): A Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2011, 377, 1846–1854. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.; Stern, H.; Amler, L.; Otterson, G.; Lin, M.; O’Connor, P.; Hainsworth, J. Biomarker Evaluation in the Phase III, Placebo (P)-Controlled, Randomized BeTa Trial of Bevacizumab (B) and Erlotinib (E) for Patients (Pts) with Advanced Non-Small Cell Lung Cancer (NSCLC) after Failure of Standard 1st-Line Chemotherapy: Correlation with Treatment Outcomes. J. Thorac. Oncol. 2009, 4, 530. [Google Scholar]
- Stinchcombe, T.E.; Jänne, P.A.; Wang, X.; Bertino, E.M.; Weiss, J.; Bazhenova, L.; Gu, L.; Lau, C.; Paweletz, C.; Jaslowski, A.; et al. Effect of Erlotinib Plus Bevacizumab vs Erlotinib Alone on Progression-Free Survival in Patients with Advanced EGFR -Mutant Non–Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 1448–1455. [Google Scholar] [CrossRef]
- Blencke, S.; Zech, B.; Engkvist, O.; Greff, Z.; Őrfi, L.; Horváth, Z.; Kéri, G.; Ullrich, A.; Daub, H. Characterization of a Conserved Structural Determinant Controlling Protein Kinase Sensitivity to Selective Inhibitors. Chem. Biol. 2004, 11, 691–701. [Google Scholar] [CrossRef]
- Dungo, R.T.; Keating, G.M. Afatinib: First Global Approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef]
- Abdallah, S.M.; Hirsh, V. Irreversible Tyrosine Kinase Inhibition of Epidermal Growth Factor Receptor with Afatinib in Egfr Activating Mutation–Positive Advanced Non-Small-Cell Lung Cancer. Curr. Oncol. 2018, 25, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Langer, C.J. Epidermal Growth Factor Receptor Inhibition in Mutation-Positive Non–Small-Cell Lung Cancer: Is Afatinib Better or Simply Newer? J. Clin. Oncol. 2013, 31, 3303–3306. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients with Metastatic Lung Adenocarcinoma with EGFR Mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-L.; Zhou, C.; Hu, C.-P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus Cisplatin plus Gemcitabine for First-Line Treatment of Asian Patients with Advanced Non-Small-Cell Lung Cancer Harbouring EGFR Mutations (LUX-Lung 6): An Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Wu, Y.-L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.-P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus Cisplatin-Based Chemotherapy for EGFR Mutation-Positive Lung Adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of Overall Survival Data from Two Randomised, Phase 3 Trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Smaill, J.B.; Gonzales, A.J.; Spicer, J.A.; Lee, H.; Reed, J.E.; Sexton, K.; Althaus, I.W.; Zhu, T.; Black, S.L.; Blaser, A.; et al. Tyrosine Kinase Inhibitors. 20. Optimization of Substituted Quinazoline and Pyrido[3,4-d]Pyrimidine Derivatives as Orally Active, Irreversible Inhibitors of the Epidermal Growth Factor Receptor Family. J. Med. Chem. 2016, 59, 8103–8124. [Google Scholar] [CrossRef]
- Ou, S.-H.; Soo, R. Dacomitinib in Lung Cancer: A “Lost Generation” EGFR Tyrosine-Kinase Inhibitor from a Bygone Era? Drug Des. Dev. Ther. 2015, 9, 5641–5653. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Lee, M.; Linke, R.; Rosell, R.; Corral, J.; et al. Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib With Gefitinib in Patients with Advanced Non–Small-Cell Lung Cancer and EGFR-Activating Mutations. J. Clin. Oncol. 2018, 36, 2244–2250. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.-M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an Irreversible Pan-ERBB Inhibitor, Is Effective in Lung Cancer Models with EGFR and ERBB2 Mutations That Are Resistant to Gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Fujino, T.; Nishino, M.; Koga, T.; Chiba, M.; Sesumi, Y.; Ohara, S.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. EGFR T790M and C797S Mutations as Mechanisms of Acquired Resistance to Dacomitinib. J. Thorac. Oncol. 2018, 13, 727–731. [Google Scholar] [CrossRef] [Green Version]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non–Small Cell Lung Cancer Harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus Gefitinib as First-Line Treatment for Patients with EGFR-Mutation-Positive Non-Small-Cell Lung Cancer (ARCHER 1050): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L.; et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finlay, M.R.V.; Anderton, M.; Ashton, S.; Ballard, P.; Bethel, P.A.; Box, M.R.; Bradbury, R.H.; Brown, S.J.; Butterworth, S.; Campbell, A.; et al. Discovery of a Potent and Selective EGFR Inhibitor (AZD9291) of Both Sensitizing and T790M Resistance Mutations That Spares the Wild Type Form of the Receptor. J. Med. Chem. 2014, 57, 8249–8267. [Google Scholar] [CrossRef]
- Yan, X.-E.; Ayaz, P.; Zhu, S.-J.; Zhao, P.; Liang, L.; Zhang, C.H.; Wu, Y.-C.; Li, J.-L.; Choi, H.G.; Huang, X.; et al. Structural Basis of AZD9291 Selectivity for EGFR T790M. J. Med. Chem. 2020, 63, 8502–8511. [Google Scholar] [CrossRef]
- Yosaatmadja, Y.; Silva, S.; Dickson, J.M.; Patterson, A.V.; Smaill, J.B.; Flanagan, J.U.; McKeage, M.J.; Squire, C.J. Binding Mode of the Breakthrough Inhibitor AZD9291 to Epidermal Growth Factor Receptor Revealed. J. Struct. Biol. 2015, 192, 539–544. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus Standard Chemotherapy as First-Line Treatment for European Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (EURTAC): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.-H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.-H.; Lee, K.H.; Lu, S.; et al. Afatinib versus Gefitinib as First-Line Treatment of Patients with EGFR Mutation-Positive Non-Small-Cell Lung Cancer (LUX-Lung 7): A Phase 2B, Open-Label, Randomised Controlled Trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Walter, A.O.; Sjin, R.T.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a Mutant-Selective Covalent Inhibitor of EGFR That Overcomes T790M-Mediated Resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Soria, J.-C.; Goldman, J.W.; Wakelee, H.A.; Gadgeel, S.M.; Varga, A.; Papadimitrakopoulou, V.; Solomon, B.J.; Oxnard, G.R.; Dziadziuszko, R.; et al. Rociletinib in EGFR -Mutated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-E.; Zhu, S.-J.; Liang, L.; Zhao, P.; Choi, H.G.; Yun, C.-H. Structural Basis of Mutant-Selectivity and Drug-Resistance Related to CO-1686. Oncotarget 2017, 8, 53508–53517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.C.-H.; Reckamp, K.L.; Kim, Y.-C.; Novello, S.; Smit, E.F.; Lee, J.-S.; Su, W.-C.; Akerley, W.L.; Blakely, C.M.; Groen, H.J.M.; et al. Efficacy and Safety of Rociletinib Versus Chemotherapy in Patients With EGFR-Mutated NSCLC: The Results of TIGER-3, a Phase 3 Randomized Study. JTO Clin. Res. Rep. 2021, 2, 100114. [Google Scholar] [CrossRef]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [Green Version]
- Ercan, D.; Choi, H.G.; Yun, C.-H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Yao, M.-Y.; Zhu, S.-J.; Chen, J.-Y.; Yun, C.-H. Crystal Structure of EGFR T790M/C797S/V948R in Complex with EAI045. Biochem. Biophys. Res. Commun. 2018, 502, 332–337. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The Fourth-Generation EGFR Inhibitor Overcoming T790M and C797S Resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- Caporuscio, F.; Tinivella, A.; Restelli, V.; Semrau, M.S.; Pinzi, L.; Storici, P.; Broggini, M.; Rastelli, G. Identification of Small-Molecule EGFR Allosteric Inhibitors by High-Throughput Docking. Future Med. Chem. 2018, 10, 1545–1553. [Google Scholar] [CrossRef]
- Sickmier, E.A.; Kurzeja, R.J.M.; Michelsen, K.; Vazir, M.; Yang, E.; Tasker, A.S. The Panitumumab EGFR Complex Reveals a Binding Mechanism That Overcomes Cetuximab Induced Resistance. PLoS ONE 2016, 11, e0163366. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Clinical Trial Identifier a | Phase | Condition c |
|---|---|---|---|
| Lazertinib (YH25448) | NCT03046992 | I/II | NSCLC |
| NCT04075396 | I/II | NSCLC | |
| NCT05167851 | II | NSCLC | |
| NCT04248829 | III | NSCLC | |
| D-0316 | NCT03452150 | I | NSCLC |
| NCT04206072 | II/III | NSCLC | |
| NCT03861156 | II | NSCLC, ST | |
| AZD3759 | NCT03360929 | I/II | NSCLC |
| NCT03653546 | II/III | NSCLC, BM | |
| FCN-411 | NCT03420079 | I | NSCLC |
| DZD9008 | NCT03974022 | I/II | NSCLC |
| Nazartinib (EGF816) | NCT03333343 | I | NSCLC |
| NCT02108964 | I/II | NSCLC | |
| NCT03292133 | II | NSCLC | |
| Icotinib | NCT05007938 | II | NSCLC |
| NCT03749213 | II | NSCLC | |
| NCT03396185 | II | NSCLC | |
| NCT03349203 | II | NSCLC | |
| NCT02737774 | II | AC | |
| Apatinib | NCT03801200 | II | NSCLC |
| NCT04824352 | II | OS | |
| NCT03913182 | II | EC | |
| NCT04253873 | II | HGG | |
| NCT03475589 | IV | GC, NSCLC. BC, OC | |
| BLU-945 | NCT04862780 | I/II | NSCLC |
| Avitinib (AC0010) | NCT02330367 | I/II | NSCLC |
| NCT03574402 | II | NSCLC | |
| Almonertinib (HS-10296) | NCT04905550 | II | NSCLC |
| NCT04952168 | II | NSCLC | |
| NCT04785742 | II | NSCLC | |
| NCT04636593 | II | NSCLC | |
| NCT04685070 | III | AC | |
| CLN-081 | NCT04036682 | I/IIa | NSCLC |
| APG-1252 | NCT04210037 | I | SCLC |
| NCT04893759 | Ib | NT | |
| NCT04001777 b | Ib | NSCLC | |
| NCT05186012 | Ib/II | NHL | |
| NCT04354727 | Ib/II | MF | |
| Furmonertinib | NCT05079022 | I/II | AC |
| NCT04858958 | Ib | NSCLC | |
| NCT04982900 | II | LC | |
| NCT04965831 | II | AC | |
| NCT04970693 | II | NSCLC | |
| NCT04895930 b | II | NSCLC | |
| NCT04853342 | III | NSCLC | |
| HBI-2376 | NCT05163028 | I | NSCLC, CC, PC, ST, PC |
| ABT-414 | NCT02573324 | III | GS |
| Dasatinib (BMS-354825) | NCT02954523 | I/II | NSCLC |
| NCT00529763 | II | LK | |
| NCT01471106 | II | BC | |
| Repotrectinib (TPX-0005) | NCT05004116 b | I | ST |
| NCT04772235 | I | NSCLC | |
| NCT03093116 | I/II | ST | |
| NCT04094610 | I/II | ST | |
| NCT05071183 b | Ib/II | ST | |
| Poziotinib (NOV120101) | NCT03066206 | II | NSCLC |
| NCT03744715 | II | NSCLC, BC | |
| NCT03066206 | II | NSCLC | |
| NCT03318939 | II | NSCLC | |
| NCT04172597 | II | BC, CC, ST, HGG | |
| Larotinib (Z650) | NCT04131192 | Ib | PC |
| NCT03888092 | Ib | EC | |
| NCT04415853 | III | EC | |
| Mobocertinib (TAK-788) | NCT04056455 | I | HV, RI |
| NCT04056468 | I | HV, HI | |
| NCT04051827 | I | NSCLC | |
| NCT03807778 | I/II | NSCLC | |
| NCT04129502 | III | NSCLC | |
| Vandetanib (ZD6474) | NCT00537095 | II | TC |
| NCT00410761 | III | TC | |
| NCT01876784 | III | TC | |
| NCT00418886 | III | NSCLC | |
| BBT-176 | NCT04820023 | I/II | NSCLC |
| Brigatinib (AP26113) | NCT04634110 | II | NSCLC, BM |
| NCT04223596 | II | NSCLC | |
| NCT03535740 | II | NSCLC | |
| NCT04074993 | II | NSCLC | |
| NCT03596866 | III | NSCLC | |
| Pyrotinib | NCT04680091 | I | HV |
| NCT00600496 | I | BC, CC, LC, KC | |
| NCT04960943 | II | GT | |
| NCT04380012 | II | CC | |
| NCT04646759 | III | BC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amelia, T.; Kartasasmita, R.E.; Ohwada, T.; Tjahjono, D.H. Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors. Molecules 2022, 27, 819. https://doi.org/10.3390/molecules27030819
Amelia T, Kartasasmita RE, Ohwada T, Tjahjono DH. Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors. Molecules. 2022; 27(3):819. https://doi.org/10.3390/molecules27030819
Chicago/Turabian StyleAmelia, Tasia, Rahmana Emran Kartasasmita, Tomohiko Ohwada, and Daryono Hadi Tjahjono. 2022. "Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors" Molecules 27, no. 3: 819. https://doi.org/10.3390/molecules27030819
APA StyleAmelia, T., Kartasasmita, R. E., Ohwada, T., & Tjahjono, D. H. (2022). Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors. Molecules, 27(3), 819. https://doi.org/10.3390/molecules27030819