
Targeted Drug Delivery for the Treatment of Blood Cancers


Abstract
:1. Introduction
2. Targeting Delivery Strategy
2.1. Targeting Bone Marrow and Its Microenvironment
2.1.1. Passive Targeting Strategy
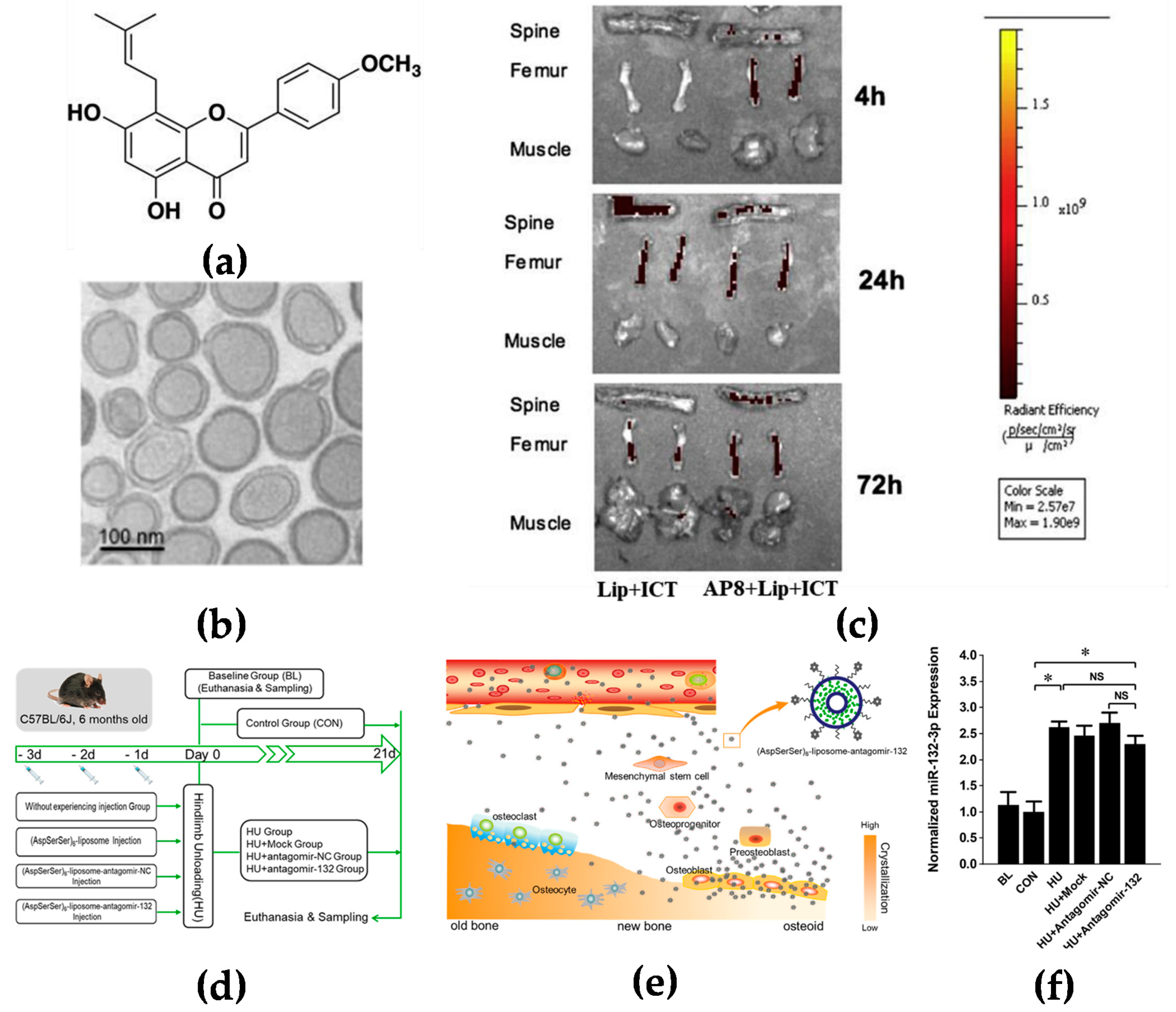
2.1.2. Targeting Bone Surface-Mediated Bone Marrow
2.1.3. Active Targeting
2.2. Targeting Spleen and Lymphoid Nodes
2.3. Targeting Vascular System
3. Nanomedicines for Blood Cancers
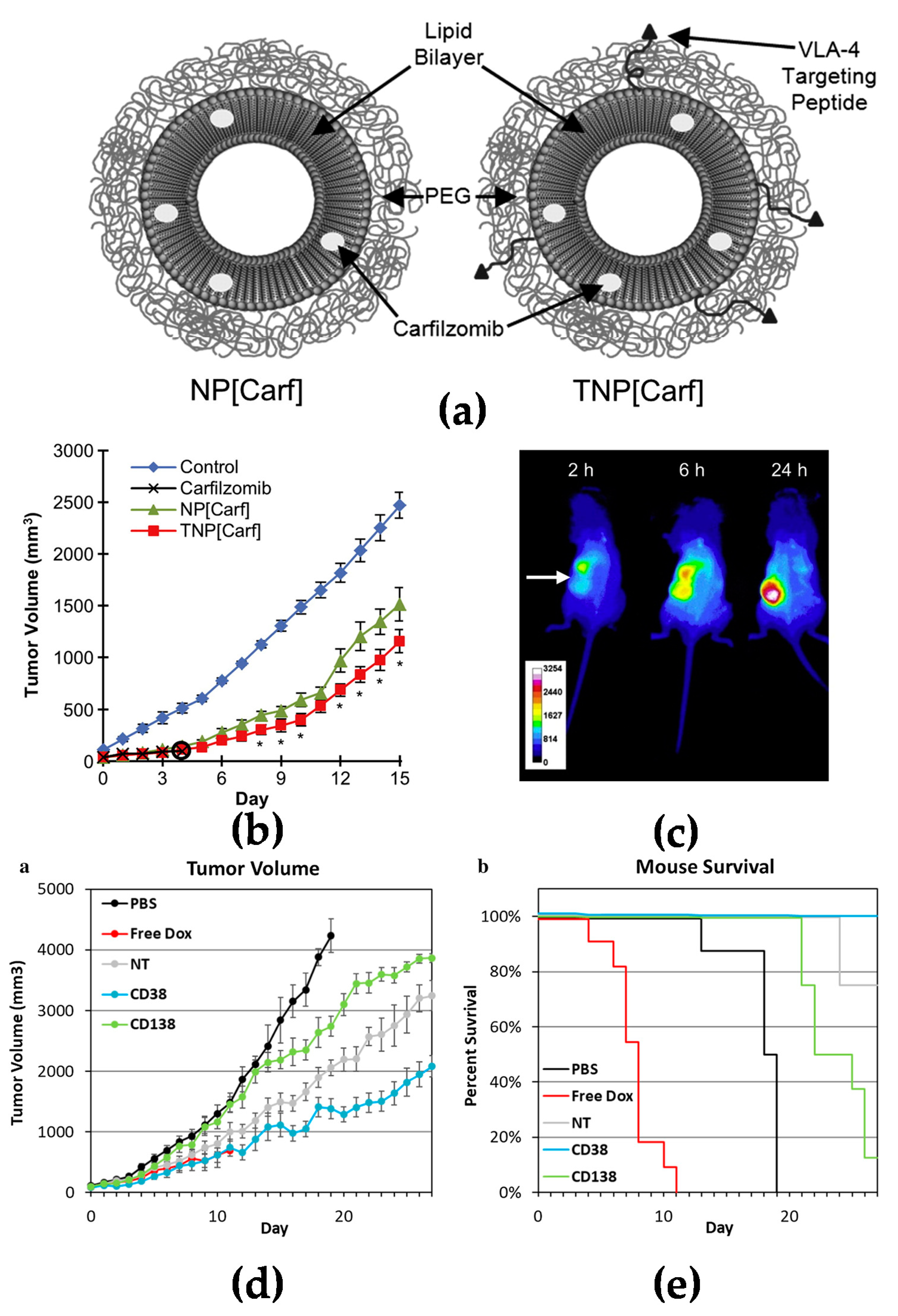
3.1. Multiple Myeloma
3.2. Acute Myeloid Leukemia
3.3. B Cell Lymphomas
4. Challenges in Drug Delivery Systems for Treating Blood Cancers
4.1. Biological Challenges
4.1.1. Characterization of Nano-Based Medicines
4.1.2. Toxicity and Side Effects of Nano-Based Medicines
4.1.3. Circulation and Clearance
4.1.4. Translational Study in the In Vivo Model
4.1.5. Interfere with the Bone Marrow for Blood Cancer Drug Delivery Systems
4.2. Non-Biological Challenges
4.2.1. Commercialized Challenges
4.2.2. Policy/Regulation Challenges
5. Summary
Funding
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Rowe, J.M. Perspectives on current survival and new developments in AML. Best Pract. Res. Clin. Haematol. 2021, 34, 101248. [Google Scholar] [CrossRef]
- Tuazon, S.A.; Holmberg, L.A.; Nadeem, O.; Richardson, P.G. A clinical perspective on plasma cell leukemia; current status and future directions. Blood Cancer J. 2021, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; June, C.H. Going viral: Chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. 2015, 263, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Swami, A.; Reagan, M.R.; Basto, P.; Mishima, Y.; Kamaly, N.; Glavey, S.; Zhang, S.; Moschetta, M.; Seevaratnam, D.; Zhang, Y.; et al. Engineered nanomedicine for myeloma and bone microenvironment targeting. Proc. Natl. Acad. Sci. USA 2014, 111, 10287–10292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adjei, I.M.; Sharma, B.; Peetla, C.; Labhasetwar, V. Inhibition of bone loss with surface-modulated, drug-loaded nanoparticles in an intraosseous model of prostate cancer. J. Control. Release 2016, 232, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Yu, X.; Carbone, E.J.; Nelson, C.; Kan, H.M.; Lo, K.W. Poly aspartic acid peptide-linked PLGA based nanoscale particles: Potential for bone-targeting drug delivery applications. Int. J. Pharm. 2014, 475, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Iannazzo, D.; Ettari, R.; Giofrè, S.; Eid, A.H.; Bitto, A. Recent Advances in Nanotherapeutics for Multiple Myeloma. Cancers 2020, 12, 3144. [Google Scholar] [CrossRef]
- Beloqui, A.; Solinís, M.A.; Delgado, A.; Evora, C.; del Pozo-Rodríguez, A.; Rodríguez-Gascón, A. Biodistribution of Nanostructured Lipid Carriers (NLCs) after intravenous administration to rats: Influence of technological factors. Eur. J. Pharm. Biopharm. 2013, 84, 309–314. [Google Scholar] [CrossRef]
- Vinhas, R.; Mendes, R.; Fernandes, A.R.; Baptista, P.V. Nanoparticles-Emerging Potential for Managing Leukemia and Lymphoma. Front. Bioeng. Biotechnol. 2017, 5, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sou, K.; Goins, B.; Oyajobi, B.O.; Travi, B.L.; Phillips, W.T. Bone marrow-targeted liposomal carriers. Expert Opin. Drug Deliv. 2011, 8, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghimi, S.M. Exploiting bone marrow microvascular structure for drug delivery and future therapies. Adv. Drug Deliv. Rev. 1995, 17, 61–73. [Google Scholar] [CrossRef]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes Res. 2010, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, H.A.; Warner, K.J.; Li, M.C.; Hunter, G.K. Binding of bone sialoprotein, osteopontin and synthetic polypeptides to hydroxyapatite. Connect. Tissue Res. 2001, 42, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Miller, S.C.; Shlyakhtenko, L.S.; Portillo, A.M.; Liu, X.M.; Papangkorn, K.; Kopecková, P.; Lyubchenko, Y.; Higuchi, W.I.; Kopecek, J. Osteotropic Peptide that differentiates functional domains of the skeleton. Bioconjug. Chem. 2007, 18, 1375–1378. [Google Scholar] [CrossRef]
- Huang, L.; Wang, X.; Cao, H.; Li, L.; Chow, D.H.; Tian, L.; Wu, H.; Zhang, J.; Wang, N.; Zheng, L.; et al. A bone-targeting delivery system carrying osteogenic phytomolecule icaritin prevents osteoporosis in mice. Biomaterials 2018, 182, 58–71. [Google Scholar] [CrossRef]
- Yang, Y.S.; Xie, J.; Chaugule, S.; Wang, D.; Kim, J.M.; Kim, J.; Tai, P.W.L.; Seo, S.K.; Gravallese, E.; Gao, G.; et al. Bone-Targeting AAV-Mediated Gene Silencing in Osteoclasts for Osteoporosis Therapy. Mol. Ther. Methods Clin. Dev. 2020, 17, 922–935. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, L.; Wang, H.; Wang, Y.; Tan, Y.; Dang, L.; Wang, K.; Sun, Z.; Li, G.; Cao, X.; et al. Targeted silencing of miRNA-132-3p expression rescues disuse osteopenia by promoting mesenchymal stem cell osteogenic differentiation and osteogenesis in mice. Stem Cell Res. Ther. 2020, 11, 58. [Google Scholar] [CrossRef]
- Shi, Y.; Su, Z.; Li, S.; Chen, Y.; Chen, X.; Xiao, Y.; Sun, M.; Ping, Q.; Zong, L. Multistep targeted nano drug delivery system aiming at leukemic stem cells and minimal residual disease. Mol. Pharm. 2013, 10, 2479–2489. [Google Scholar] [CrossRef]
- Santini, D.; Caraglia, M.; Vincenzi, B.; Holen, I.; Scarpa, S.; Budillon, A.; Tonini, G. Mechanisms of disease: Preclinical reports of antineoplastic synergistic action of bisphosphonates. Nat. Clin. Pract. Oncol. 2006, 3, 325–338. [Google Scholar] [CrossRef]
- Tian, Z.; Wu, L.; Yu, C.; Chen, Y.; Xu, Z.; Bado, I.; Loredo, A.; Wang, L.; Wang, H.; Wu, K.L.; et al. Harnessing the power of antibodies to fight bone metastasis. Sci. Adv. 2021, 7, 26. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, N.; Zhang, H.; Sun, B.; Hou, C.; Ji, C.; Zheng, J.; Liu, Y.; Zuo, P. Enhanced in vivo antitumor efficacy of dual-functional peptide-modified docetaxel nanoparticles through tumor targeting and Hsp90 inhibition. J. Control. Release 2016, 221, 26–36. [Google Scholar] [CrossRef]
- Yang, N.; Jiang, Y.; Zhang, H.; Sun, B.; Hou, C.; Zheng, J.; Liu, Y.; Zuo, P. Active targeting docetaxel-PLA nanoparticles eradicate circulating lung cancer stem-like cells and inhibit liver metastasis. Mol. Pharm. 2015, 12, 232–239. [Google Scholar] [CrossRef]
- Mahmoudi, R.; Ashraf Mirahmadi-Babaheidri, S.; Delaviz, H.; Fouani, M.H.; Alipour, M.; Jafari Barmak, M.; Christiansen, G.; Bardania, H. RGD peptide-mediated liposomal curcumin targeted delivery to breast cancer cells. J. Biomater. Appl. 2021, 35, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Ashley, J.D.; Stefanick, J.F.; Schroeder, V.A.; Suckow, M.A.; Alves, N.J.; Suzuki, R.; Kikuchi, S.; Hideshima, T.; Anderson, K.C.; Kiziltepe, T.; et al. Liposomal carfilzomib nanoparticles effectively target multiple myeloma cells and demonstrate enhanced efficacy in vivo. J. Control. Release 2014, 196, 113–121. [Google Scholar] [CrossRef]
- Kim, D.; Park, C.Y.; Medeiros, B.C.; Weissman, I.L. CD19-CD45 low/-CD38 high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia 2012, 26, 2530–2537. [Google Scholar] [CrossRef]
- Omstead, D.T.; Mejia, F.; Sjoerdsma, J.; Kim, B.; Shin, J.; Khan, S.; Wu, J.; Kiziltepe, T.; Littlepage, L.E.; Bilgicer, B. In vivo evaluation of CD38 and CD138 as targets for nanoparticle-based drug delivery in multiple myeloma. J. Hematol. Oncol. 2020, 13, 145. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Sato, T.; Iyama, S.; Tatekoshi, A.; Hashimoto, A.; Kamihara, Y.; Horiguchi, H.; Kikuchi, S.; Kawano, Y.; Takada, K.; et al. A novel strategy inducing autophagic cell death in Burkitt’s lymphoma cells with anti-CD19-targeted liposomal rapamycin. Blood Cancer J. 2014, 4, e180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shen, D.; Jia, M.; Zhao, H.; Tang, Y. The targeting effect of Hm2E8b-NCTD-liposomes on B-lineage leukaemia stem cells is associated with the HLF-SLUG axis. J. Drug Target. 2018, 26, 55–65. [Google Scholar] [CrossRef]
- Bronte, V.; Pittet, M.J. The spleen in local and systemic regulation of immunity. Immunity 2013, 39, 806–818. [Google Scholar] [CrossRef] [Green Version]
- Saboo, S.S.; Krajewski, K.M.; O’Regan, K.N.; Giardino, A.; Brown, J.R.; Ramaiya, N.; Jagannathan, J.P. Spleen in haematological malignancies: Spectrum of imaging findings. Br. J. Radiol. 2012, 85, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, S.K.; Jittiwat, J.; Manikandan, J.; Ong, C.N.; Yu, L.E.; Ong, W.Y. Biodistribution of gold nanoparticles and gene expression changes in the liver and spleen after intravenous administration in rats. Biomaterials 2010, 31, 2034–2042. [Google Scholar] [CrossRef]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef]
- Jindal, A.B. Nanocarriers for spleen targeting: Anatomo-physiological considerations, formulation strategies and therapeutic potential. Drug Deliv. Transl. Res. 2016, 6, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.F.; Shen, J.; Liang, J.; Zheng, H.S.; Xiong, Y.; Wei, Y.H.; Li, F. Targeted drug delivery for tumor therapy inside the bone marrow. Biomaterials 2018, 155, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Park, C.S.; Choi, Y.S. How do follicular dendritic cells interact intimately with B cells in the germinal centre? Immunology 2005, 114, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Shamay, Y.; Golan, M.; Tyomkin, D.; David, A. Assessing the therapeutic efficacy of VEGFR-1-targeted polymer drug conjugates in mouse tumor models. J. Control. Release 2016, 229, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Runbeck, E.; Crescioli, S.; Karagiannis, S.N.; Papa, S. Utilizing Immunocytokines for Cancer Therapy. Antibodies 2021, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Schliemann, C.; Gutbrodt, K.L.; Kerkhoff, A.; Pohlen, M.; Wiebe, S.; Silling, G.; Angenendt, L.; Kessler, T.; Mesters, R.M.; Giovannoni, L.; et al. Targeting interleukin-2 to the bone marrow stroma for therapy of acute myeloid leukemia relapsing after allogeneic hematopoietic stem cell transplantation. Cancer Immunol. Res. 2015, 3, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.; Videira, P.A.; Sackstein, R. E-Selectin Ligands in the Human Mononuclear Phagocyte System: Implications for Infection, Inflammation, and Immunotherapy. Front. Immunol. 2017, 8, 1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholizadeh, S.; Visweswaran, G.R.R.; Storm, G.; Hennink, W.E.; Kamps, J.; Kok, R.J. E-selectin targeted immunoliposomes for rapamycin delivery to activated endothelial cells. Int. J. Pharm. 2018, 548, 759–770. [Google Scholar] [CrossRef]
- Ning, Y.M.; He, K.; Dagher, R.; Sridhara, R.; Farrell, A.T.; Justice, R.; Pazdur, R. Liposomal doxorubicin in combination with bortezomib for relapsed or refractory multiple myeloma. Oncology 2007, 21, 1503–1508. [Google Scholar]
- Orlowski, R.Z.; Nagler, A.; Sonneveld, P.; Bladé, J.; Hajek, R.; Spencer, A.; Robak, T.; Dmoszynska, A.; Horvath, N.; Spicka, I.; et al. Final overall survival results of a randomized trial comparing bortezomib plus pegylated liposomal doxorubicin with bortezomib alone in patients with relapsed or refractory multiple myeloma. Cancer 2016, 122, 2050–2056. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhuang, J. Major advances in the treatment of multiple myeloma in American Society of Hematology annual meeting 2020. Chronic Dis. Transl. Med. 2021, 7, 220–226. [Google Scholar] [CrossRef]
- Lee, A.L.Z.; Voo, Z.X.; Chin, W.; Ono, R.J.; Yang, C.; Gao, S.; Hedrick, J.L.; Yang, Y.Y. Injectable Coacervate Hydrogel for Delivery of Anticancer Drug-Loaded Nanoparticles in vivo. ACS Appl Mater. Interfaces 2018, 10, 13274–13282. [Google Scholar] [CrossRef]
- Huang, Y.H.; Vakili, M.R.; Molavi, O.; Morrissey, Y.; Wu, C.; Paiva, I.; Soleimani, A.H.; Sanaee, F.; Lavasanifar, A.; Lai, R. Decoration of Anti-CD38 on Nanoparticles Carrying a STAT3 Inhibitor Can Improve the Therapeutic Efficacy Against Myeloma. Cancers 2019, 11, 248. [Google Scholar] [CrossRef] [Green Version]
- Löwenberg, B.; Downing, J.R.; Burnett, A. Acute myeloid leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef]
- Huang, X.; Lin, H.; Huang, F.; Xie, Y.; Wong, K.H.; Chen, X.; Wu, D.; Lu, A.; Yang, Z. Targeting Approaches of Nanomedicines in Acute Myeloid Leukemia. Dose Response 2019, 17, 1559325819887048. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Dombret, H.; Gardin, C. An update of current treatments for adult acute myeloid leukemia. Blood 2016, 127, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; Rosenblat, T.; Arellano, M.; Gobbi, M.; Altman, J.K.; Montesinos, P.; O’Connell, C.; Solomon, S.R.; Pigneux, A.; Vey, N.; et al. International randomized phase III study of elacytarabine versus investigator choice in patients with relapsed/refractory acute myeloid leukemia. J. Clin. Oncol 2014, 32, 1919–1926. [Google Scholar] [CrossRef]
- Alakhova, D.Y.; Zhao, Y.; Li, S.; Kabanov, A.V. Effect of doxorubicin/pluronic SP1049C on tumorigenicity, aggressiveness, DNA methylation and stem cell markers in murine leukemia. PLoS ONE 2013, 8, e72238. [Google Scholar] [CrossRef]
- Tardi, P.; Wan, C.P.; Mayer, L. Passive and semi-active targeting of bone marrow and leukemia cells using anionic low cholesterol liposomes. J. Drug Target. 2016, 24, 797–804. [Google Scholar] [CrossRef]
- Krauss, A.C.; Gao, X.; Li, L.; Manning, M.L.; Patel, P.; Fu, W.; Janoria, K.G.; Gieser, G.; Bateman, D.A.; Przepiorka, D.; et al. FDA Approval Summary: (Daunorubicin and Cytarabine) Liposome for Injection for the Treatment of Adults with High-Risk Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 2685–2690. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol 2018, 36, 2684–2692. [Google Scholar] [CrossRef]
- Floc’h, N.; Ashton, S.; Taylor, P.; Trueman, D.; Harris, E.; Odedra, R.; Maratea, K.; Derbyshire, N.; Caddy, J.; Jacobs, V.N.; et al. Optimizing Therapeutic Effect of Aurora B Inhibition in Acute Myeloid Leukemia with AZD2811 Nanoparticles. Mol. Cancer Ther. 2017, 16, 1031–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novo, M.; Castellino, A.; Nicolosi, M.; Santambrogio, E.; Vassallo, F.; Chiappella, A.; Vitolo, U. High-grade B-cell lymphoma: How to diagnose and treat. Expert Rev. Hematol. 2019, 12, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Deshantri, A.K.; Varela Moreira, A.; Ecker, V.; Mandhane, S.N.; Schiffelers, R.M.; Buchner, M.; Fens, M. Nanomedicines for the treatment of hematological malignancies. J. Control. Release 2018, 287, 194–215. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Radford, J.; Van Hoof, A.; Vitolo, U.; Soubeyran, P.; Tilly, H.; Huijgens, P.C.; Kolstad, A.; d’Amore, F.; Gonzalez Diaz, M.; et al. Phase III trial of consolidation therapy with yttrium-90-ibritumomab tiuxetan compared with no additional therapy after first remission in advanced follicular lymphoma. J. Clin. Oncol. 2008, 26, 5156–5164. [Google Scholar] [CrossRef]
- Nevala, W.K.; Butterfield, J.T.; Sutor, S.L.; Knauer, D.J.; Markovic, S.N. Antibody-targeted paclitaxel loaded nanoparticles for the treatment of CD20(+) B-cell lymphoma. Sci. Rep. 2017, 7, 45682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martucci, N.M.; Migliaccio, N.; Ruggiero, I.; Albano, F.; Calì, G.; Romano, S.; Terracciano, M.; Rea, I.; Arcari, P.; Lamberti, A. Nanoparticle-based strategy for personalized B-cell lymphoma therapy. Int. J. Nanomed. 2016, 11, 6089–6101. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.Y.; Correa, S.; Min, J.; Li, J.; Roy, S.; Laccetti, K.H.; Dreaden, E.; Kong, S.; Heo, R.; Roh, Y.H.; et al. Binary Targeting of siRNA to Hematologic Cancer Cells In Vivo using Layer-by-Layer Nanoparticles. Adv. Funct. Mater. 2019, 29, 1900018. [Google Scholar] [CrossRef]
- Roscigno, R.F.; Vaughn, T.; Parsley, E.; Hunt, T.; Eldon, M.A.; Rubin, L.J. Comparative bioavailability of inhaled treprostinil administered as LIQ861 and Tyvaso® in healthy subjects. Vascul. Pharmacol. 2021, 138, 106840. [Google Scholar] [CrossRef]
- Petersen, R.S.; Boisen, A.; Keller, S.S. Micromechanical Punching: A Versatile Method for Non-Spherical Microparticle Fabrication. Polymers 2020, 13, 83. [Google Scholar] [CrossRef]
- Perry, J.L.; Tian, S.; Sengottuvel, N.; Harrison, E.B.; Gorentla, B.K.; Kapadia, C.H.; Cheng, N.; Luft, J.C.; Ting, J.P.; DeSimone, J.M.; et al. Pulmonary Delivery of Nanoparticle-Bound Toll-like Receptor 9 Agonist for the Treatment of Metastatic Lung Cancer. ACS Nano 2020, 14, 7200–7215. [Google Scholar] [CrossRef]
- Dumont, E.F.; Oliver, A.J.; Ioannou, C.; Billiard, J.; Dennison, J.; van den Berg, F.; Yang, S.; Chandrasekaran, V.; Young, G.C.; Lahiry, A.; et al. A Novel Inhaled Dry-Powder Formulation of Ribavirin Allows for Efficient Lung Delivery in Healthy Participants and Those with Chronic Obstructive Pulmonary Disease in a Phase 1 Study. Antimicrob. Agents Chemother. 2020, 64, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, E.M.; Luft, J.C.; DeSimone, J.M. Formulation of High-Performance Dry Powder Aerosols for Pulmonary Protein Delivery. Pharm. Res. 2018, 35, 195. [Google Scholar] [CrossRef]
- Metselaar, J.M.; Lammers, T. Challenges in nanomedicine clinical translation. Drug Deliv. Transl. Res. 2020, 10, 721–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilnawaz, F.; Acharya, S.; Sahoo, S.K. Recent trends of nanomedicinal approaches in clinics. Int. J. Pharm. 2018, 538, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Dilnawaz, F.; Sahoo, S.K. Challenges of moving theranostic nanomedicine into the clinic. Nanomedicine 2020, 15, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Hua, S.; de Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharmacol. 2018, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.A.; Mészáros, T.; Fülöp, T.; Urbanics, R.; Szebeni, J.; Cho, N.J. Comparison of complement activation-related pseudoallergy in miniature and domestic pigs: Foundation of a validatable immune toxicity model. Nanomedicine 2016, 12, 933–943. [Google Scholar] [CrossRef]
- Szebeni, J.; Storm, G. Complement activation as a bioequivalence issue relevant to the development of generic liposomes and other nanoparticulate drugs. Biochem. Biophys. Res. Commun. 2015, 468, 490–497. [Google Scholar] [CrossRef]
- Hu, X.; Dong, M.; Liang, X.; Liu, Z.; Li, Q. Reactive Oxygen Species-Mediated Inflammation and Apoptosis in Hand-Foot Syndrome Induced by PEGylated Liposomal Doxorubicin. Int. J. Nanomed. 2021, 16, 471–480. [Google Scholar] [CrossRef]
- Ni, C.; Fang, J.; Qian, H.; Xu, Q.; Shen, F. Liposomal doxorubicin-related palmar-plantar erythrodysesthesia (hand-foot syndrome): A case report. J. Int. Med. Res. 2020, 48, 300060520974854. [Google Scholar] [CrossRef]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Butcher, N.J.; Mortimer, G.M.; Minchin, R.F. Drug delivery: Unravelling the stealth effect. Nat. Nanotechnol. 2016, 11, 310–311. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jantawong, C.; Priprem, A.; Intuyod, K.; Pairojkul, C.; Pinlaor, P.; Waraasawapati, S.; Mongkon, I.; Chamgramol, Y.; Pinlaor, S. Curcumin-loaded nanocomplexes: Acute and chronic toxicity studies in mice and hamsters. Toxicol. Rep. 2021, 8, 1346–1357. [Google Scholar] [CrossRef] [PubMed]
- Cicuéndez, M.; Casarrubios, L.; Barroca, N.; Silva, D.; Feito, M.J.; Diez-Orejas, R.; Marques, P.; Portolés, M.T. Benefits in the Macrophage Response Due to Graphene Oxide Reduction by Thermal Treatment. Int. J. Mol. Sci. 2021, 22, 6701. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Ma, J.; Ji, Z.; Shen, J.; Wang, Q. Recent Advances of Cell Membrane Coated Nanoparticles in Treating Cardiovascular Disorders. Molecules 2021, 26, 3428. [Google Scholar] [CrossRef]
- Chen, T.Y.; Chen, M.R.; Liu, S.W.; Lin, J.Y.; Yang, Y.T.; Huang, H.Y.; Chen, J.K.; Yang, C.S.; Lin, K.M. Assessment of Polyethylene Glycol-Coated Gold Nanoparticle Toxicity and Inflammation In Vivo Using NF-κB Reporter Mice. Int. J. Mol. Sci. 2020, 21, 8158. [Google Scholar] [CrossRef] [PubMed]
- Pandita, D.; Munjal, A.; Poonia, N.; Awasthi, R.; Kalonia, H.; Lather, V. Albumin-Coated Mesoporous Silica Nanoparticles of Docetaxel: Preparation, Characterization, and Pharmacokinetic Evaluation. Assay Drug Dev. Technol. 2021, 19, 226–236. [Google Scholar] [CrossRef]
- Zou, Y.; Ito, S.; Yoshino, F.; Suzuki, Y.; Zhao, L.; Komatsu, N. Polyglycerol Grafting Shields Nanoparticles from Protein Corona Formation to Avoid Macrophage Uptake. ACS Nano 2020, 14, 7216–7226. [Google Scholar] [CrossRef]
- Tavano, R.; Gabrielli, L.; Lubian, E.; Fedeli, C.; Visentin, S.; Polverino De Laureto, P.; Arrigoni, G.; Geffner-Smith, A.; Chen, F.; Simberg, D.; et al. C1q-Mediated Complement Activation and C3 Opsonization Trigger Recognition of Stealth Poly(2-methyl-2-oxazoline)-Coated Silica Nanoparticles by Human Phagocytes. ACS Nano 2018, 12, 5834–5847. [Google Scholar] [CrossRef]
- Lassenberger, A.; Scheberl, A.; Stadlbauer, A.; Stiglbauer, A.; Helbich, T.; Reimhult, E. Individually Stabilized, Superparamagnetic Nanoparticles with Controlled Shell and Size Leading to Exceptional Stealth Properties and High Relaxivities. ACS Appl. Mater. Interfaces 2017, 9, 3343–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.E.; Wang, Y.Y.; Yang, Q.; Hoang, T.; Chattopadhyay, S.; Hoen, T.; Ensign, L.M.; Nunn, K.L.; Schroeder, H.; McCallen, J.; et al. Anti-PEG antibodies alter the mobility and biodistribution of densely PEGylated nanoparticles in mucus. Acta Biomater. 2016, 43, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Li, M.; Qi, W.; Bai, H.; Nie, Z.; Hu, Z.; Xiao, Y.; de Bruijn, J.D.; Bao, C.; Yuan, H. Serial cellular events in bone formation initiated by calcium phosphate ceramics. Acta Biomater. 2021, 134, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yang, J.; Sun, G.; Hu, J.; Zhang, Q.; Cai, J.; Yuan, D.; Li, H.; Hei, Z.; Yao, W. Macrophage extracellular traps aggravate iron overload-related liver ischaemia/reperfusion injury. Br. J. Pharmacol. 2021, 178, 3783–3796. [Google Scholar] [CrossRef] [PubMed]
- Le, K.; Sun, J.; Khawaja, H.; Shibata, M.; Maggirwar, S.B.; Smith, M.R.; Gupta, M. Mantle cell lymphoma polarizes tumor-associated macrophages into M2-like macrophages, which in turn promote tumorigenesis. Blood Adv. 2021, 5, 2863–2878. [Google Scholar] [CrossRef]
- Li, J.; Sun, Z.; Luo, G.; Wang, S.; Cui, H.; Yao, Z.; Xiong, H.; He, Y.; Qian, Y.; Fan, C. Quercetin Attenuates Trauma-Induced Heterotopic Ossification by Tuning Immune Cell Infiltration and Related Inflammatory Insult. Front. Immunol. 2021, 12, 649285. [Google Scholar] [CrossRef]
- Dora, D.; Ferenczi, S.; Stavely, R.; Toth, V.E.; Varga, Z.V.; Kovacs, T.; Bodi, I.; Hotta, R.; Kovacs, K.J.; Goldstein, A.M.; et al. Evidence of a Myenteric Plexus Barrier and its Macrophage-Dependent Degradation during Murine Colitis: Implications in Enteric Neuroinflammation. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 1617–1641. [Google Scholar] [CrossRef]
- Schlundt, C.; El Khassawna, T.; Serra, A.; Dienelt, A.; Wendler, S.; Schell, H.; van Rooijen, N.; Radbruch, A.; Lucius, R.; Hartmann, S. Macrophages in bone fracture healing: Their essential role in endochondral ossification. Bone 2018, 106, 78–89. [Google Scholar] [CrossRef]
- Narla, R.K.; Modi, H.; Bauer, D.; Abbasian, M.; Leisten, J.; Piccotti, J.R.; Kopytek, S.; Eckelman, B.P.; Deveraux, Q.; Timmer, J.; et al. Modulation of CD47-SIRPα innate immune checkpoint axis with Fc-function detuned anti-CD47 therapeutic antibody. Cancer Immunol. Immunother. 2021, 71, 473–489. [Google Scholar] [CrossRef]
- Alaranji, G.; Goyal, A.; Bansal, P. Bisphosphonate Toxicity. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- McCloskey, E.; Paterson, A.H.; Powles, T.; Kanis, J.A. Clodronate. Bone 2021, 143, 115715. [Google Scholar] [CrossRef]
- Hanna, B.; McClanahan, F.; Yazdanparast, H.; Zaborsky, N.; Kalter, V.; Rößner, P.; Benner, A.; Dürr, C.; Egle, A.; Gribben, J. Depletion of CLL-associated patrolling monocytes and macrophages controls disease development and repairs immune dysfunction in vivo. Leukemia 2016, 30, 570–579. [Google Scholar] [CrossRef]
- Galletti, G.; Scielzo, C.; Barbaglio, F.; Rodriguez, T.V.; Riba, M.; Lazarevic, D.; Cittaro, D.; Simonetti, G.; Ranghetti, P.; Scarfò, L.; et al. Targeting Macrophages Sensitizes Chronic Lymphocytic Leukemia to Apoptosis and Inhibits Disease Progression. Cell Rep. 2016, 14, 1748–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piaggio, F.; Kondylis, V.; Pastorino, F.; Di Paolo, D.; Perri, P.; Cossu, I.; Schorn, F.; Marinaccio, C.; Murgia, D.; Daga, A.; et al. A novel liposomal Clodronate depletes tumor-associated macrophages in primary and metastatic melanoma: Anti-angiogenic and anti-tumor effects. J. Control. Release 2016, 223, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Braham, M.V.; Minnema, M.C.; Aarts, T.; Sebestyen, Z.; Straetemans, T.; Vyborova, A.; Kuball, J.; Öner, F.C.; Robin, C.; Alblas, J. Cellular immunotherapy on primary multiple myeloma expanded in a 3D bone marrow niche model. Oncoimmunology 2018, 7, e1434465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanz, H.L.; Saleh, A.; Kramer, B.; Cairns, J.; Ng, C.P.; Yu, J.; Trietsch, S.J.; Hankemeier, T.; Joore, J.; Vulto, P. Therapy response testing of breast cancer in a 3D high-throughput perfused microfluidic platform. BMC Cancer 2017, 17, 709. [Google Scholar] [CrossRef] [Green Version]
- Gould, S.E.; Junttila, M.R.; de Sauvage, F.J. Translational value of mouse models in oncology drug development. Nat. Med. 2015, 21, 431–439. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef]
- Song, G.; Suzuki, O.T.; Santos, C.M.; Lucas, A.T.; Wiltshire, T.; Zamboni, W.C. Gulp1 is associated with the pharmacokinetics of PEGylated liposomal doxorubicin (PLD) in inbred mouse strains. Nanomedicine 2016, 12, 2007–2017. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal. Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Fairfield, H.; Falank, C.; Avery, L.; Reagan, M.R. Multiple myeloma in the marrow: Pathogenesis and treatments. Ann. N. Y. Acad. Sci. 2016, 1364, 32–51. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Sun, W.; Wang, C.; Gu, Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv. Drug Deliv. Rev. 2016, 98, 19–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, L.; Guo, S.; Lin, C.M.; Liu, Q.; Huang, L. Nanoformulations for combination or cascade anticancer therapy. Adv. Drug Deliv. Rev. 2017, 115, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, S.; Wu, S.Y. Editorial: Advances and Challenges in Nanomedicine. Front. Pharmacol. 2018, 9, 1397. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Havel, H.; Finch, G.; Strode, P.; Wolfgang, M.; Zale, S.; Bobe, I.; Youssoufian, H.; Peterson, M.; Liu, M. Nanomedicines: From Bench to Bedside and Beyond. AAPS J. 2016, 18, 1373–1378. [Google Scholar] [CrossRef]
- Sainz, V.; Conniot, J.; Matos, A.I.; Peres, C.; Zupancic, E.; Moura, L.; Silva, L.C.; Florindo, H.F.; Gaspar, R.S. Regulatory aspects on nanomedicines. Biochem. Biophys. Res. Commun. 2015, 468, 504–510. [Google Scholar] [CrossRef]
- Grossman, J.H.; Crist, R.M.; Clogston, J.D. Early Development Challenges for Drug Products Containing Nanomaterials. AAPS J. 2017, 19, 92–102. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar]
- Tinkle, S.; McNeil, S.E.; Mühlebach, S.; Bawa, R.; Borchard, G.; Barenholz, Y.C.; Tamarkin, L.; Desai, N. Nanomedicines: Addressing the scientific and regulatory gap. Ann. N. Y. Acad. Sci. 2014, 1313, 35–56. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Types of Blood Cancers | Origin | New Cases | Deaths |
|---|---|---|---|
| Est. in 2022 | Est. in 2022 | ||
| Multiple myeloma (MM) | B cells (plasma cells) | 34,470 | 12,640 |
| Acute myeloid leukaemia (AML) | Myeloid lineage hematopoietic precursors | 20,050 | 11,540 |
| B cell lymphoma (BL) | B cells and lymphocytes | 80,910 | 21,170 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Lin, W.; Zhu, L. Targeted Drug Delivery for the Treatment of Blood Cancers. Molecules 2022, 27, 1310. https://doi.org/10.3390/molecules27041310
Jiang Y, Lin W, Zhu L. Targeted Drug Delivery for the Treatment of Blood Cancers. Molecules. 2022; 27(4):1310. https://doi.org/10.3390/molecules27041310
Chicago/Turabian StyleJiang, Yao, Weifeng Lin, and Linyi Zhu. 2022. "Targeted Drug Delivery for the Treatment of Blood Cancers" Molecules 27, no. 4: 1310. https://doi.org/10.3390/molecules27041310
APA StyleJiang, Y., Lin, W., & Zhu, L. (2022). Targeted Drug Delivery for the Treatment of Blood Cancers. Molecules, 27(4), 1310. https://doi.org/10.3390/molecules27041310