
Umbrella Sampling Simulations of Carbon Nanoparticles Crossing Immiscible Solvents


Abstract
:1. Introduction
2. Materials and Methods
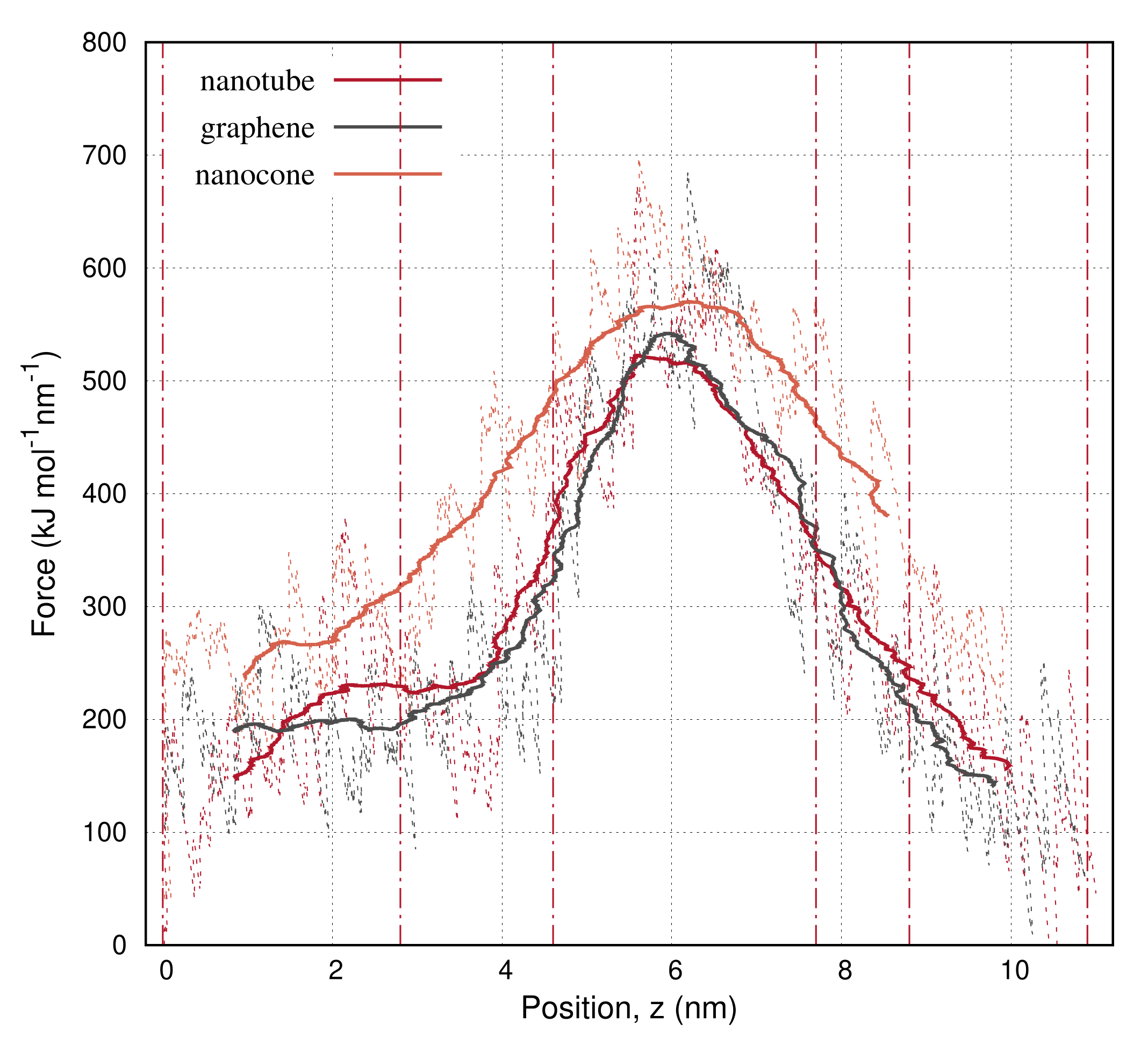
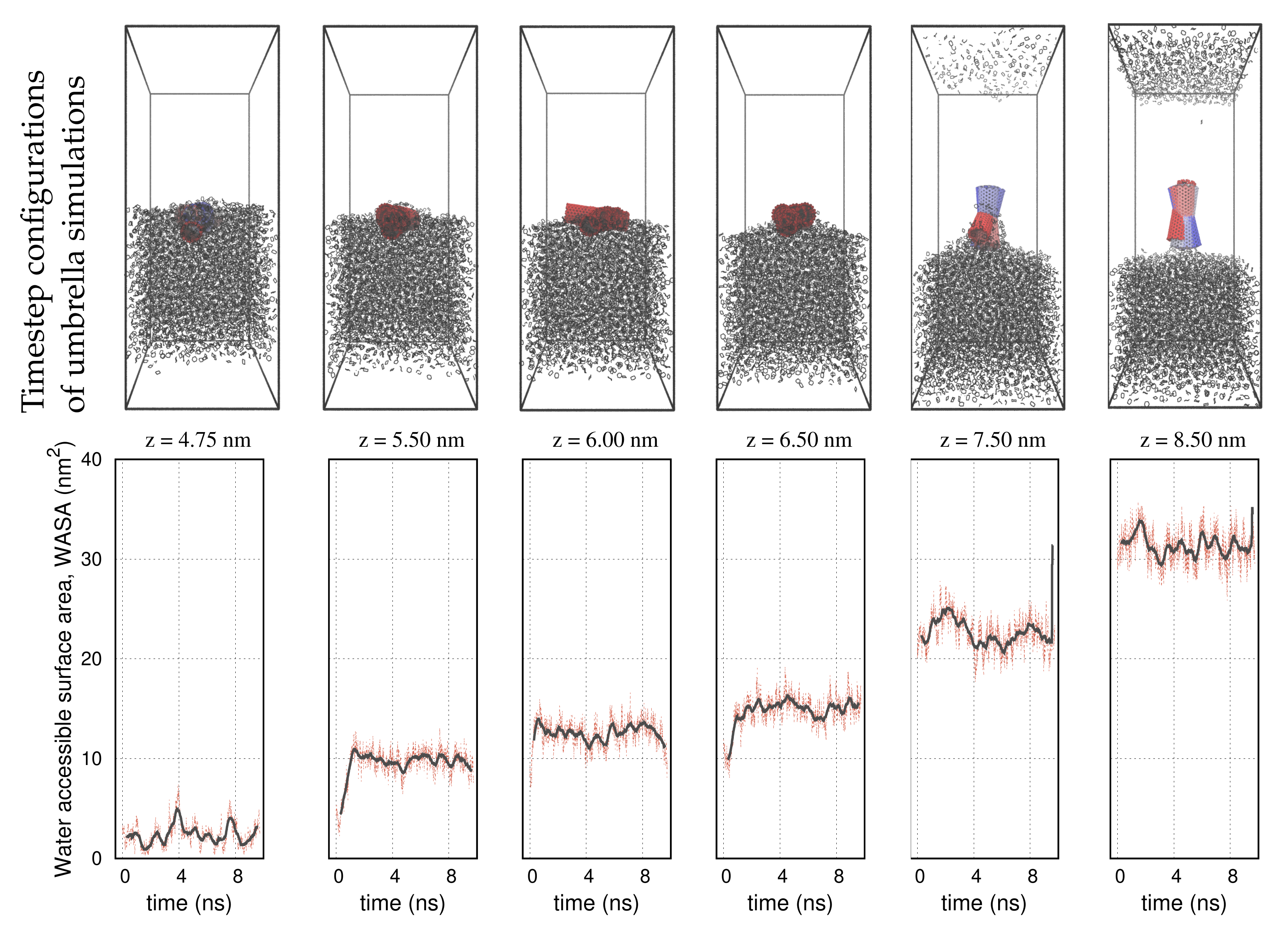
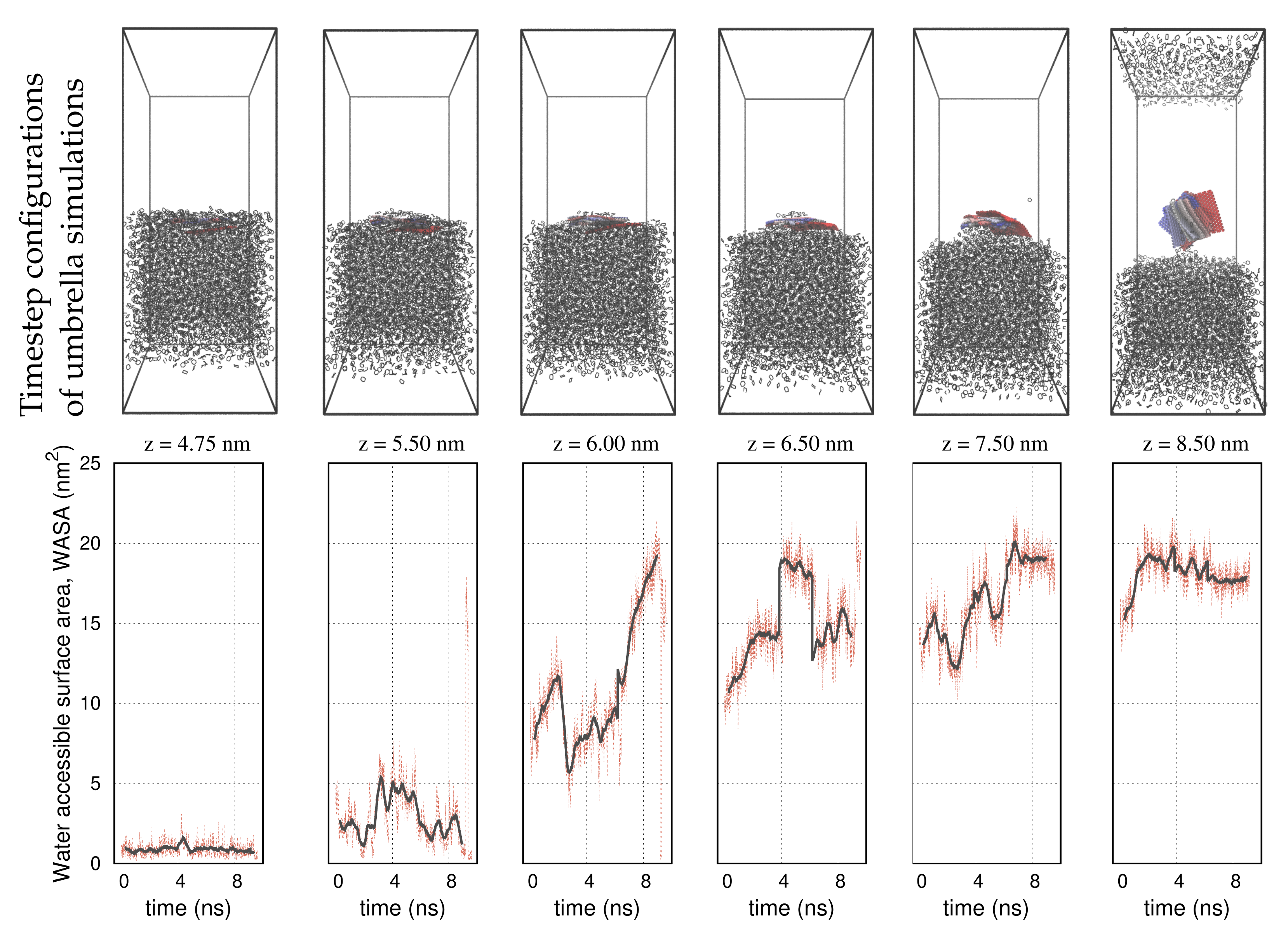
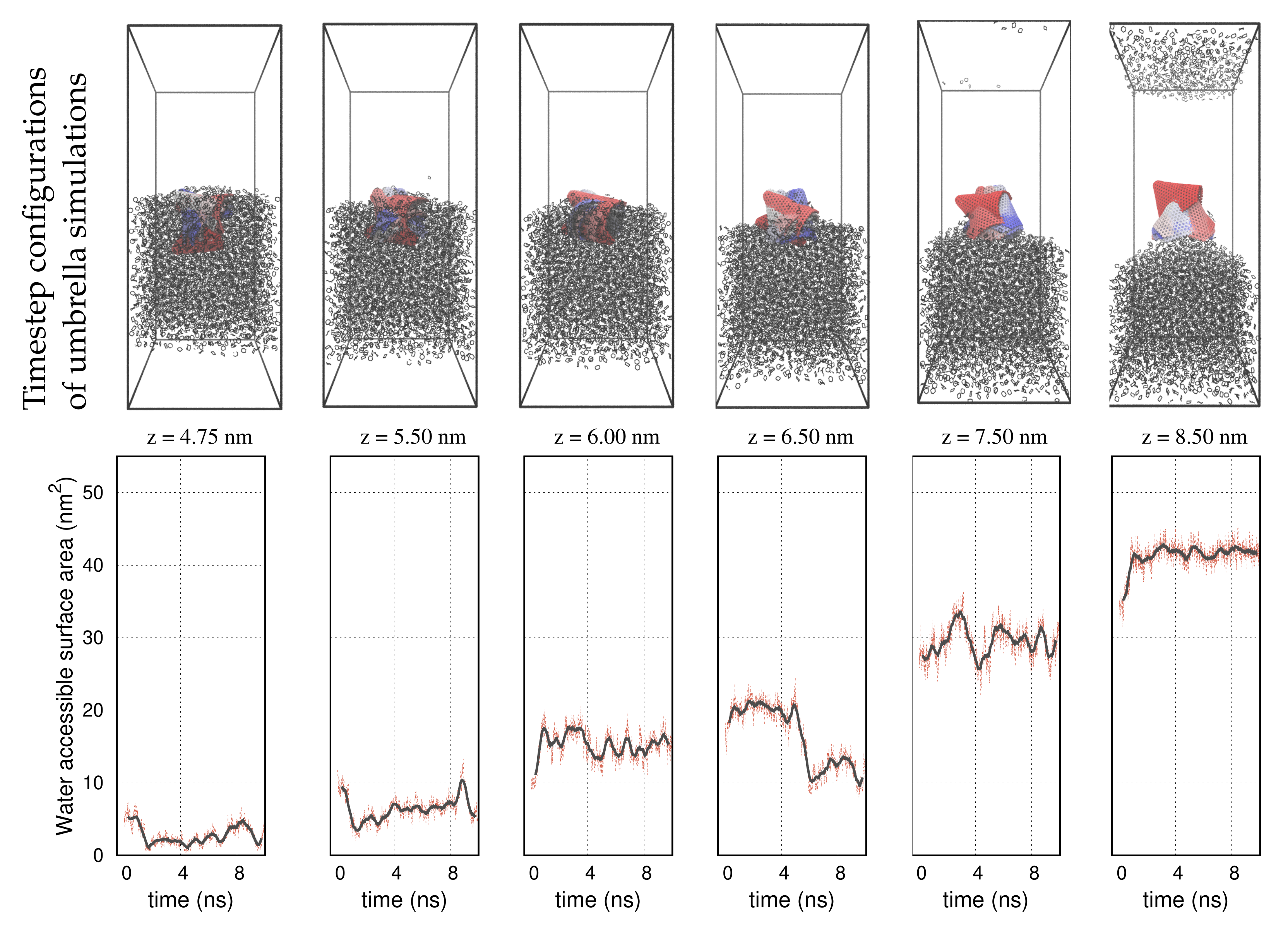
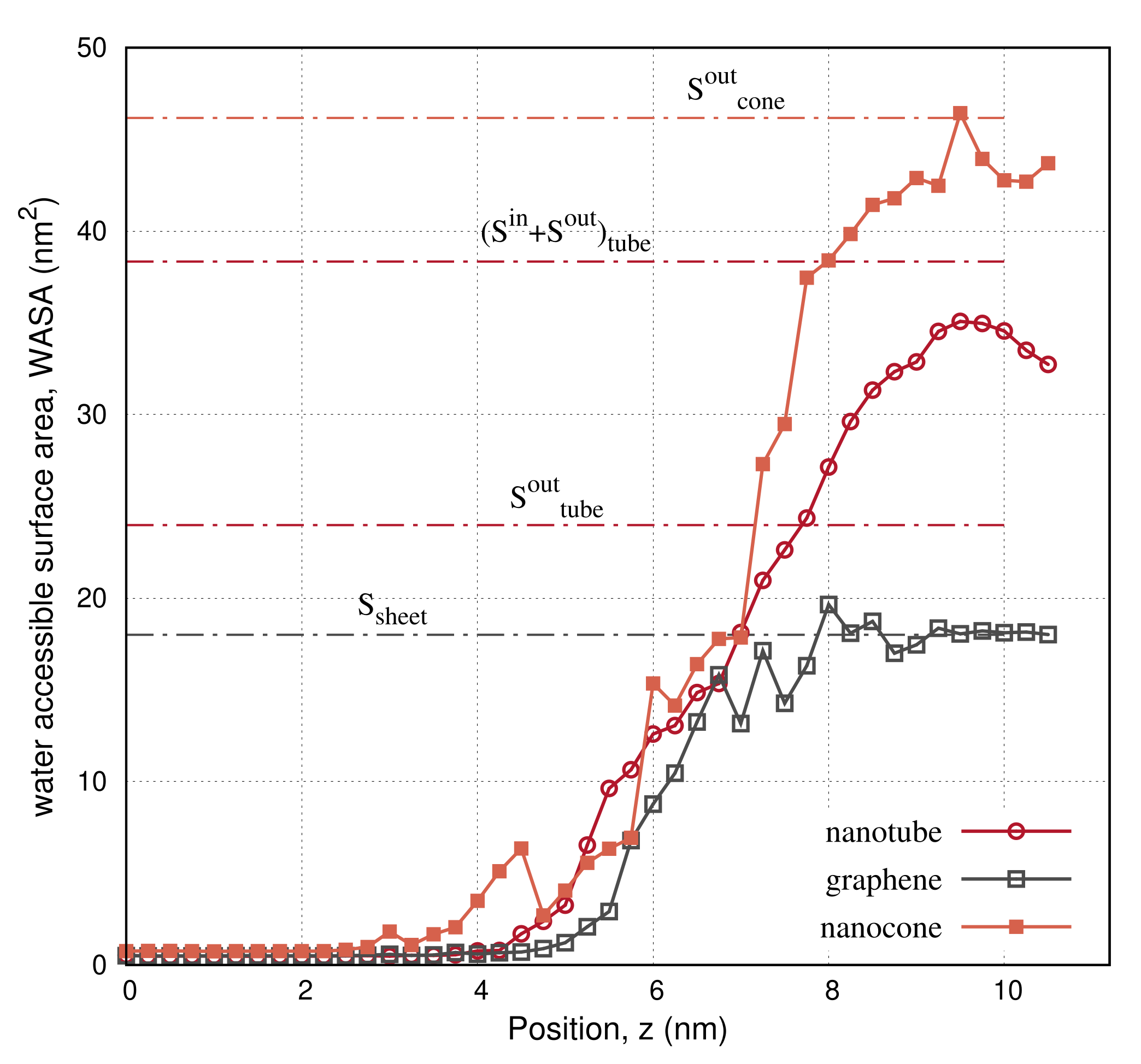
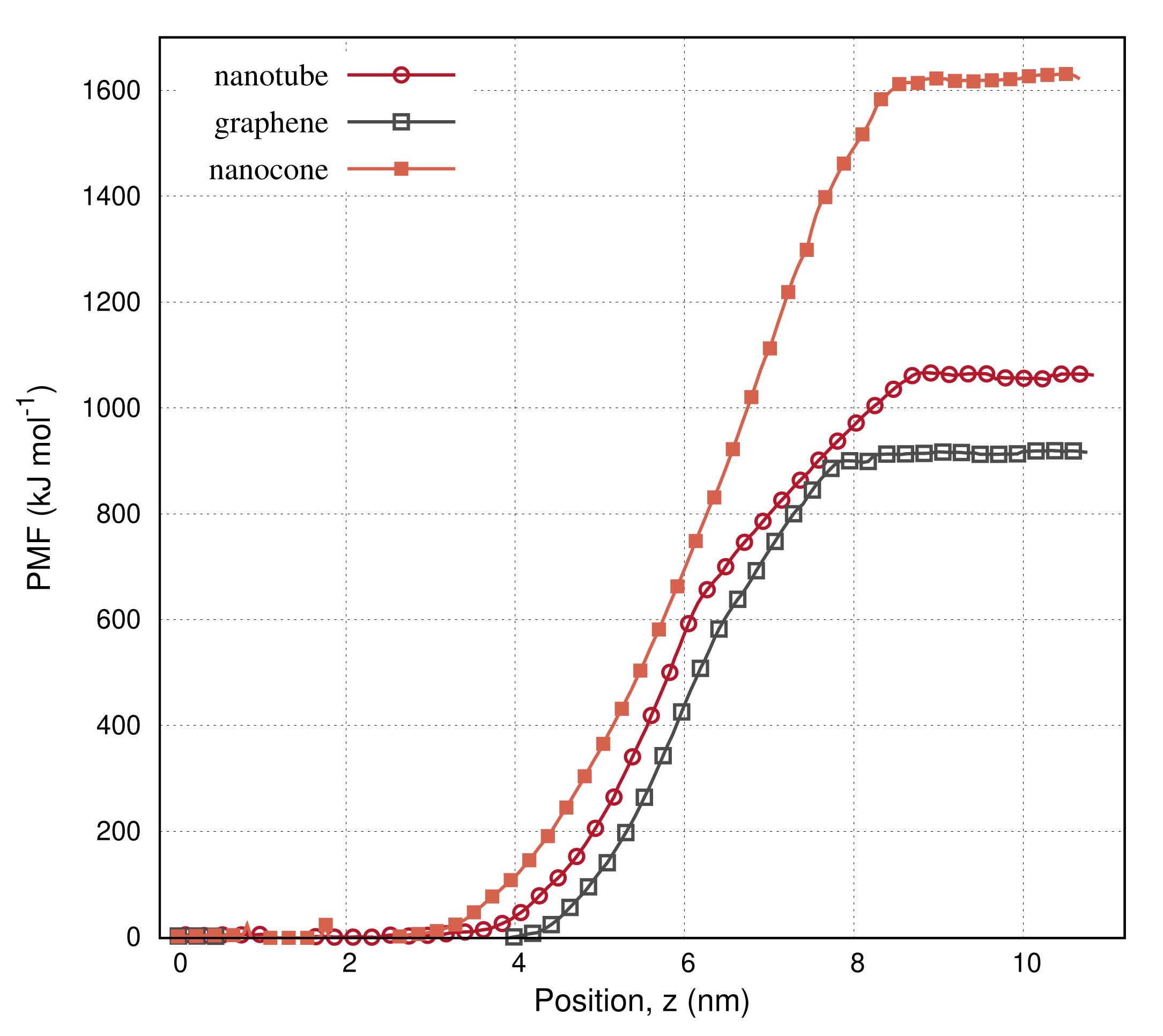
3. Results
4. Discussion
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cracknell, R.F.; Gubbins, K.E.; Maddox, M.; Nicholson, D. Modeling Fluid Behavior in Well-Characterized Porous Materials. Accounts Chem. Res. 1995, 28, 281–288. [Google Scholar] [CrossRef]
- Bojan, M.J.; van Slooten, R.; Steele, W. Computer Simulation Studies of the Storage of Methane in Microporous Carbons. Sep. Sci. Technol. 1992, 27, 1837–1856. [Google Scholar] [CrossRef]
- Bojan, M.J.; Vernov, A.V.; Steele, W.A. Simulation studies of adsorption in rough-walled cylindrical pores. Langmuir 1992, 8, 901–908. [Google Scholar] [CrossRef]
- Maddox, M.W.; Gubbins, K.E. Molecular simulation of fluid adsorption in buckytubes and MCM-41. Int. J. Thermophys. 1994, 15, 1115–1123. [Google Scholar] [CrossRef]
- Sweatman, M.B.; Quirke, N. Characterization of Porous Materials by Gas Adsorption Comparison of Nitrogen at 77 K and Carbon Dioxide at 298 K for Activated Carbon. Langmuir 2001, 17, 5011–5020. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Gun’ko, V.M.; Terzyk, A.P.; Gauden, P.A.; Rong, H.; Ryu, Z.; Do, D.D. The comparative characterization of structural heterogeneity of mesoporous activated carbon fibers (ACFs). Appl. Surf. Sci. 2003, 206, 67–77. [Google Scholar] [CrossRef]
- Gotzias, A.; Charalambopoulou, G.; Steriotis, T. On the orientation of N2 and CO2 molecules adsorbed in slit pore models with oxidised graphitic surface. Mol. Simul. 2016, 42, 186–195. [Google Scholar] [CrossRef]
- Gotzias, A.; Sapalidis, A. Pulling Simulations and Hydrogen Sorption Modelling on Carbon Nanotube Bundles. C 2020, 6. [Google Scholar] [CrossRef] [Green Version]
- Neimark, A.V.; Lin, Y.; Ravikovitch, P.I.; Thommes, M. Quenched solid density functional theory and pore size analysis of micro-mesoporous carbons. Carbon 2009, 47, 1617–1628. [Google Scholar] [CrossRef]
- Thomson, K.T.; Gubbins, K.E. Modeling Structural Morphology of Microporous Carbons by Reverse Monte Carlo. Langmuir 2000, 16, 5761–5773. [Google Scholar] [CrossRef]
- Gotzias, A. Calculating adsorption isotherms using Lennard Jones particle density distributions. J. Phys. Condens. Matter 2019, 31, 435901. [Google Scholar] [CrossRef]
- Denoyel, R.; Fernandez-Colinas, J.; Grillet, Y.; Rouquerol, J. Assessment of the surface area and microporosity of activated charcoals from immersion calorimetry and nitrogen adsorption data. Langmuir 1993, 9, 515–518. [Google Scholar] [CrossRef]
- Silvestre-Albero, J.; Gómez de Salazar, C.; Sepúlveda-Escribano, A.; Rodriguez-Reinoso, F. Characterization of microporous solids by immersion calorimetry. Colloids Surfaces Physicochem. Eng. Asp. 2001, 187–188, 151–165. [Google Scholar] [CrossRef]
- Dziatko, R.A.; Janicek, B.E.; Karten, J.L.; Harris, K.A.; Gibson, M.M.; Crowther, A.C. Solvent-Mediated Chemical Hole Doping of Graphene by Iodine. J. Phys. Chem. C 2020, 124, 3827–3834. [Google Scholar] [CrossRef]
- Hernandez, Y.; Nicolosi, V.; Lotya, M.; Blighe, F.M.; Sun, Z.; De, S.; McGovern, I.T.; Holland, B.; Byrne, M.; Gun’Ko, Y.K.; et al. High-yield production of graphene by liquid-phase exfoliation of graphite. Nat. Nanotechnol. 2008, 3, 563–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.N.; Lotya, M.; O’Neill, A.; Bergin, S.D.; King, P.J.; Khan, U.; Young, K.; Gaucher, A.; De, S.; Smith, R.J.; et al. Two-Dimensional Nanosheets Produced by Liquid Exfoliation of Layered Materials. Science 2011, 331, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Zhao, Y.; Yang, X.; Liu, D.; Shan, S.; Cui, F.; Xing, B. Understanding the pH-dependent adsorption of ionizable compounds on graphene oxide using molecular dynamics simulations. Environ. Sci. Nano 2017, 4, 1935–1943. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Allen, W.J.; Bevan, D.R. Practical Considerations for Building GROMOS-Compatible Small-Molecule Topologies. J. Chem. Inf. Model. 2010, 50, 2221–2235. [Google Scholar] [CrossRef]
- Hinkle, K.R.; Phelan, F.R. Solvation of Carbon Nanoparticles in Water/Alcohol Mixtures: Using Molecular Simulation To Probe Energetics, Structure, and Dynamics. J. Phys. Chem. C 2017, 121, 22926–22938. [Google Scholar] [CrossRef] [PubMed]
- Schneemilch, M.; Quirke, N. Free energy of adhesion of lipid bilayers on silica surfaces. J. Chem. Phys. 2018, 148, 194704. [Google Scholar] [CrossRef] [PubMed]
- Aragones, J.L.; Noya, E.G.; Valeriani, C.; Vega, C. Free energy calculations for molecular solids using GROMACS. J. Chem. Phys. 2013, 139, 034104. [Google Scholar] [CrossRef] [PubMed]
- Perthold, J.W.; Petrov, D.; Oostenbrink, C. Toward Automated Free Energy Calculation with Accelerated Enveloping Distribution Sampling (A-EDS). J. Chem. Inf. Model. 2020, 60, 5395–5406. [Google Scholar] [CrossRef] [PubMed]
- Gotzias, A. Binding Free Energy Calculations of Bilayer Graphenes Using Molecular Dynamics. J. Chem. Inf. Model. 2021, 61, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Gotzias, A.; Tocci, E.; Sapalidis, A. On the Consistency of the Exfoliation Free Energy of Graphenes by Molecular Simulations. Int. J. Mol. Sci. 2021, 22, 8291. [Google Scholar] [CrossRef] [PubMed]
- Christ, C.D.; van Gunsteren, W.F. Multiple free energies from a single simulation: Extending enveloping distribution sampling to nonoverlapping phase-space distributions. J. Chem. Phys. 2008, 128, 174112. [Google Scholar] [CrossRef]
- Tang, X.; Koenig, P.H.; Larson, R.G. Molecular Dynamics Simulations of Sodium Dodecyl Sulfate Micelles in Water—The Effect of the Force Field. J. Phys. Chem. B 2014, 118, 3864–3880. [Google Scholar] [CrossRef] [PubMed]
- Bohlén, M.; Bolton, K. Molecular dynamics studies of the influence of single wall carbon nanotubes on the mechanical properties of Poly(vinylidene fluoride). Comput. Mater. Sci. 2013, 68, 73–80. [Google Scholar] [CrossRef]
- Bandosz, T.; Biggs, M.; Gubbins, K.; Hattori, Y.; Liyama, T.; Kaneko, K.; Pikunic, J.; Thomson, K. Molecular models of porous carbons. In Chemistry & Physics of Carbon, 1st ed.; Radovic, L., Ed.; CRC Press: Boca Raton, FL, USA, 2003; pp. 41–218. [Google Scholar] [CrossRef]
- Bock, H.; Gubbins, K.E.; Pikunic, J. Chapter Five—Models of Porous Carbons. In Adsorption by Carbons; Elsevier: Amsterdam, The Netherlands, 2008; pp. 103–132. [Google Scholar]
- Gao, Y.; Maruyama, M.; Okada, S. Influence of interlayer stacking arrangements on carrier accumulation in bilayer graphene field effect transistors. Appl. Phys. Express 2020, 13, 065006. [Google Scholar] [CrossRef]
- Vögele, M.; Köfinger, J.; Hummer, G. Molecular dynamics simulations of carbon nanotube porins in lipid bilayers. Faraday Discuss. 2018, 209, 341–358. [Google Scholar] [CrossRef] [Green Version]
- Puigpelat, E.; Ignés-Mullol, J.; Sagués, F.; Reigada, R. Interaction of Graphene Nanoparticles and Lipid Membranes Displaying Different Liquid Orderings: A Molecular Dynamics Study. Langmuir 2019, 35, 16661–16668. [Google Scholar] [CrossRef]
- Arjmandi-Tash, H.; Lima, L.M.C.; Belyaeva, L.A.; Mukhina, T.; Fragneto, G.; Kros, A.; Charitat, T.; Schneider, G.F. Encapsulation of Graphene in the Hydrophobic Core of a Lipid Bilayer. Langmuir 2020, 36, 14478–14482. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Mao, J.; Yan, L.T. Computer simulation of cell entry of graphene nanosheet. Biomaterials 2013, 34, 4296–4301. [Google Scholar] [CrossRef] [PubMed]
- Fedel, M. Hemocompatibility of Carbon Nanostructures. C 2020, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Gotzias, A. Injecting Carbon Nanostructures in Living Cells. In Proceedings of the Workshops of the 11th EETN Conference on Artificial Intelligence 2020 (SETN2020 Workshops), Athens, Greece, 2–4 September 2020. [Google Scholar]
- Kordzadeh, A.; Amjad-Iranagh, S.; Zarif, M.; Modarress, H. Adsorption and encapsulation of the drug doxorubicin on covalent functionalized carbon nanotubes: A scrutinized study by using molecular dynamics simulation and quantum mechanics calculation. J. Mol. Graph. Model. 2019, 88, 11–22. [Google Scholar] [CrossRef]
- Narjabadifam, A.; Vakili-Tahami, F.; Zehsaz, M. Modal analysis of multi-walled carbon nanocones using molecular dynamics simulation. Comput. Mater. Sci. 2017, 137, 55–66. [Google Scholar] [CrossRef]
- Wohner, N.; Lam, P.; Sattler, K. Energetic stability of graphene nanoflakes and nanocones. Carbon 2014, 67, 721–735. [Google Scholar] [CrossRef]
- Karataraki, G.; Sapalidis, A.; Tocci, E.; Gotzias, A. Molecular Dynamics of Water Embedded Carbon Nanocones: Surface Waves Observation. Computation 2019, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Ge, M.; Sattler, K. Observation of fullerene cones. Chem. Phys. Lett. 1994, 220, 192–196. [Google Scholar] [CrossRef]
- Krishnan, A.; Dujardin, E.; Treacy, M.M.J.; Hugdahl, J.; Lynum, S.; Ebbesen, T.W. Graphitic cones and the nucleation of curved carbon surfaces. Nature 1997, 388, 451–454. [Google Scholar] [CrossRef]
- Naess, S.N.; Elgsaeter, A.; Helgesen, G.; Knudsen, K.D. Carbon nanocones: Wall structure and morphology. Sci. Technol. Adv. Mater. 2009, 10, 065002. [Google Scholar] [CrossRef]
- Heiberg-Andersen, H.; Skjeltorp, A.T. Stability of Conjugated Carbon Nanocones. J. Math. Chem. 2005, 38, 589–604. [Google Scholar] [CrossRef]
- Bultheel, A.; Ori, O. Topological modeling of 1-Pentagon carbon nanocones—Topological efficiency and magic sizes. Fullerenes Nanotub. Carbon Nanostruct. 2018, 26, 291–302. [Google Scholar] [CrossRef]
- Nazeer, W.; Farooq, A.; Younas, M.; Munir, M.; Kang, S.M. On Molecular Descriptors of Carbon Nanocones. Biomolecules 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apriliyanto, Y.B.; Battaglia, S.; Evangelisti, S.; Faginas-Lago, N.; Leininger, T.; Lombardi, A. Toward a Generalized Hückel Rule: The Electronic Structure of Carbon Nanocones. J. Phys. Chem. A 2021, 125, 9819–9825. [Google Scholar] [CrossRef] [PubMed]
- Gotzias, A.; Heiberg-Andersen, H.; Kainourgiakis, M.; Steriotis, T. A grand canonical Monte Carlo study of hydrogen adsorption in carbon nanohorns and nanocones at 77K. Carbon 2011, 49, 2715–2724. [Google Scholar] [CrossRef]
- Gotzias, A.; Heiberg-Andersen, H.; Kainourgiakis, M.; Steriotis, T. Grand canonical Monte Carlo simulations of hydrogen adsorption in carbon cones. Appl. Surf. Sci. 2010, 256, 5226–5231. [Google Scholar] [CrossRef]
- Ulloa, P.; Pacheco, M.; Latgé, A. Optical properties of graphene nanocones under electric and magnetic fields. J. Phys. Condens. Matter 2017, 29, 455304. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.C.; Chen, L.H.; Ye, X.R.; Daraio, C.; Jin, S.; Orme, C.A.; Quist, A.; Lal, R. Extremely sharp carbon nanocone probes for atomic force microscopy imaging. Appl. Phys. Lett. 2006, 88, 153102. [Google Scholar] [CrossRef] [Green Version]
- Furmaniak, S.; Gauden, P.A.; Patrykiejew, A.; Miśkiewicz, R.; Kowalczyk, P. Carbon Nanohorns as Reaction Nanochambers—A Systematic Monte Carlo Study. Sci. Rep. 2018, 8, 15407. [Google Scholar] [CrossRef] [Green Version]
- Jarzynski, C. Equilibrium free-energy differences from nonequilibrium measurements: A master-equation approach. Phys. Rev. E 1997, 56, 5018–5035. [Google Scholar] [CrossRef] [Green Version]
- Lemkul, J.A.; Bevan, D.R. Assessing the Stability of Alzheimer’s Amyloid Protofibrils Using Molecular Dynamics. J. Phys. Chem. B 2010, 114, 1652–1660. [Google Scholar] [CrossRef] [PubMed]
- Minoia, A.; Chen, L.; Beljonne, D.; Lazzaroni, R. Molecular modeling study of the structure and stability of polymer/carbon nanotube interfaces. Polymer 2012, 53, 5480–5490. [Google Scholar] [CrossRef]
- Available online: http://chembytes.wikidot.com (accessed on 25 January 2022).
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Eisenhaber, F.; Lijnzaad, P.; Argos, P.; Sander, C.; Scharf, M. The double cubic lattice method: Efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 1995, 16, 273–284. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Nanotube | Graphene | Nanocone |
|---|---|---|---|
| carbon atoms | 760 | 416 | 1077 |
| hydrogen atoms | 40 | 58 | 57 |
| edge, , or base radius, r (nm) | 0.68 | 3 | 2.09 |
| height, h (nm) | 4.5 | 3 | 3.55 |
| geometrical surface, S (nm) | |||
| () | 23.98 | 9.0 | 46.16 |
| () | 14.36 | - | 36.13 |
| Structure | Nanotube | Graphene | Nanocone |
|---|---|---|---|
| Volume X × Y × Z (nm) | 9.01 × 9.01 × 22.53 | 9.09 × 9.09 × 22.73 | 8.91 × 8.91 × 22.27 |
| cycloexane molecules | 4293 | 4338 | 4235 |
| water molecules (spc) | 34,235 | 35,410 | 32,702 |
| Structure | Nanotube | Graphene | Nanocone |
|---|---|---|---|
| ns/day | 5.278 | 5.183 | 5.433 |
| hours/ns | 4.547 | 4.631 | 4.417 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gotzias, A. Umbrella Sampling Simulations of Carbon Nanoparticles Crossing Immiscible Solvents. Molecules 2022, 27, 956. https://doi.org/10.3390/molecules27030956
Gotzias A. Umbrella Sampling Simulations of Carbon Nanoparticles Crossing Immiscible Solvents. Molecules. 2022; 27(3):956. https://doi.org/10.3390/molecules27030956
Chicago/Turabian StyleGotzias, Anastasios. 2022. "Umbrella Sampling Simulations of Carbon Nanoparticles Crossing Immiscible Solvents" Molecules 27, no. 3: 956. https://doi.org/10.3390/molecules27030956
APA StyleGotzias, A. (2022). Umbrella Sampling Simulations of Carbon Nanoparticles Crossing Immiscible Solvents. Molecules, 27(3), 956. https://doi.org/10.3390/molecules27030956