
Identification of α-Glucosidase Inhibitors from Scutellaria edelbergii: ESI-LC-MS and Computational Approach

 ,
, 
 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Result and Discussion
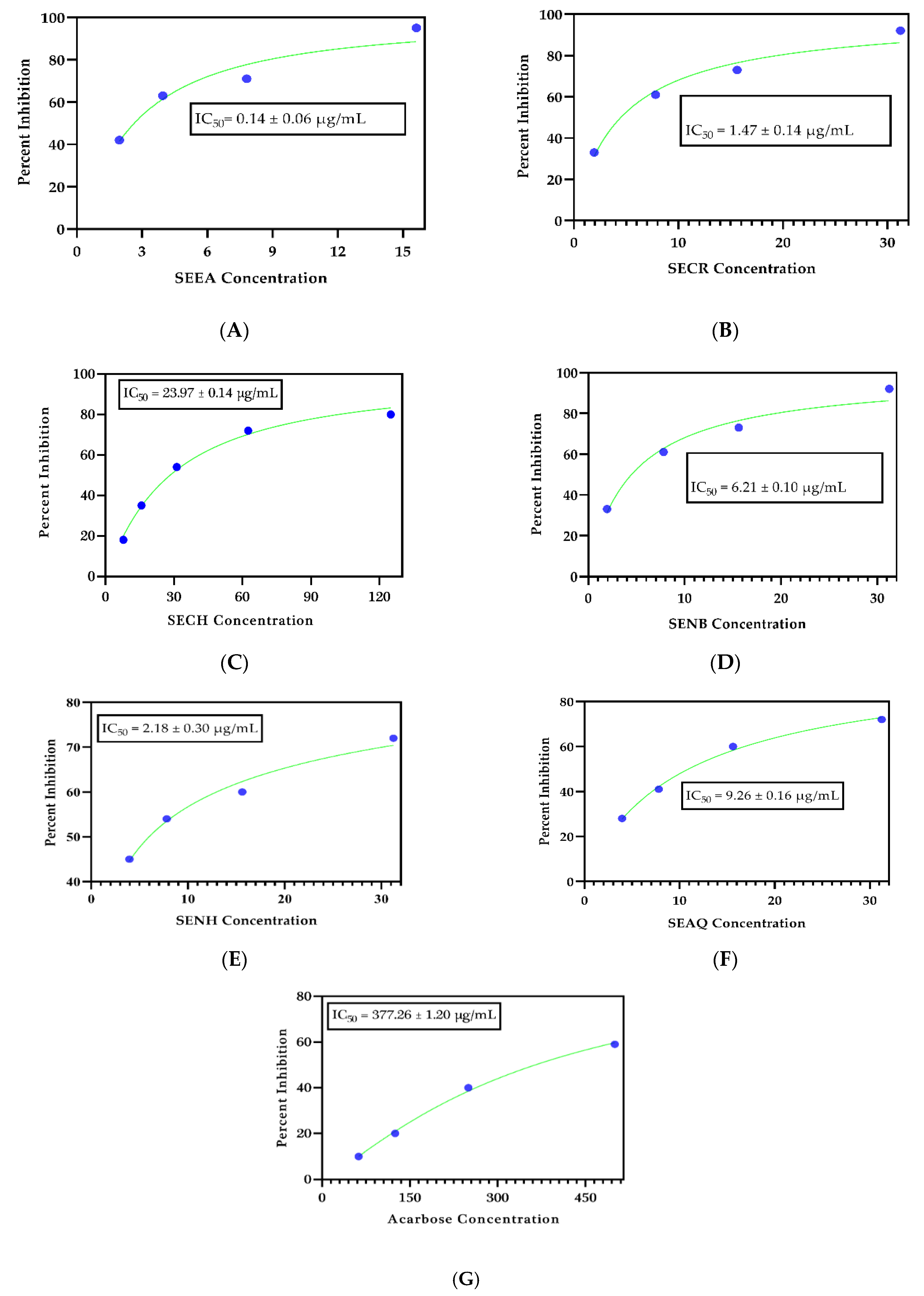
2.1. Antidiabetic Significance
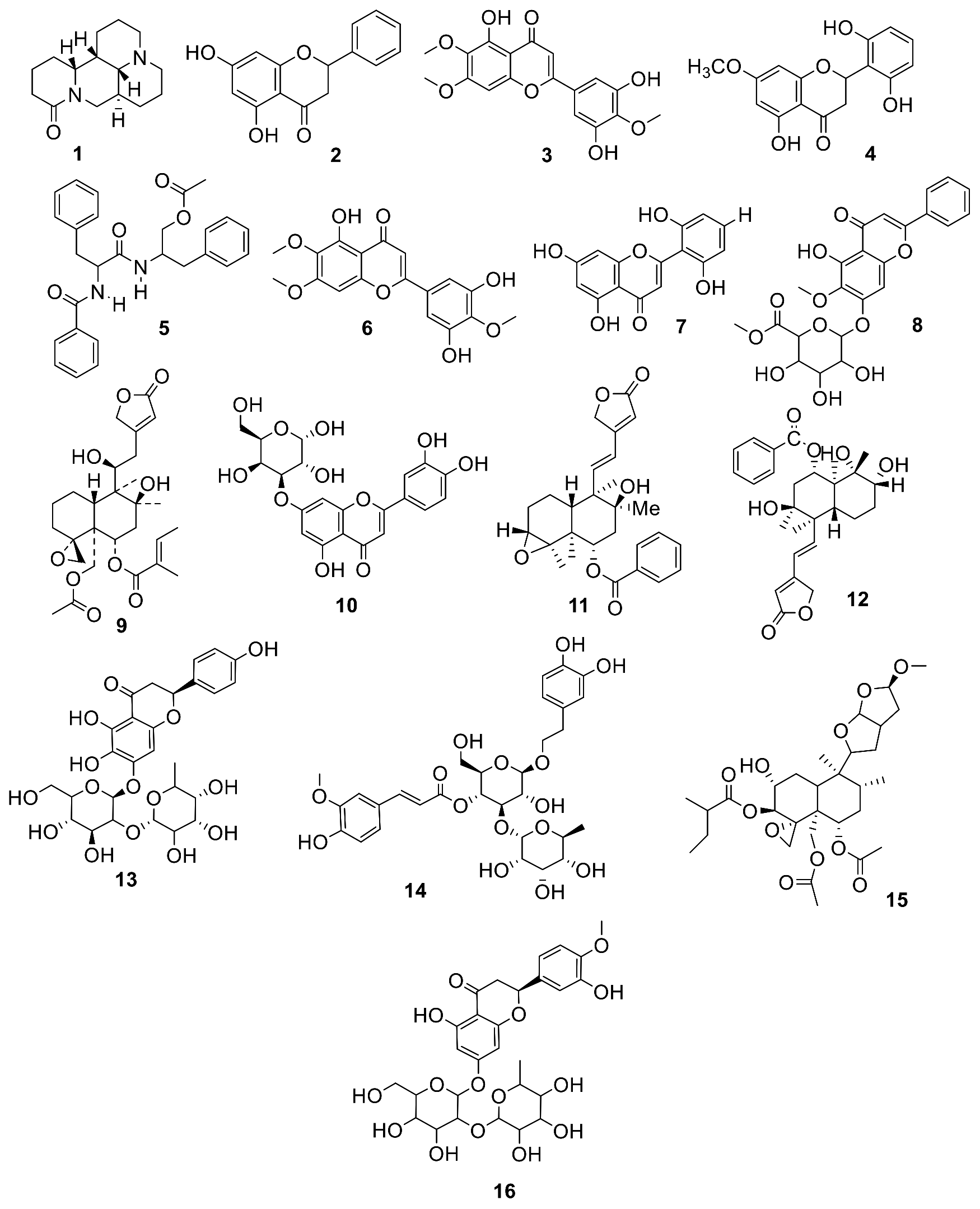
ESI-LC-MS Analysis
2.2. Computational Analysis
2.2.1. Database of the Compounds Identified in EtOAc Extract
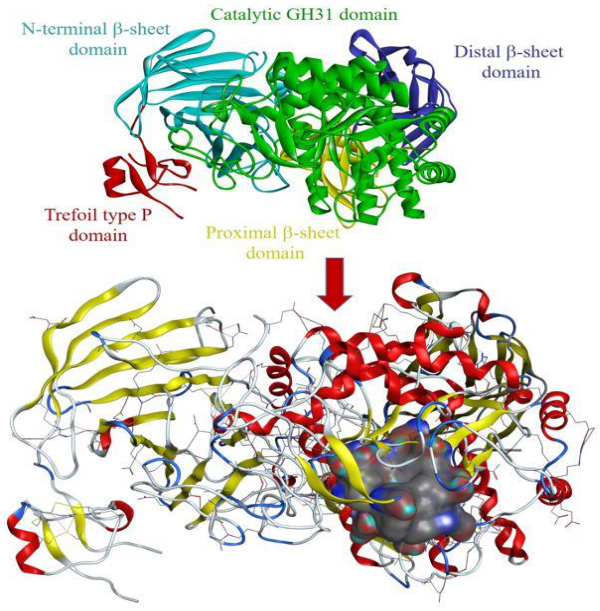
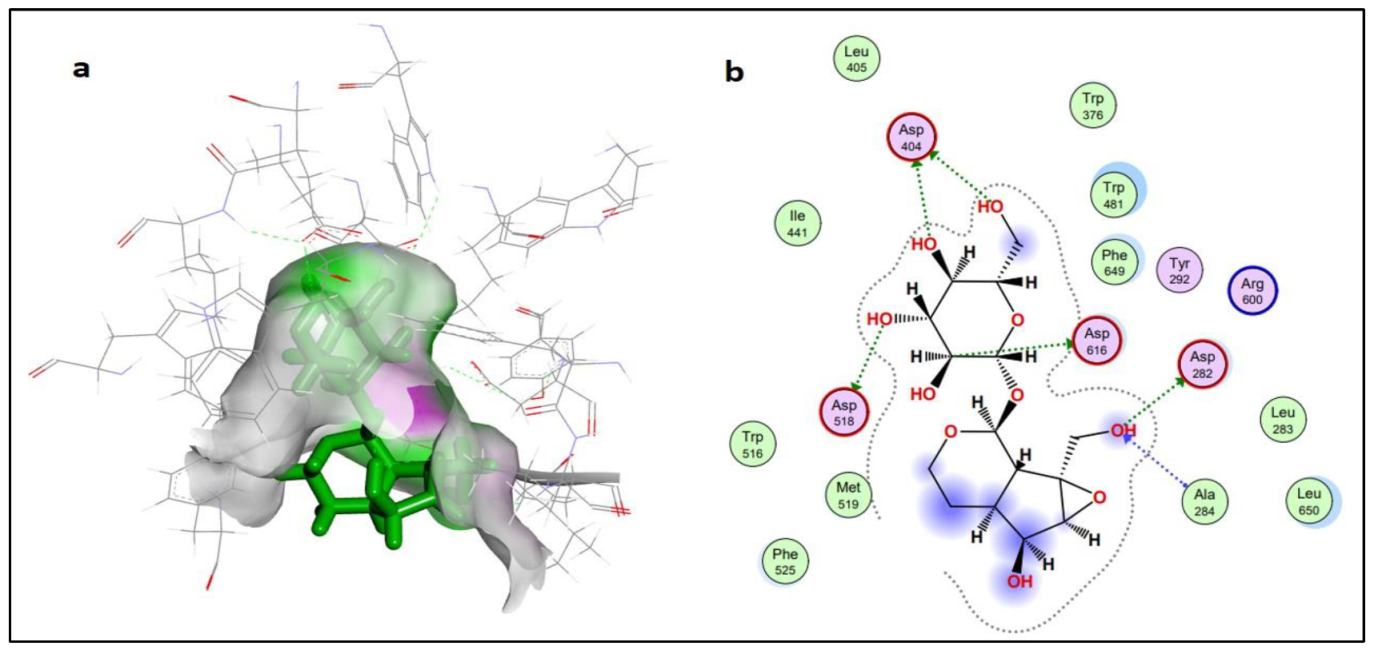
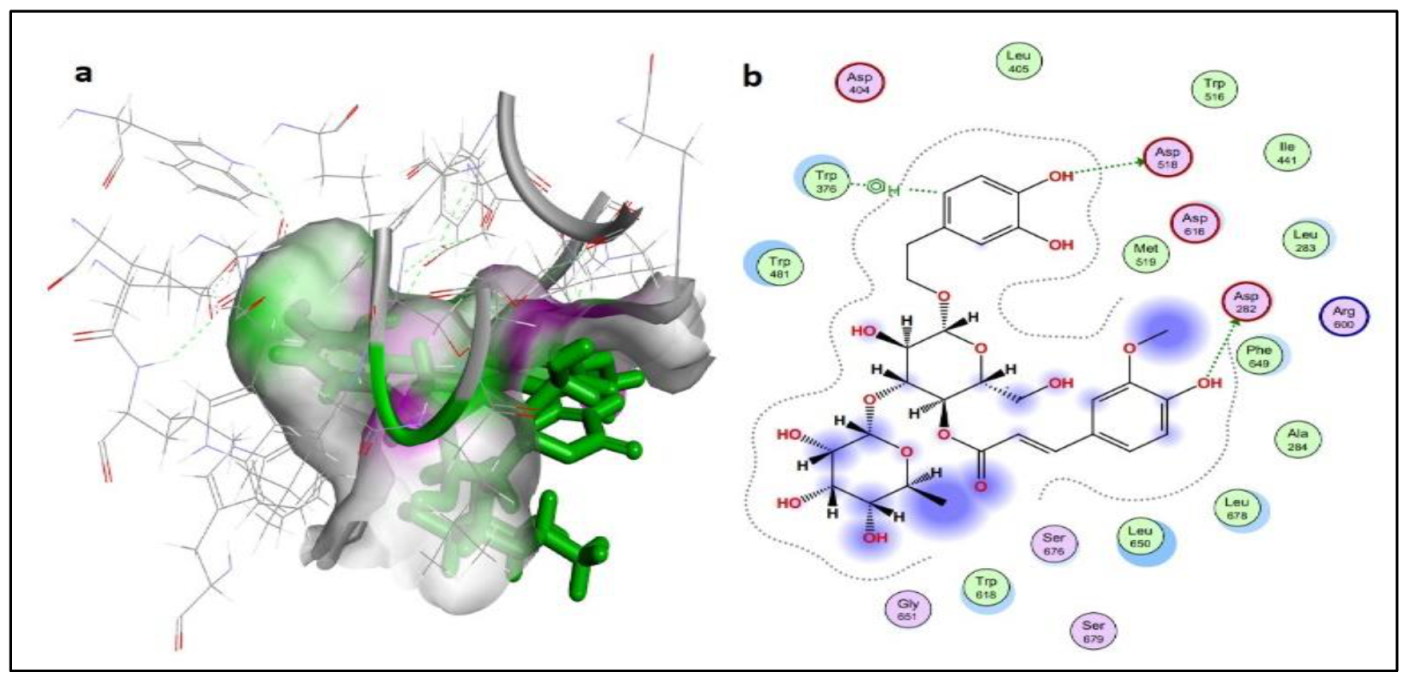
2.2.2. Protein-Ligand Interaction Analysis
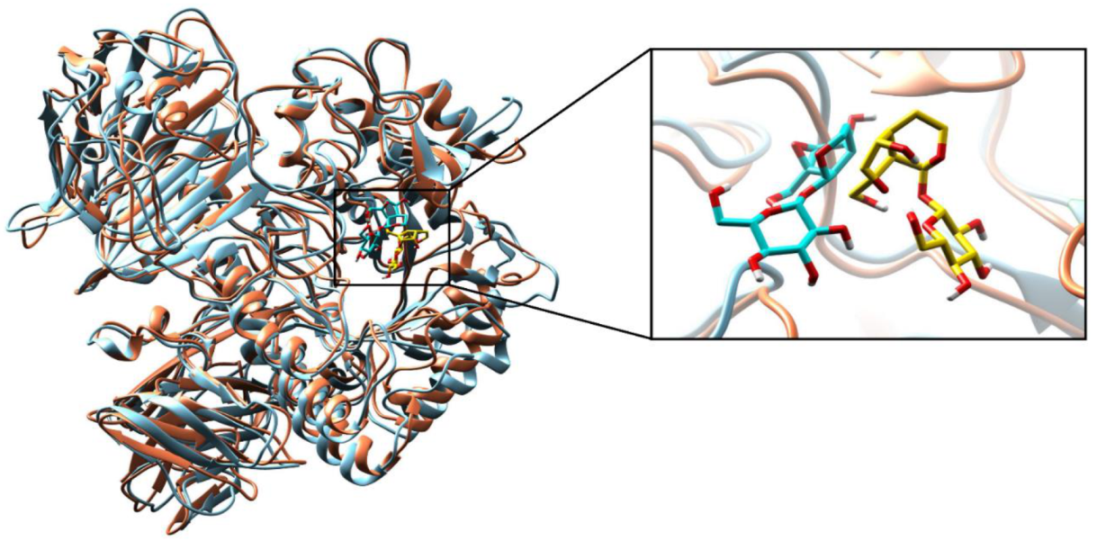
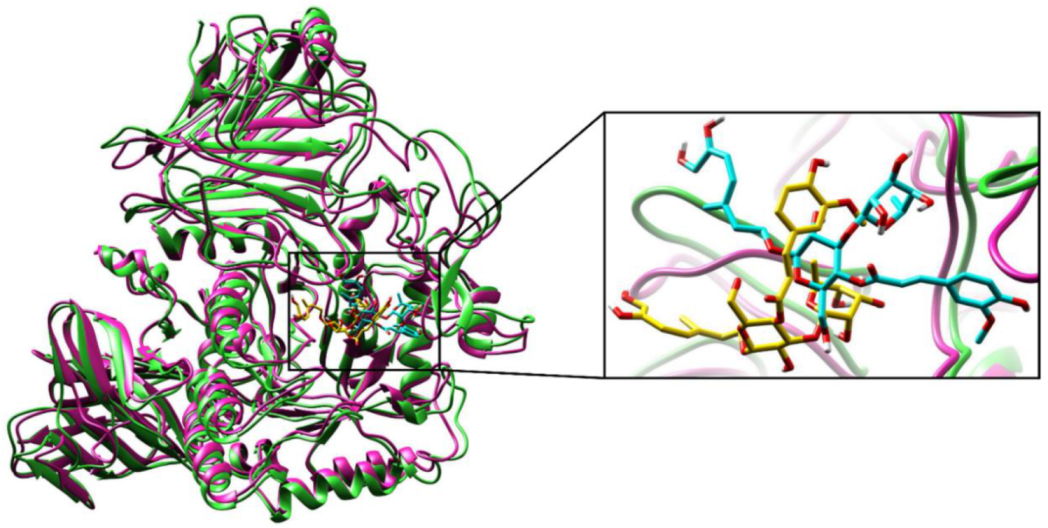
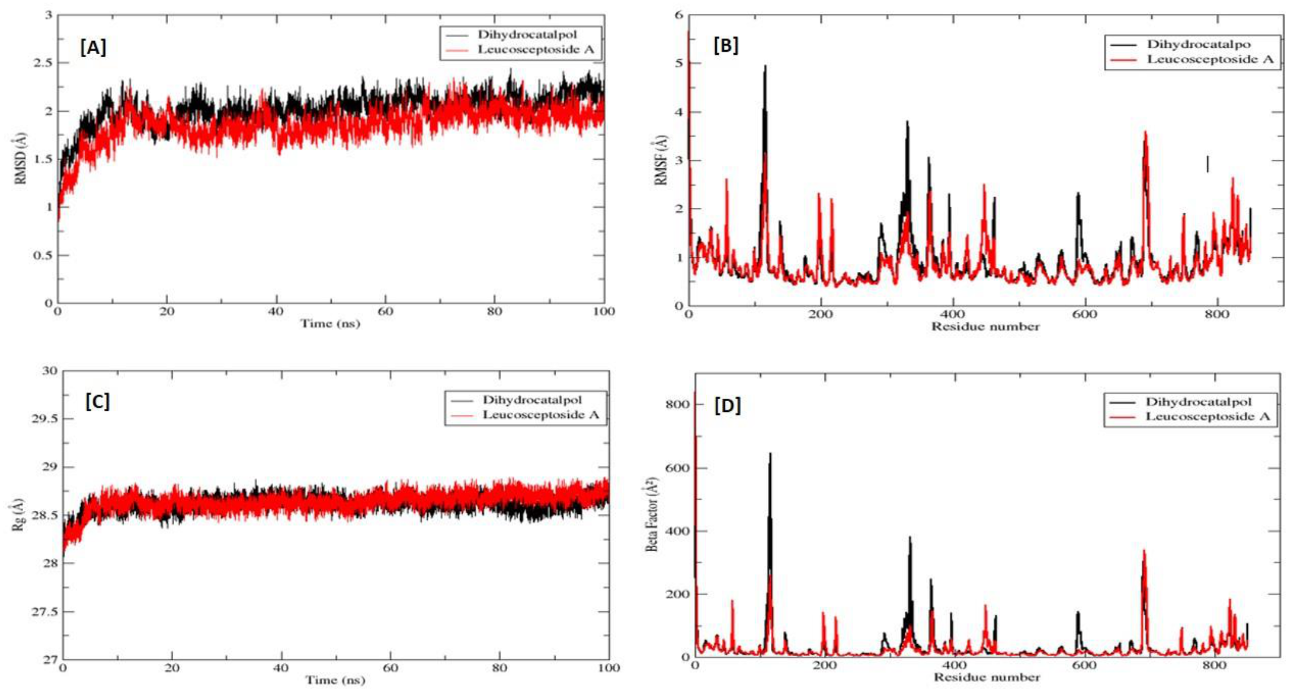
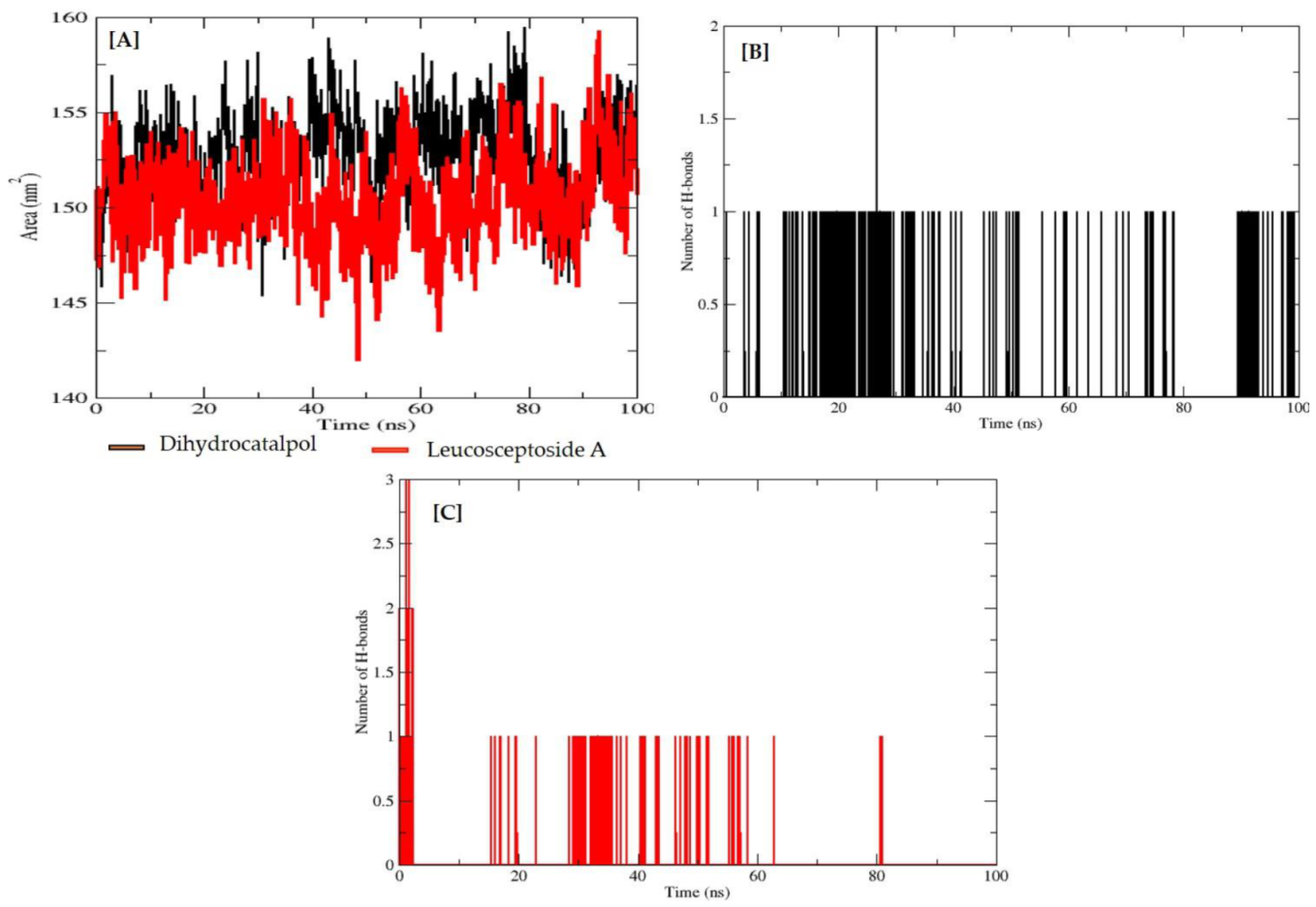
2.2.3. Molecular Dynamics (MD) Simulations
2.2.4. Binding Free Energy Calculations (MM-GBSA/ MM-PBSA)
2.2.5. Pharmacokinetic /ADMET Profile Estimation
3. Material and Methods
3.1. Apparatus Used
3.2. Plant Collection and Identification
3.3. Extraction and Fractionation
3.4. α-Glucosidase Inhibitory Assay
3.5. LC-MS/MS Analysis
3.6. Computational Analysis
3.6.1. Construction of Chemical Database for In Silico Screening
3.6.2. Selection of Target Protein
3.6.3. Protein-Ligand Docking and Interactions Analysis
3.6.4. Molecular Dynamics Simulations
3.6.5. Binding Free Energy (BFE) Estimation
3.6.6. Pharmacokinetic /ADMET Profile Estimation
3.7. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bruno, M.; Piozzi, F.; Rosselli, S. Natural and hemisynthetic neoclerodane diterpenoids from Scutellaria and their antifeedant activity. Nat. Prod. Rep. 2002, 19, 357–378. [Google Scholar] [CrossRef]
- Shang, X.; He, X.; He, X.; Li, M.; Zhang, R.; Fan, P.; Zhang, Q.; Jia, Z. The genus Scutellaria an ethnopharmacological and phytochemical review. J. Ethnopharmacol. 2010, 128, 279–313. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Murad, W.; Ur Rehman, N.; Halim, S.A.; Ahmed, M.; Rehman, H.; Zahoor, M.; Mubin, S.; Khan, A.; Nassan, M.A. Biomedical applications of Scutellaria edelbergii Rech. f.: In vitro and in vivo approach. Molecules 2021, 26, 3740. [Google Scholar] [CrossRef] [PubMed]
- Majid, A.; Ahmad, H.; Saqib, Z.; Ali, H. Potential distribution of endemic Scutellaria chamaedrifolia; Geographic Information System and statistical model approach. Pak. J. Bot. 2015, 47, 51–56. [Google Scholar]
- Asgarpanah, J.; Kazemivash, N. Phytochemistry, pharmacology and medicinal properties of Carthamus tinctorius L. Chin. J. Integr. Med. 2013, 19, 153–159. [Google Scholar] [CrossRef]
- Rasul, A.; Millimouno, F.M.; Ali Eltayb, W.; Ali, M.; Li, J.; Li, X. Pinocembrin: A novel natural compound with versatile pharmacological and biological activities. BioMed Res. Int. 2013, 2013, 379850. [Google Scholar] [CrossRef] [PubMed]
- Krishna, P.M.; KNV, R.; Banji, D. A review on phytochemical, ethnomedical and pharmacological studies on genus Sophora, Fabaceae. Rev. Bras. Farmacogn. 2012, 22, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Waisundara, V.Y.; Hsu, A.; Huang, D.; Tan, B.K.-H. Scutellaria baicalensis enhances the anti-diabetic activity of metformin in streptozotocin-induced diabetic Wistar rats. Am. J. Chinese Med. 2008, 36, 517–540. [Google Scholar] [CrossRef]
- Salehi, B.; Ata, A.; V Anil Kumar, N.; Sharopov, F.; Ramírez-Alarcón, K.; Ruiz-Ortega, A.; Abdulmajid Ayatollahi, S.; Valere Tsouh Fokou, P.; Kobarfard, F.; Amiruddin Zakaria, Z. Antidiabetic potential of medicinal plants and their active components. Biomolecules 2019, 9, 551. [Google Scholar] [CrossRef] [Green Version]
- Tomimori, T.; Miyaichi, Y.; Imoto, Y.; Kizu, H.; Namba, T. Studies on the nepese crude drugs. VI.: On the flavonoid constituents of the root of Scutellaria discolor Colebr.(2). Chem. Pharm. Bull. 1986, 34, 406–408. [Google Scholar] [CrossRef] [Green Version]
- De Smet, P. Scutellaria Species. In Adverse Effects of Herbal Drugs 2; Springer: Berlin/Heidelberg, Germany, 1993; pp. 289–296. [Google Scholar]
- Hu, B.; Liu, Y. Studies on the structures of new flavonoids from the root of Scutellaria amoena. Acta pharm. Sin. 1989, 24, 200–206. [Google Scholar]
- Kikuchi, Y.; Miyaichi, Y.; Yamaguchi, Y.; Kizu, H.; Tomimori, T. Studies on the nepalese crude drugs. XII. On the phenolic compounds from the root of Scutellaria prostrata Jacq. ex Benth. Chem. Pharm. Bull. 1991, 39, 1047–1050. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-H.; Zhang, Y.-J.; Yang, C.-R. New flavonoid glycosides from Scutellaria amoena. Plant Sci. J. 1999, 6, 305–310. [Google Scholar]
- Dehkordi, F.J.; Kharazian, N.; Lorigooini, Z. Characterization of flavonoid components in Scutellaria L. species (Lamiaceae) using finger-printing analysis. Acta Biol. Crac. Ser. Bot. 2020, 62, 79–96. [Google Scholar]
- Rashid, M.; Fareed, M.; Rashid, H.; Aziz, H.; Ehsan, N.; Khalid, S.; Ghaffar, I.; Ali, R.; Gul, A.; Hakeem, K.R. Flavonoids, and their biological secrets. Plant and Hum. Health 2019, 2, 579–605. [Google Scholar]
- Lin, Y.-L.; Kuo, Y.-H.; Cheng, M.-C.; Wang, Y. Structures of scutellones D and E determined from X-ray diffraction, spectral and chemical evidence. Neoclerodane-type diterpenoids from Scutellaria rivularis WALL. Chem. Pharm. Bull. 1988, 36, 2642–2646. [Google Scholar] [CrossRef] [Green Version]
- Hussain, H.; Ahmad, V.U.; Anwar, S.; Miana, G.A.; Krohn, K. Chemical constituents of Scutellaria linearis. Biochem. Syst. Ecol. 2008, 5, 490–492. [Google Scholar] [CrossRef]
- Muñoz, D.M.; Maria, C.; Rodríguez, B.; Simmonds, M.S.; Blaney, W.M. Neo-clerodane insect antifeedants from Scutellaria alpina subsp. javalambrensis. Phytochemistry 1997, 44, 593–597. [Google Scholar] [CrossRef]
- Chen, Q.; Rahman, K.; Wang, S.-J.; Zhou, S.; Zhang, H. Scutellaria barbata: A review on chemical constituents, pharmacological activities, and clinical applications. Curr. Pharm. Des. 2020, 26, 160–175. [Google Scholar] [CrossRef]
- Denaro, M.; Smeriglio, A.; Trombetta, D. Antioxidant and anti-inflammatory activity of citrus flavanones mix and its stability after in vitro simulated digestion. Antioxidant 2021, 10, 140. [Google Scholar] [CrossRef]
- Ma, S.-C.; Du, J.; But, P.P.-H.; Deng, X.-L.; Zhang, Y.-W.; Ooi, V.E.-C.; Xu, H.-X.; Lee, S.H.-S.; Lee, S.F. Antiviral Chinese medicinal herbs against respiratory syncytial virus. J. Ethnopharmacol. 2002, 79, 205–211. [Google Scholar] [CrossRef]
- Lin, Y.-L. Aurantiamide from the aerial parts of Scutellaria rivularis. Planta Med. 1987, 53, 507–508. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.W.; Kim, Y.T. Dried root of Rehmannia glutinosa prevents bone loss in ovariectomized rats. Molecules 2013, 18, 5804–5813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, A.; Ramirez, J.E.; Areche, C.; Sepúlveda, B.; Simirgiotis, M.J. HPLC-UV-MS profiles of phenolic compounds and antioxidant activity of fruits from three citrus species consumed in Northern Chile. Molecules 2014, 19, 17400–17421. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hirotani, M.; Yoshikawa, T.; Furuya, T. Flavonoids and phenylethanoids from hairy root cultures of Scutellaria baicalensis. Phytochemistry 1997, 44, 83–87. [Google Scholar] [CrossRef]
- Ismail, S.; Ahmad, S.; Azam, S.S. Immunoinformatics characterization of SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine. J. Mol. Liq. 2020, 314, 113612. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; Sakata, K. Current status of computer-aided drug design for type 2 diabetes. Curr. Comp. Aided Drug Des. 2016, 12, 167–177. [Google Scholar] [CrossRef]
- Shaker, B.; Ahmad, S.; Thai, T.D.; Eyun, S.-i.; Na, D. Rational drug design for Pseudomonas aeruginosa PqsA enzyme: An in silico guided study to block biofilm formation. Front. Mol. Biosci. 2020, 7, 284. [Google Scholar] [CrossRef]
- Kausar, N.; Ullah, S.; Khan, M.A.; Zafar, H.; Choudhary, M.I.; Yousuf, S. Celebrex derivatives: Synthesis, α-glucosidase inhibition, crystal structures and molecular docking studies. Bioorg. Chem. 2021, 106, 104499. [Google Scholar] [CrossRef]
- Khan, I.; Rahman, H.; Abd El-Salam, N.M.; Tawab, A.; Hussain, A.; Khan, T.A.; Khan, U.A.; Qasim, M.; Adnan, M.; Azizullah, A. Punica granatum peel extracts: HPLC fractionation and LC-MS analysis to quest compounds having activity against multidrug resistant bacteria. BMC Complement. Altern. Med. 2017, 17, 1–6. [Google Scholar] [CrossRef]
- De Sousa Luisa, J.A.; Barrosb, R.P.C.; de Sousab, N.F.; Muratovc, E.; Scottib, L.; Scottib, M.T. Virtual screening of natural products database. Mini Rev. Med. Chem. 2021, 21, 2657–2730. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, 1202–1213. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Dirir, A.M.; Daou, M.; Yousef, A.F.; Yousef, L.F. A review of alpha-glucosidase inhibitors from plants as potential candidates for the treatment of type-2 diabetes. Phytochem. Rev. 2021, 1–31. [Google Scholar] [CrossRef]
- Roig-Zamboni, V.; Cobucci-Ponzano, B.; Iacono, R.; Ferrara, M.C.; Germany, S.; Bourne, Y.; Parenti, G.; Moracci, M.; Sulzenbacher, G. Structure of human lysosomal acid α-glucosidase–a guide for the treatment of Pompe disease. Nat. Comm. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry, and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Yousafi, Q.; Batool, J.; Khan, M.S.; Perveen, T.; Sajid, M.W.; Hussain, A.; Mehmood, A.; Saleem, S. In Silico Evaluation of Food Derived Bioactive Peptides as Inhibitors of Angiotensin Converting Enzyme (ACE). Int. J. Pept. Res. Ther. 2021, 27, 341–349. [Google Scholar] [CrossRef]
- Aldeghi, M.; Heifetz, A.; Bodkin, M.J.; Knapp, S.; Biggin, P.C. Accurate calculation of the absolute free energy of binding for drug molecules. Chem. Sci. 2016, 7, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Ranaghan, K.E.; Azam, S. Combating tigecycline resistant Acinetobacter baumannii: A leap forward towards multi-epitope-based vaccine discovery. Eur. J. Pharm. Sci. 2019, 132, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.-S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; McGee Jr, T.D.; Pearlman, D.A.; Radak, B.K.; Tao, Y. Alchemical binding free energy calculations in AMBER20: Advances and best practices for drug discovery. J. Chem. Inf. Model. 2020, 60, 5595–5623. [Google Scholar] [CrossRef]
- Bergonzo, C.; Cheatham III, T.E. Improved force field parameters lead to a better description of RNA structure. J. Chem. Theory Comput. 2015, 11, 3969–3972. [Google Scholar] [CrossRef]
- Hyun, T.K.; Eom, S.H.; Kim, J.-S. Molecular docking studies for discovery of plant-derived a-glucosidase inhibitors. Plant Omics 2014, 7, 166–170. [Google Scholar]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. Ptraj and Cpptraj: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Grap. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miller III, B.R.; McGee Jr, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Khan, M.S.; Mehmood, B.; Yousafi, Q.; Bibi, S.; Fazal, S.; Saleem, S.; Sajid, M.W.; Ihsan, A.; Azhar, M.; Kamal, M.A. Molecular Docking Studies Reveal Rhein from rhubarb (Rheum rhabarbarum) as a Putative Inhibitor of ATP-binding Cassette Super-family G member 2. Med. Chem. 2021, 17, 273–288. [Google Scholar] [CrossRef]
- Avdović, E.H.; Petrović, I.P.; Stevanović, M.J.; Saso, L.; Dimitrić Marković, J.M.; Filipović, N.D.; Živić, M.Ž.; Cvetić Antić, T.N.; Žižić, M.V.; Todorović, N.V. Synthesis and Biological Screening of New 4-Hydroxycoumarin Derivatives and Their Palladium (II) Complexes. Oxid. Med. Cell. Longev. 2021, 2021, 8849568. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. Data Warrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Avdović, E.H.; Milanović, Ž.B.; Molčanov, K.; Roca, S.; Vikić-Topić, D.; Mrkalić, E.M.; Jelić, R.M.; Marković, Z.S. Synthesis, characterization and investigating the binding mechanism of novel coumarin derivatives with human serum albumin: Spectroscopic and computational approach. J. Mol. Struct. 2022, 1254, 132366. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | RT (Min) | MW IM | Fragmentation | Tentative Identification | Reference Species | Class |
|---|---|---|---|---|---|---|
| 1 | 4.87 | [249]+ | 240.92, 234.17 | Isomatrine | S. flavescens [22] | Alkaloid |
| 2 | 5.15 | [255]− | 245.25, 237.17 | Pinocembrin | S. altissima [10] | Flavonoid |
| 3 | 5.35 | [285]− | 270.08, 267.08 | 5,7,2,6-Tetrahydroxy-flavone | S. baicalensis [11] | Flavonoid |
| 4 | 5.50 | [301]− | 286.08, 284.0 | Scuteamoenin | S. amoena [12] | Flavonoid |
| 5 | 5.74 | [365]+ | 350.08, 347.25 | Dihydrocatalpol | S. albida [18] | Terpenoid |
| 6 | 6.08 | [359]− | 344.08, 315.17 | 5,2,5-Trihydroxy-6,7,8-trimethoxyflavone | S. baicalensis [11] | Flavonoid |
| 7 | 6.43 | [445]+ | 430.17, 427.25 | Aurantiamide acetate | S. rivularis [23] | Alkaloid |
| 8 | 6.53 | [475]+ | 460.25, 457.17 | OroxylinA-7-O-d-glucuronopyranosidemethylester | S. amoena [14] | Flavonoid |
| 9 | 6.97 | [507]+ | 489.17, 475.25 | Scutalpin C | S. alpina [19] | Terpenoid |
| 10 | 7.04 | [447]− | 429.17, 403.17 | Luteloin-7-O-d-glucuronopyranoside | S. prostrata [13] | Flavonoid |
| 11 | 7.17 | [451]− | 433.25, 407.25 | Scutellone F | S. rivularis [17] | Terpenoid |
| 12 | 7.31 | [469]− | 451.17, 425.17 | Scutellone D | S. rivularis [17] | Terpenoid |
| 13 | 7.43 | [595]+ | 565.25, 523.42 | Scutellarein-7-O-neohesperidoside | S. multicaulis [15] | Flavonoid |
| 14 | 7.72 | [639]+ | 621.42, 567.42 | Leucosceptoside A | S. baicalensis [26] | Phenol |
| 15 | 8.02 | [581]− | 545.08, 519.25 | Lupulin A | S. linearis [18] | Terpenoid |
| 16 | 8.24 | [609]− | 577.33, 565.08 | Hesperetin-7-O-neohesperidoside | S. multicaulis [15] | Flavonoid |
| No. | Dock Score | RMSD | Interacting Residues in the Binding Pocket | ||||
|---|---|---|---|---|---|---|---|
| Ligand | Receptor | Interaction | Distance | E (kcal/mol) | |||
| 1 | −5.7671 | 0.81 | - | - | - | - | - |
| 2 | −5.6933 | 0.97 | C6 O12 | SD:MET519(A) OD1:ASP404(A) | H-donor H-donor | 3.76 3.06 | −0.5 −3.0 |
| 3 | −5.6735 | 2.40 | O20 | OD1:ASP518(A) | H-donor | 2.93 | −3.2 |
| 4 | −6.1364 | 1.22 | O20 | OD1:ASP518(A) | H-donor | 2.87 | −2.1 |
| 5 | −6.6079 | 1.09 | O14 O20 O21 O22 O28 O28 | OD2:ASP616(A) OD1:ASP518(A) OD1:ASP404(A) OD1:ASP404(A) OD1:ASP282(A) N:ALA284(A) | H-donor H-donor H-donor H-donor H-donor H-acceptor | 3.15 2.99 3.03 3.00 2.93 3.12 | −0.5 −1.1 −0.9 −3.0 −0.9 −0.7 |
| 6 | −6.523 | 1.64 | - | - | - | - | - |
| 7 | −7.1585 | 1.63 | O27 | N:ALA284(A) | H-acceptor | 3.17 | −0.8 |
| 8 | −7.1405 | 1.09 | C17 O19 | OD1:ASP282(A) N:ALA284(A) | H-donor H-acceptor | 3.28 3.04 | −0.7 −1.6 |
| 9 | −7.0130 | 1.95 | - | - | - | - | - |
| 10 | −6.9602 | 1.15 | O20 O30 O19 | O:ASN524(A) OD2:ASP616(A) N:ALA284(A) | H-donor H-donor H-acceptor | 3.00 2.97 3.23 | −2.7 −2.3 −1.0 |
| 11 | −6.8364 | 1.60 | O20 | OD1:ASP282(A) | H-donor | 3.02 | −1.9 |
| 12 | −6.4003 | 1.95 | O33 | NH1:ARG600(A) | H-acceptor | 3.16 | −1.4 |
| 13 | −8.2278 | 2.17 | O38 | OD2:ASP282(A) | H-donor | 3.35 | −0.5 |
| 14 | −7.5862 | 2.08 | O29 O45 C28 | OD1:ASP518(A) OD1:ASP282(A) 6ring:TRP376(A) | H-donor H-donor H-pi | 3.32 2.78 4.54 | −0.5 −3.4 −0.6 |
| 15 | −77456 | 2.33 | - | - | - | - | - |
| 16 | −8.169 | 2.96 | O35 | OD1:ASP518(A) | H-donor | 2.17 | −1.2 |
| C. No | MM/GBSA Model | MM/PBSA Model | ||||||
|---|---|---|---|---|---|---|---|---|
| 5 | VDWAALS | −33.8515 | 2.3527 | 0.2353 | VDWAALS | −33.8515 | 2.3527 | 0.2353 |
| EEL | −44.2017 | 85.0494 | 0.5049 | EEL | −44.2017 | 5.0494 | 0.5049 | |
| EGB | −56.4345 | 4.7492 | 0.4749 | EPB | 71.4721 | 6.3198 | 0.6321 | |
| ESURF | −4.6737 | 0.3065 | 0.0306 | ENPOLAR | −3.9887 | 0.1867 | 0.0187 | |
| EDISPER | 0 | 0 | 0 | |||||
| ΔG gas | −78.0531 | 5.3641 | 0.5364 | ΔG gas | −78.0531 | 5.3641 | 0.5364 | |
| ΔG solv | 51.7608 | 4.6677 | 0.4668 | ΔG solv | 67.4834 | 6.2590 | 0.6259 | |
| ΔTOTAL | −26.2923 | 2.3706 | 0.2371 | ΔTOTAL | −10.5697 | 4.2572 | 0.4257 | |
| 14 | VDWAALS | −26.1053 | 1.7151 | 0.1715 | VDWAALS | −26.1053 | 1.7151 | 0.1715 |
| EEL | −14.9926 | 4.3080 | 0.4308 | EEL | −14.9926 | 4.3080 | 0.4308 | |
| EGB | 25.7016 | 4.7242 | 0.4724 | EPB | 28.1595 | 5.1235 | 0.5123 | |
| ESURF | −2.9626 | 0.1831 | 0.0183 | ENPOLAR | −2.4337 | 0.0970 | 0.0097 | |
| EDISPER | 0 | 0 | 0 | |||||
| ΔG gas | −41.0979 | 5.3198 | 0.5320 | ΔG gas | −41.0979 | 5.3198 | 0.5320 | |
| ΔG solv | 22.7391 | 4.6019 | 0.4602 | ΔG solv | 25.7258 | 5.0694 | 0.5069 | |
| ΔTOTAL | −18.3588 | 1.6221 | 0.1622 | ΔTOTAL | −15.3721 | 2.0689 | 0.2069 | |
| Chemical Parameters | Dihydrocatalpol | Leucosceptoside A |
|---|---|---|
| Physicochemical Properties | ||
| Molecular weight (MW) (g/mol) | 364.35 | 638.61 |
| Rotatable bonds | 4 | 12 |
| Hydrogen bond acceptors (HBA) | 10 | 15 |
| Hydrogen bond donors (HBD) | 6 | 8 |
| Molar Refractivity (MR) | 77.11 | 152.89 |
| Total polar surface area (TPSA) (Å) | 161.60 | 234.29 |
| Bioavailability Score | 0.55 | 0.17 |
| Lipophilicity | ||
| Log Po/w (iLOGP) | 1.38 | 2.78 |
| LogPo/w (XLOGP3) | −3.34 | −0.18 |
| LogPo/w (WLOGP) | −3.71 | −0.82 |
| Lpg Po/w (MLOGP) | −2.81 | −2.18 |
| LogPo/w (SILICOS-IT) | −2.50 | −0.58 |
| Consensus Log Po/w | −2.20 | −0.20 |
| Water Solubility | ||
| Class | Highly soluble | Soluble |
| Pharmacokinetics | ||
| GI absorption | Low | Low |
| BBB permeant | No | No |
| P-gp substrate | Yes | Yes |
| CYP1A2 inhibitor | No | No |
| CYP2C19 Inhibitor | No | No |
| CYP2C9 inhibitor | No | No |
| CYP2D6 inhibitor | No | No |
| CYP3A4 inhibitor | No | No |
| Log Kp (skin permeation) (cm/s) | −10.89 | −10.32 |
| Toxicity estimation | ||
| Mutagenicity | Toxic effects | No toxic effects |
| Tumorigenicity | No toxic effects | No toxic effects |
| Reproductive effects | No toxic effects | No toxic effects |
| Irritant effects | No toxic effects | No toxic effects |
| Medicinal chemistry-related properties | ||
| PAINS | No alert | I alert Catechol_A |
| Brenk | 1 alert Three_membered_ heterocycle | 2 alert Catechol, Micheal_acceptor_1 |
| Synthetic accessibility | 5.39 | 6.47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, M.; Rahman, H.; Khan, A.; Bibi, S.; Ullah, O.; Ullah, S.; Ur Rehman, N.; Murad, W.; Al-Harrasi, A. Identification of α-Glucosidase Inhibitors from Scutellaria edelbergii: ESI-LC-MS and Computational Approach. Molecules 2022, 27, 1322. https://doi.org/10.3390/molecules27041322
Shah M, Rahman H, Khan A, Bibi S, Ullah O, Ullah S, Ur Rehman N, Murad W, Al-Harrasi A. Identification of α-Glucosidase Inhibitors from Scutellaria edelbergii: ESI-LC-MS and Computational Approach. Molecules. 2022; 27(4):1322. https://doi.org/10.3390/molecules27041322
Chicago/Turabian StyleShah, Muddaser, Hazir Rahman, Ajmal Khan, Shabana Bibi, Obaid Ullah, Saeed Ullah, Najeeb Ur Rehman, Waheed Murad, and Ahmed Al-Harrasi. 2022. "Identification of α-Glucosidase Inhibitors from Scutellaria edelbergii: ESI-LC-MS and Computational Approach" Molecules 27, no. 4: 1322. https://doi.org/10.3390/molecules27041322
APA StyleShah, M., Rahman, H., Khan, A., Bibi, S., Ullah, O., Ullah, S., Ur Rehman, N., Murad, W., & Al-Harrasi, A. (2022). Identification of α-Glucosidase Inhibitors from Scutellaria edelbergii: ESI-LC-MS and Computational Approach. Molecules, 27(4), 1322. https://doi.org/10.3390/molecules27041322