
Halogen-Based 17β-HSD1 Inhibitors: Insights from DFT, Docking, and Molecular Dynamics Simulation Studies


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Methodology
2.1. Calculation of Quantum Chemical Descriptors
2.2. Prediction of Biological Activities of Compounds Using Molecular Docking Analysis
2.3. Assessment of the Stability of Docked Complexes Using Molecular Dynamics (MD) Simulations
3. Results and Discussion
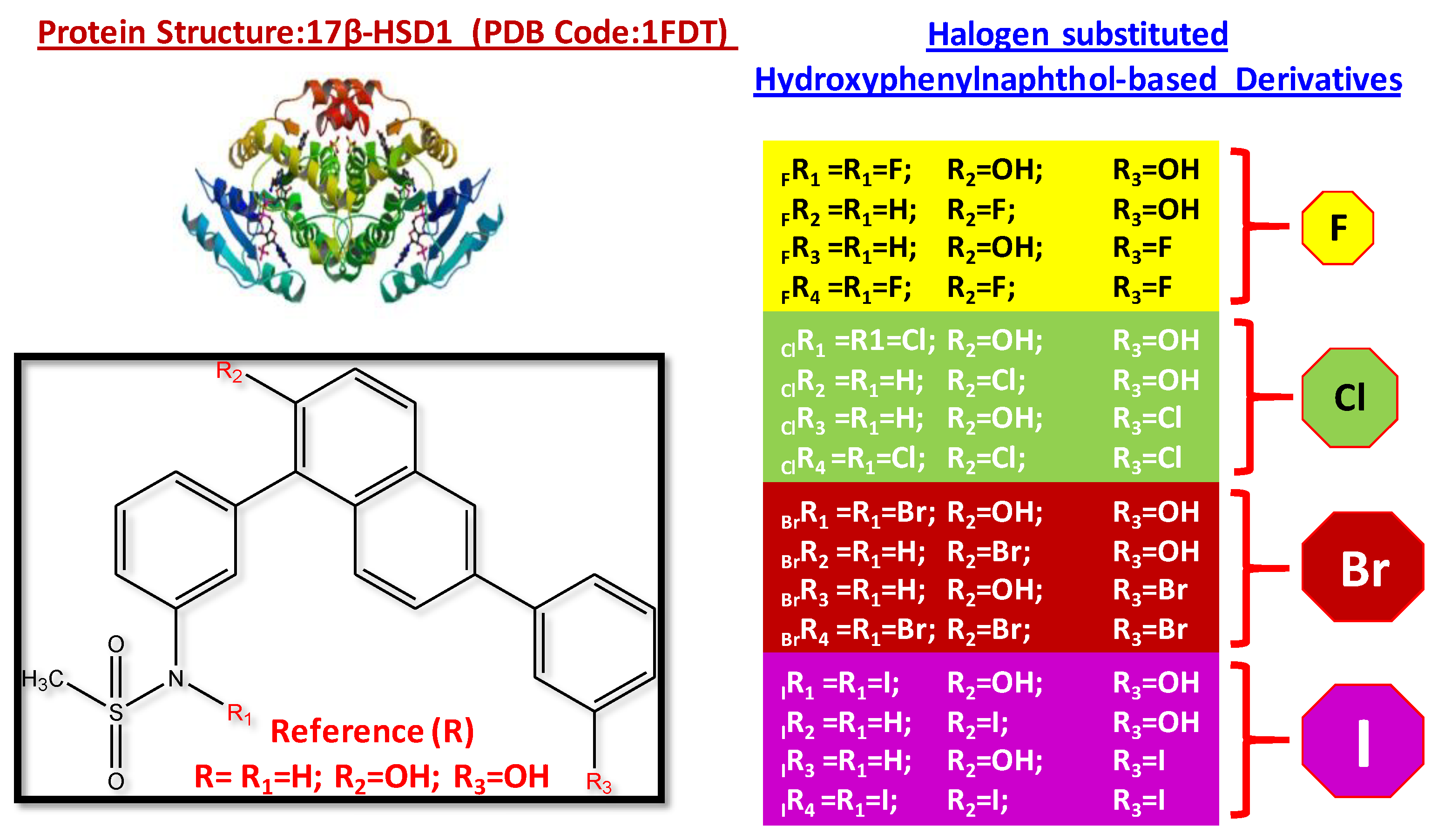
3.1. Structural Features of Halogen-Substituted Compounds against 17β-hydroxysteroid Dehydrogenase Type 1
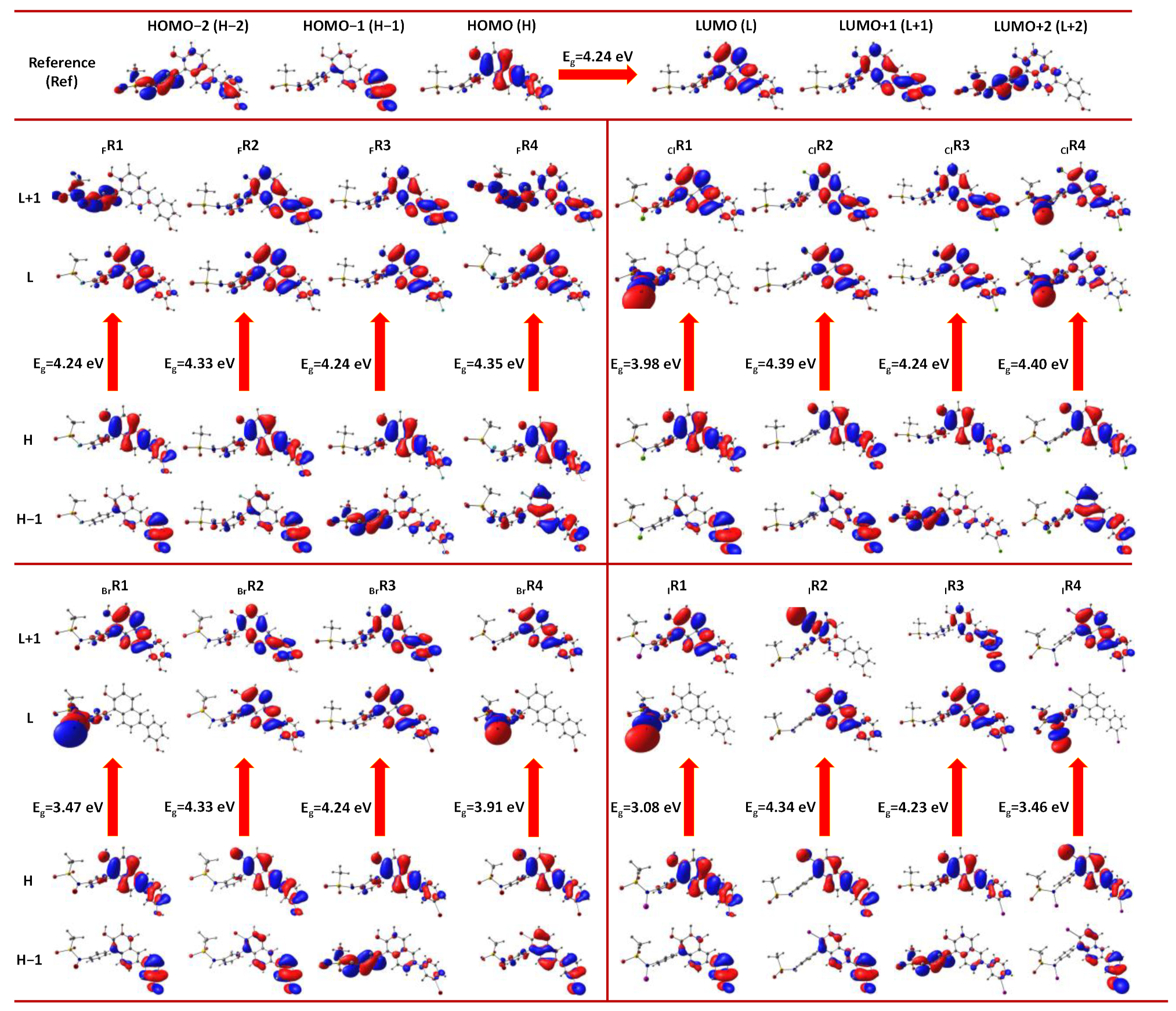
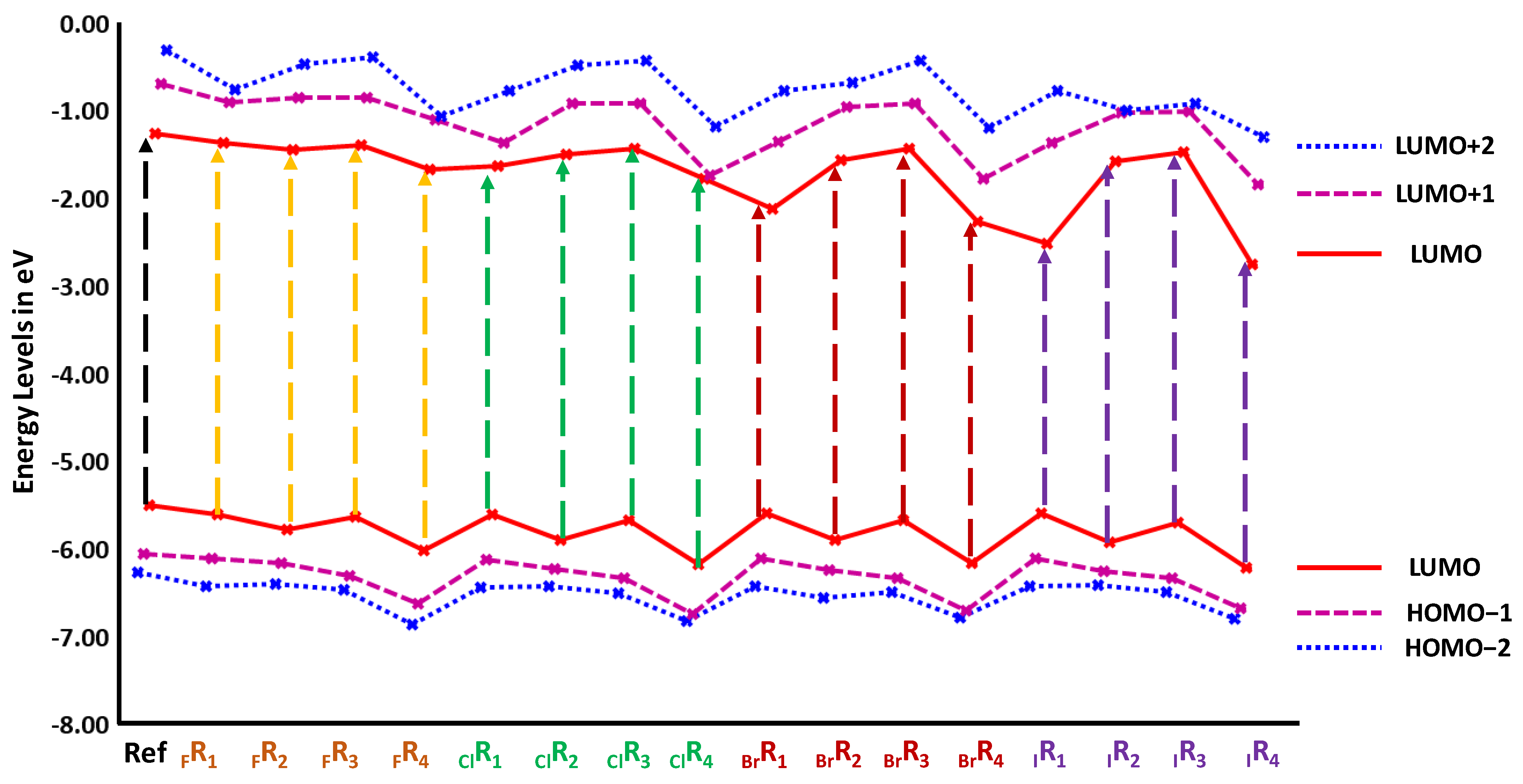
3.2. Characterization of Halogen-Derived Compounds Using Quantum Chemical Descriptors
3.2.1. Frontier Molecular Orbital Analysis
3.2.2. Hardness and Softness
3.2.3. Electrophilicity (ω) and Chemical Potential (µ)
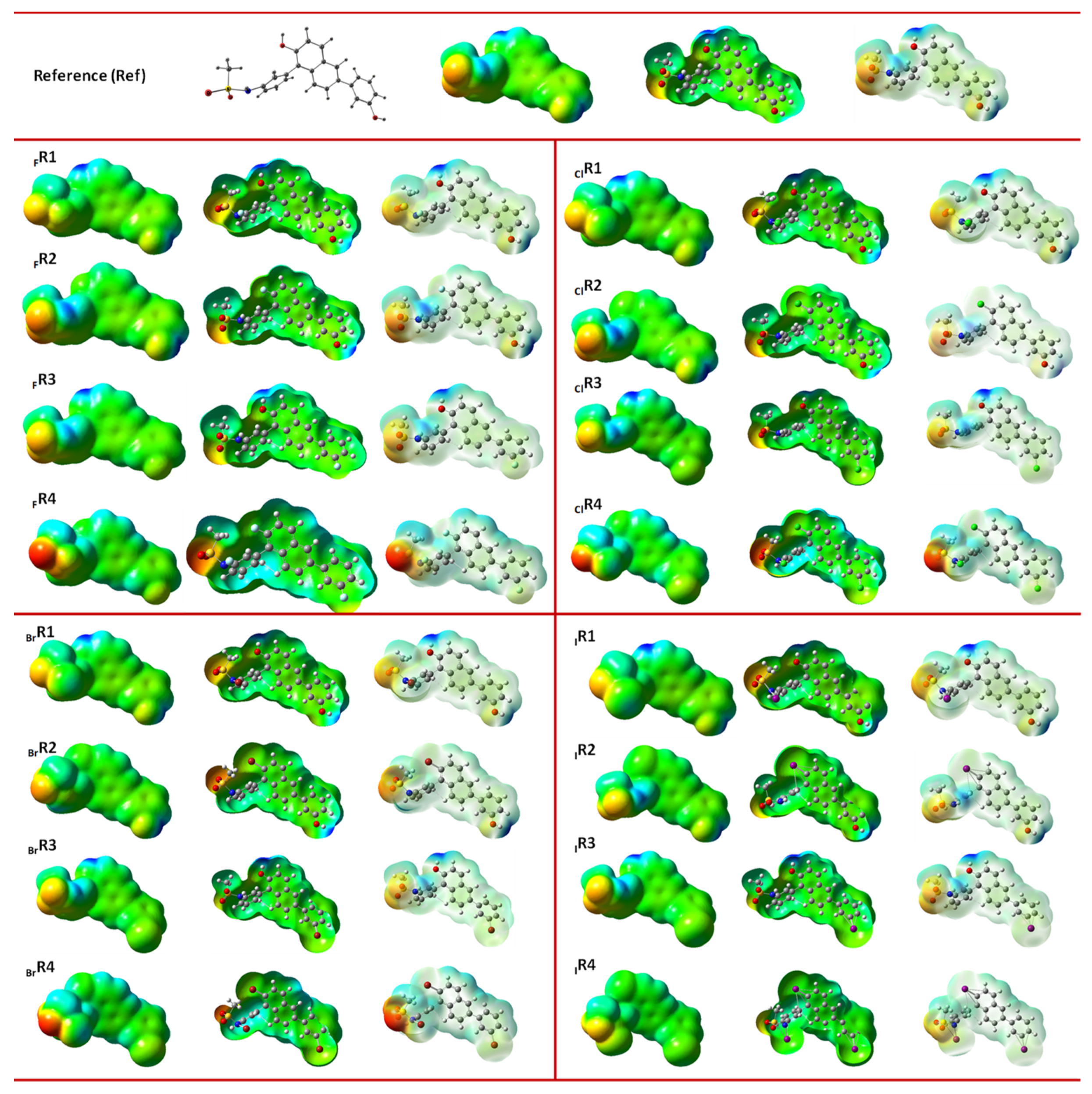
3.3. Molecular Electrostatic Potential (MESP) Analysis
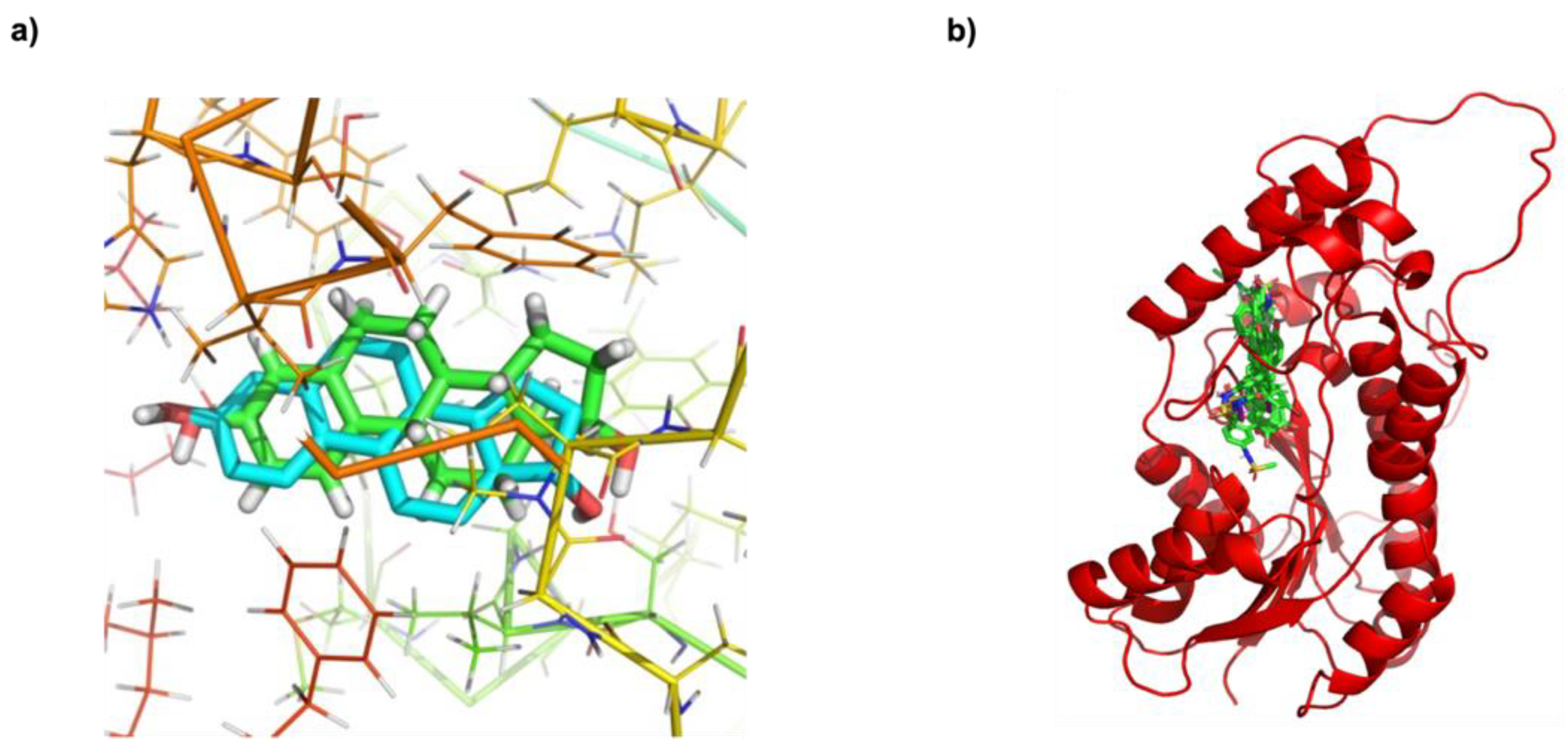
3.4. Predicting the Biological Activities of Halogen-Substituted Ligands against 17β-HSD1 Validation of Molecular Docking Using a Redocking Approach
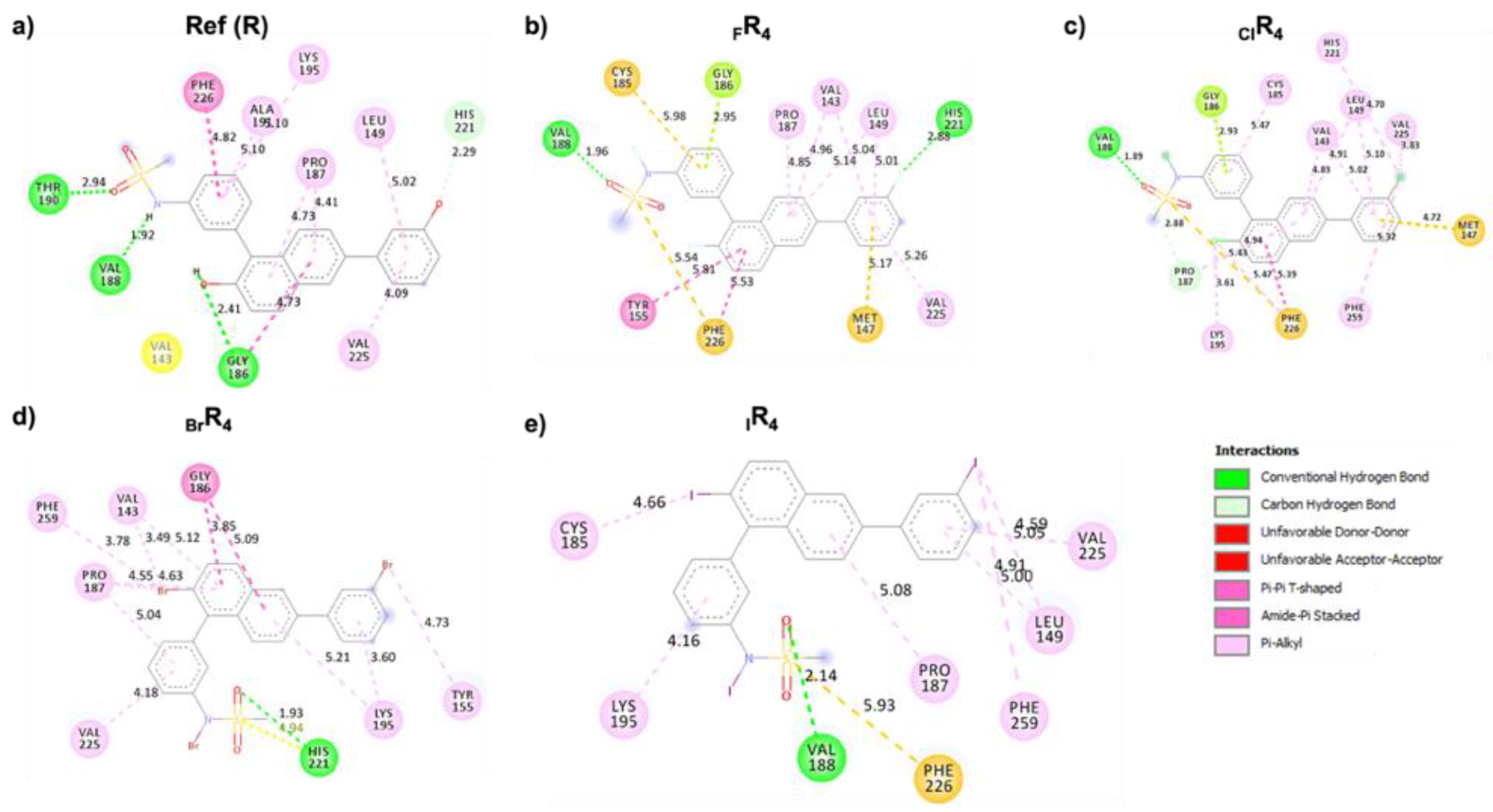
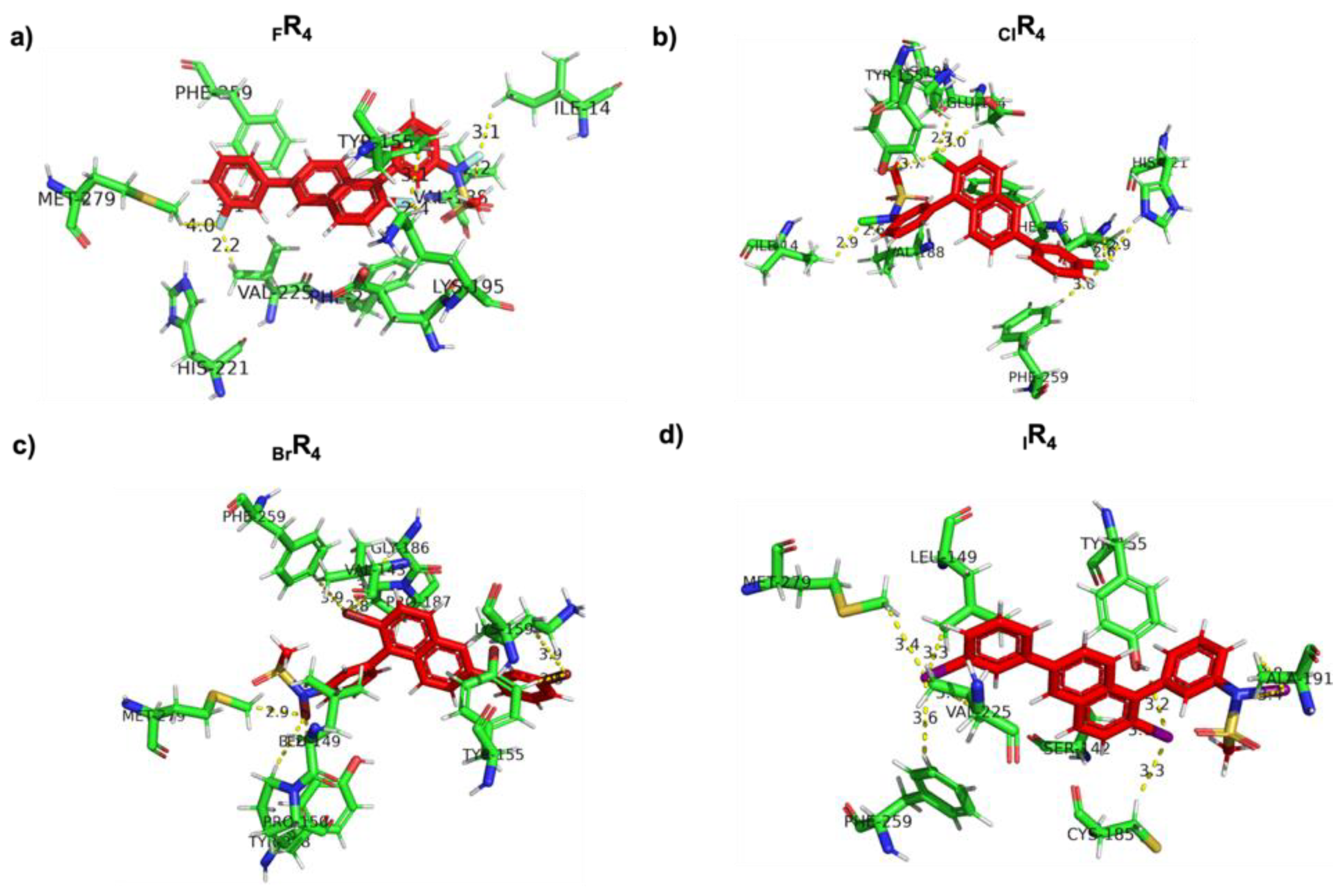
Analysis of Noncovalent Interactions between 17β-HSD1 and Halogen-Substituted Ligands
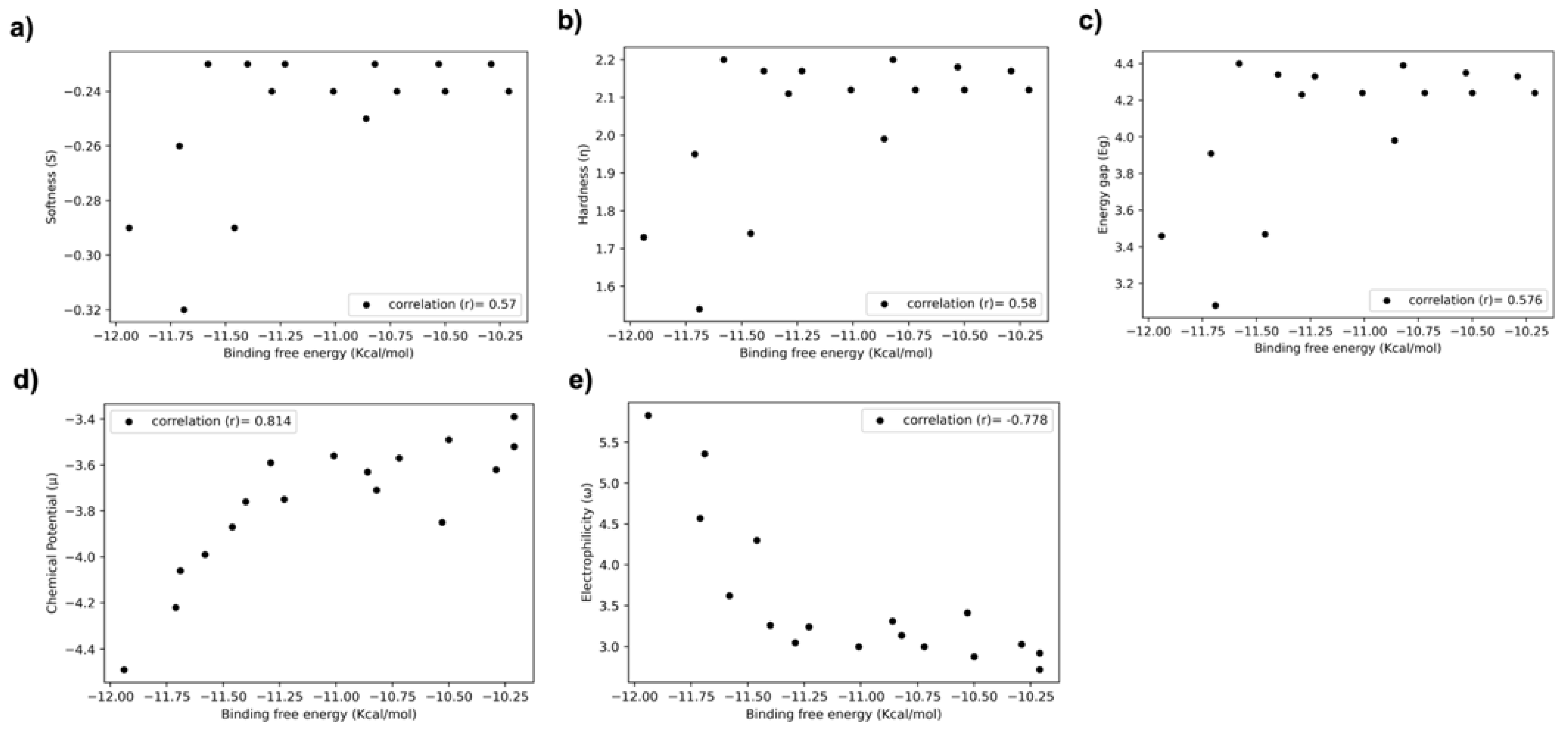
3.5. Relationship between DFT Descriptors and Predicted Biological Activities (Binding Energy/Score) of Halogen-Substituted Compounds
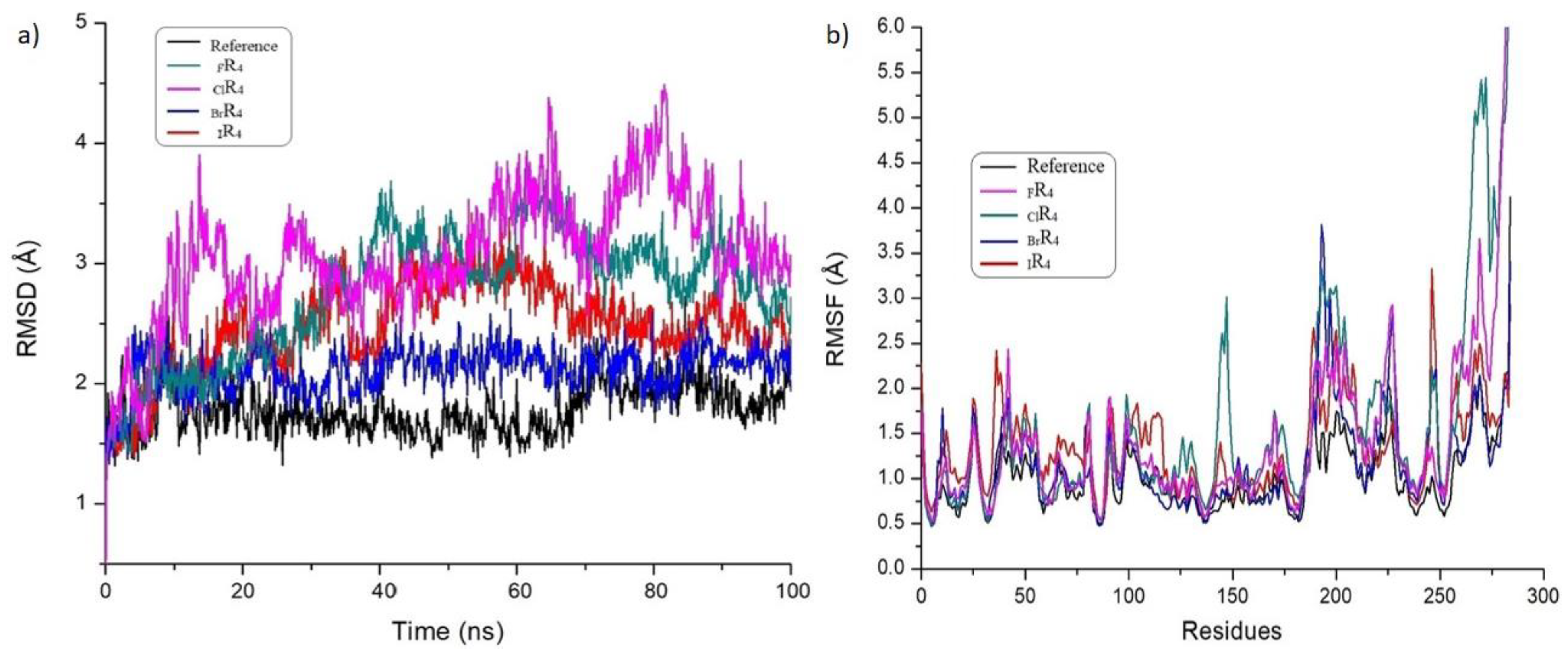
3.6. Investigation of the Structural Stability of Halogen-Substituted-Inhibitor Complexes from MD Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Pike, M.C.; Spicer, D.V.; Dahmoush, L.; Press, M.F. Estrogens progestogens normal breast cell proliferation and breast cancer risk. Endocrinol. Rev. 1993, 15, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Ferin, M.; Zimmering, P.E.; Lieberman, S.; Vande Wiele, R.L. Inactivation of the biological effects of exogenous and endogenous estrogens by antibodies to 17β-estradiol. Endocrinology 1968, 83, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Jeon, G.H.; Kim, S.H.; Yun, S.C.; Chae, H.D.; Kim, C.H.; Kang, B.M. Association between serum estradiol level and coronary artery calcification in postmenopausal women. Menopause 2010, 17, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Youn, M.Y.; Kondoh, S.; Nakamura, T.; Kouzmenko, A.; Matsumoto, T.; Takada, I.; Takaoka, K.; Kato, S. Estrogens maintain bone mass by regulating expression of genes controlling function and life span in mature osteoclasts. Ann. N. Y. Acad. Sci. 2009, 1173, E31–E39. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, A.; Negri, M.; Marchais-Oberwinkler, S.; Bey, E.; Frotscher, M. Hydroxybenzothiazoles as new nonsteroidal inhibitors of 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1). PLoS ONE 2012, 7, e29252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liehr, J.G. Is Estradiol a Genotoxic Mutagenic Carcinogen? Endocr. Rev. 2000, 21, 40–54. [Google Scholar] [PubMed] [Green Version]
- Thomas, D.B. Do hormones cause breast cancer? Cancer 1984, 53, 595–604. [Google Scholar] [CrossRef]
- Russo, J.; Fernandez, S.V.; Russo, P.A.; Fernbaugh, R.; Sheriff, F.S.; Lareef, H.M.; Garber, J.; Russo, I.H. 17-Beta-estradiol induces transformation and tumorigenesis in human breast epithelial cells. FASEB J. 2006, 20, 1622–1634. [Google Scholar] [CrossRef]
- Dizerega, G.S.; Barber, D.L.; Hodgen, G. Endometriosis: Role of ovarian steroids in initiation, maintenance, and suppression. Fertil. Steril. 1980, 33, 649–653. [Google Scholar] [CrossRef]
- Gobbi, S.; Cavalli, A.; Rampa, A.; Belluti, F.; Piazzi, L.; Paluszcak, A.; Hartmann, R.W.; Recanatini, M.; Bisi, A. Lead optimization providing a series of flavone derivatives as potent nonsteroidal inhibitors of the cytochrome P450 aromatase enzyme. J. Med. Chem. 2006, 49, 4777–4780. [Google Scholar] [CrossRef]
- Borgne, M.L.; Marchand, P.; Duflos, M.; Delevoye-Seiller, B.; Piessard-Robert, S.; Baut, G.L.; Hartmann, R.W.; Palzer, M. Synthesis and in Vitro Evaluation of 3-(1-Azolylmethyl)-1H-indoles and 3-(1-Azolyl-1-phenylmethyl)-1H-indoles as Inhibitors of P450 arom. Archiv. Pharm. 1997, 330, 141–145. [Google Scholar] [CrossRef]
- Jacobs, C.; Frotscher, M.; Dannhardt, G.; Hartmann, R.W. 1-Imidazolyl(alkyl)-substituted di- and tetrahydroquinolines and analogues: Syntheses and evaluation of dual inhibitors of thromboxane A2 synthase and aromatase. J. Med. Chem. 2000, 43, 1841–1851. [Google Scholar] [CrossRef]
- Marchais-Oberwinkler, S.; Wetzel, M.; Ziegler, E.; Kruchten, P.; Werth, R.; Henn, C.; Hartmann, R.W.; Frotscher, M. New drug-like hydroxyphenylnaphthol steroidomimetics as potent and selective 17β-hydroxysteroid dehydrogenase type 1 inhibitors for the treatment of estrogen-dependent diseases. J. Med. Chem. 2010, 54, 534–547. [Google Scholar] [CrossRef]
- Jansson, A. 17Beta-hydroxysteroid dehydrogenase enzymes and breast cancer. J. Steroid Biochem. Mol. Biol. 2009, 114, 64–67. [Google Scholar] [CrossRef]
- Zhou, P.P.; Qiu, W.Y.; Liu, S.; Jin, N.Z. Halogen as halogen-bonding donor and hydrogen-bonding acceptor simultaneously in ring-shaped H3N·X(Y)·HF (X= Cl, Br and Y= F, Cl, Br) Complexes. Phys. Chem. Chem. Phys. 2011, 13, 7408–7418. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Amezaga, N.J.M.; Pamies, S.C.; Peruchena, N.M.; Sosa, G.L. Halogen bonding: A study based on the electronic charge density. J. Phys. Chem. A 2009, 114, 552–562. [Google Scholar] [CrossRef]
- Bernal-Uruchurtu, M.I.; Hernández-Lamoneda, R.; Janda, K.C. On the Unusual Properties of Halogen Bonds: A Detailed ab Initio Study of X2−(H2O) 1−5 clusters (X = Cl and Br). J. Phys. Chem. A 2009, 113, 5496–5505. [Google Scholar] [CrossRef]
- An, X.; Jing, B.; Li, Q. Novel Halogen-Bonded Complexes H3NBH3·· XY (XY = ClF, ClCl, BrF, BrCl, and BrBr): Partially Covalent Character. J. Phys. Chem. A 2010, 114, 6438–6443. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Xu, Z.; Li, H.; Liu, H.; Zhu, W. Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug Discov. 2012, 7, 375–383. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding in supramolecular chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef]
- Derossi, S.; Brammer, L.; Hunter, C.A.; Ward, M.D. Halogen Bonded Supramolecular Assemblies of [Ru (bipy)(CN)4] 2−Anions and N-Methyl-Halopyridinium Cations in the Solid State and in Solution. Inorg. Chem. 2009, 48, 1666–1677. [Google Scholar] [CrossRef]
- Shirman, T.; Freeman, D.; Posner, Y.D.; Feldman, I.; Facchetti, A.; van der Boom, M.E. Assembly of Crystalline Halogen-Bonded Materials by Physical Vapor Deposition. J. Am. Chem. Soc. 2008, 130, 8162–8163. [Google Scholar] [CrossRef]
- Tuikka, M.; Hirva, P.; Rissanen, K.; Korppi-Tommola, J.; Haukka, M. Halogen bonding—A key step in charge recombination of the dye-sensitized solar cell. Chem. Commun. 2011, 47, 4499–4501. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Lu, Y.; Shi, T.; Wang, Y.; Yang, H.; Yan, X.; Luo, X.; Jiang, H.; Zhu, W. Halogen bonding a novel interaction for rational drug design? J. Med. Chem. 2009, 52, 2854–2862. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [Green Version]
- Parker, A.J.; Stewart, J.; Donald, K.J.; Parish, C.A. Halogen Bonding in DNA Base Pairs. J. Am. Chem. Soc. 2012, 134, 5165–5172. [Google Scholar] [CrossRef]
- Voth, A.R.; Hays, F.A.; Ho, P.S. Directing macromolecular conformation through halogen bonds. Proc. Natl. Acad. Sci. USA 2007, 104, 6188–6193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrea, V.R. The role of halogen bonding in inhibitor recognition and binding by protein kinases. Curr. Top. Med. Chem. 2007, 7, 1336–1348. [Google Scholar] [CrossRef]
- Hays, F.A.; Vargason, J.M.; Ho, P.S. Effect of sequence on the conformation of DNA Holliday junctions. Biochemistry 2003, 42, 9586–9597. [Google Scholar] [CrossRef] [Green Version]
- Benedetto, T.D.; Bagnoli, L.; Rosati, O.; Marini, F.; Sancineto, L.; Santi, C. New Halogen-Containing Drugs Approved by FDA in 2021: An Overview on Their Syntheses and Pharmaceutical Use. Molecules 2022, 27, 1643. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.C.; Ho, P.S. Computational tools to model halogen bonds in medicinal chemistry. J. Med. Chem. 2016, 59, 1655–1670. [Google Scholar] [CrossRef]
- Bhutani, P.; Joshi, G.; Raja, N.; Bachhav, N.; Rajanna, P.K.; Bhutani, H.; Paul, A.T.; Kumar, R. US FDA approved drugs from 2015–June 2020: A perspective. J. Med. Chem. 2021, 64, 2339–2381. [Google Scholar] [CrossRef]
- Uzzaman, M.; Hasan, M.K.; Mahmud, S.; Yousuf, A.; Islam, S.; Uddin, M.N.; Barua, A. Physicochemical, spectral, molecular docking and ADMET studies of Bisphenol analogues; A computational approach. Inform. Med. Unlocked. 2021, 25, 100706. [Google Scholar] [CrossRef]
- Solomon, R.V.; Vedha, S.A.; Venuvanalingam, P. A new turn in codon–anticodon selection through halogen bonds. Phys. Chem. Chem. Phys. 2014, 16, 7430–7440. [Google Scholar] [CrossRef]
- Frisch, M.J.T.G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter. 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Solomon, R.V.; Veerapandian, P.; Vedha, S.A.; Venuvanalingam, P. Tuning nonlinear optical and optoelectronic properties of vinyl coupled triazene chromophores: A density functional theory and time-dependent density functional theory investigation. J. Phys. Chem. A 2012, 116, 4667–4677. [Google Scholar] [CrossRef]
- Kathiravan, A.; Panneerselvam, M.; Sundaravel, K.; Pavithra, N.; Srinivasan, V.; Anandan, S.; Jaccob, M. Unravelling the effect of anchoring groups on the ground and excited state properties of pyrene using computational and spectroscopic methods. Phys. Chem. Chem. Phys. 2016, 18, 13332–13345. [Google Scholar] [CrossRef] [PubMed]
- Bella, A.P.; Panneerselvam, M.; Angeline, V.S.; Jaccob, M.; Vijay, S.R.; Princy, M.J. DFT-TDDFT framework of diphenylamine based mixed valence compounds for optoelectronic applications—Structural modification of π-acceptors. Comput. Mater. Sci. 2019, 162, 259–369. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.P.; Katzmann, D.J.; Sosa, C.P. Multilevel Parallelization of AutoDock 4.2. J. Cheminform. 2011, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton, R.; Housset, D.; Mazza, C.; Fontecilla-Camps, J.C. The structure of a complex of human 17β-hydroxysteroid dehydrogenase with estradiol and NADP+ identifies two principal targets for the design of inhibitors. Structure 1996, 4, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Accelrys Discovery Studio 2.5; Accelrys: San Diego, CA, USA, 2009.
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; Association for Computing Machinery: New York, NY, USA, 2006. [Google Scholar]
- Shaw, D.E. Desmond; Desmond Molecular Dynamics System: New York, NY, USA, 2021. [Google Scholar]
- Yadav, R.P.; Syed Ibrahim, K.; Gurusubramanian, G.; Senthil Kumar, N. In silico docking studies of non-azadirachtin limonoids against ecdysone receptor of Helicoverpa armigera (Hubner) (Lepidoptera: Noctuidae). Med. Chem. Res. 2015, 24, 2621–2631. [Google Scholar] [CrossRef]
- Ul-Haq, Z.; Ashraf, S.; Bkhaitan, M.M. Molecular dynamics simulations reveal structural insights into inhibitor binding modes and mechanism of casein kinase II inhibitors. J. Biomol. Struct. Dyn. 2019, 37, 1120–1135. [Google Scholar] [CrossRef]
- Kulandaisamy, A.; Lathi, V.; ViswaPoorani, K.; Yugandhar, K.; Gromiha, M.M. Important amino acid residues involved in folding and binding of protein–protein complexes. Int. J. Biol. Macromol. 2017, 94, 438–444. [Google Scholar] [CrossRef]
- Shanmugam, N.S.; Selvin, J.F.A.; Veluraja, K.; Gromiha, M.M. Identification and analysis of key residues involved in folding and binding of protein-carbohydrate complexes. Protein Pept. Lett. 2018, 25, 379–389. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulandaisamy, A.; Panneerselvam, M.; Solomon, R.V.; Jaccob, M.; Ramakrishnan, J.; Poomani, K.; Maruthamuthu, M.; Tharmalingam, N. Halogen-Based 17β-HSD1 Inhibitors: Insights from DFT, Docking, and Molecular Dynamics Simulation Studies. Molecules 2022, 27, 3962. https://doi.org/10.3390/molecules27123962
Kulandaisamy A, Panneerselvam M, Solomon RV, Jaccob M, Ramakrishnan J, Poomani K, Maruthamuthu M, Tharmalingam N. Halogen-Based 17β-HSD1 Inhibitors: Insights from DFT, Docking, and Molecular Dynamics Simulation Studies. Molecules. 2022; 27(12):3962. https://doi.org/10.3390/molecules27123962
Chicago/Turabian StyleKulandaisamy, Arulsamy, Murugesan Panneerselvam, Rajadurai Vijay Solomon, Madhavan Jaccob, Jaganathan Ramakrishnan, Kumaradhas Poomani, Muralikannan Maruthamuthu, and Nagendran Tharmalingam. 2022. "Halogen-Based 17β-HSD1 Inhibitors: Insights from DFT, Docking, and Molecular Dynamics Simulation Studies" Molecules 27, no. 12: 3962. https://doi.org/10.3390/molecules27123962
APA StyleKulandaisamy, A., Panneerselvam, M., Solomon, R. V., Jaccob, M., Ramakrishnan, J., Poomani, K., Maruthamuthu, M., & Tharmalingam, N. (2022). Halogen-Based 17β-HSD1 Inhibitors: Insights from DFT, Docking, and Molecular Dynamics Simulation Studies. Molecules, 27(12), 3962. https://doi.org/10.3390/molecules27123962