
Anti-Neuroinflammatory Components from Clausena lenis Drake




Abstract
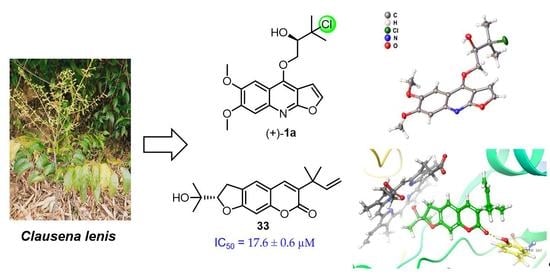
:
1. Introduction
2. Results
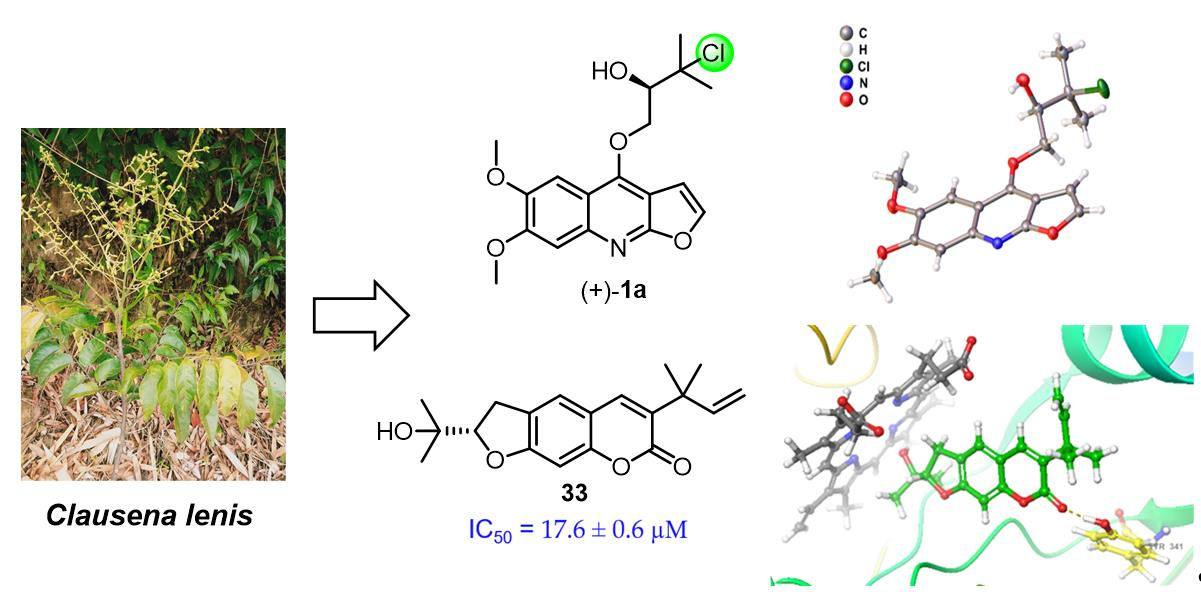
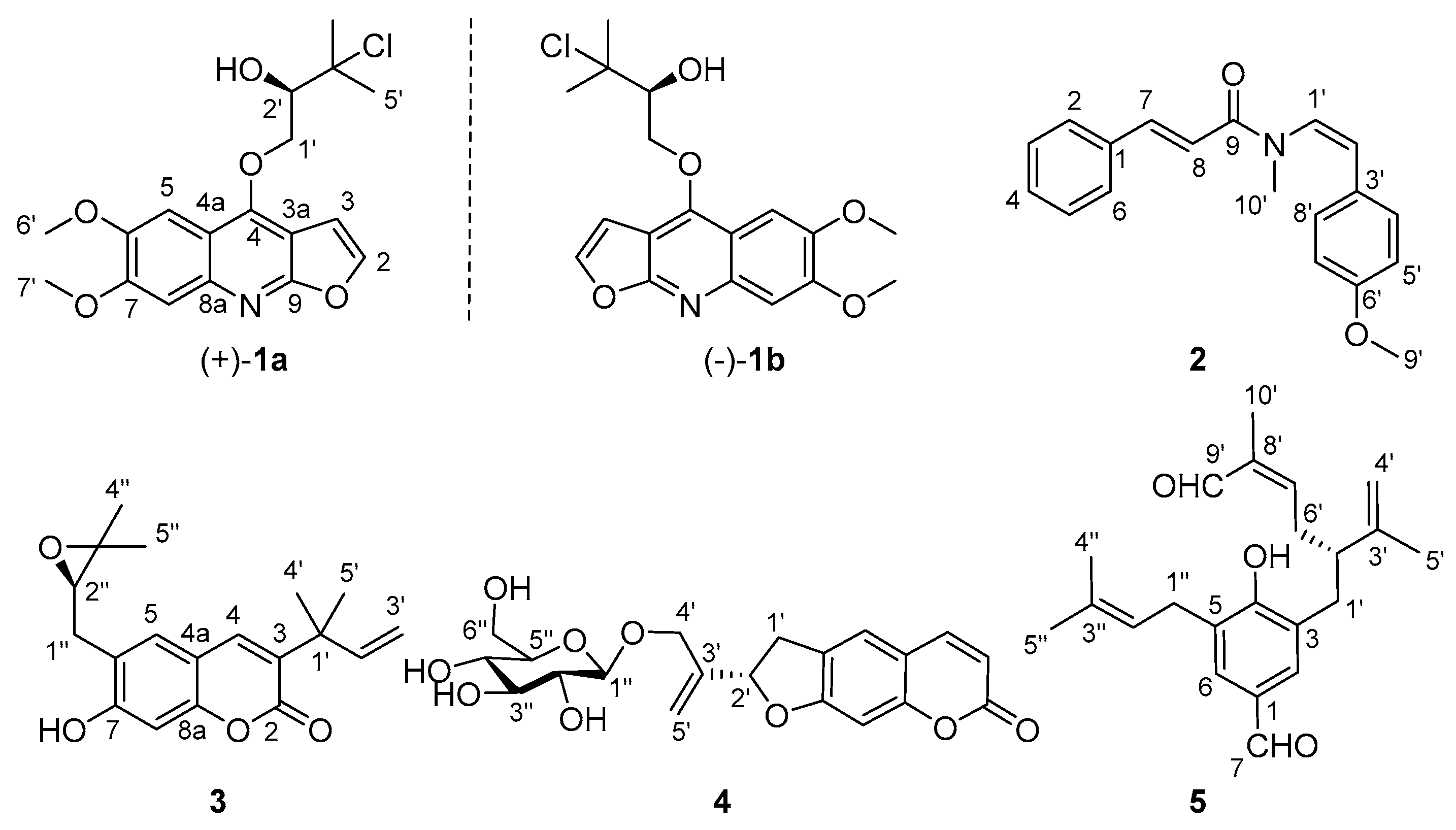
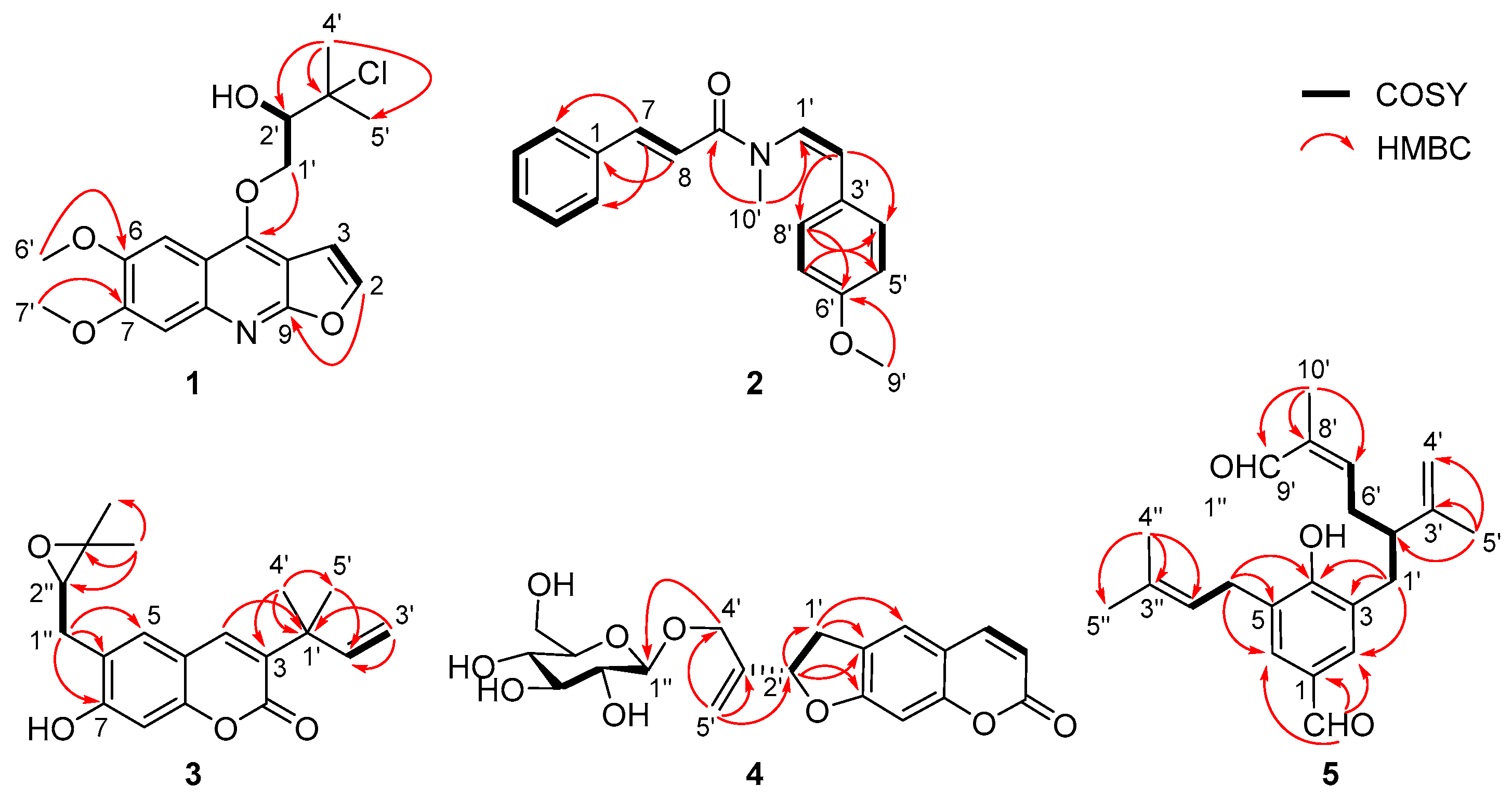
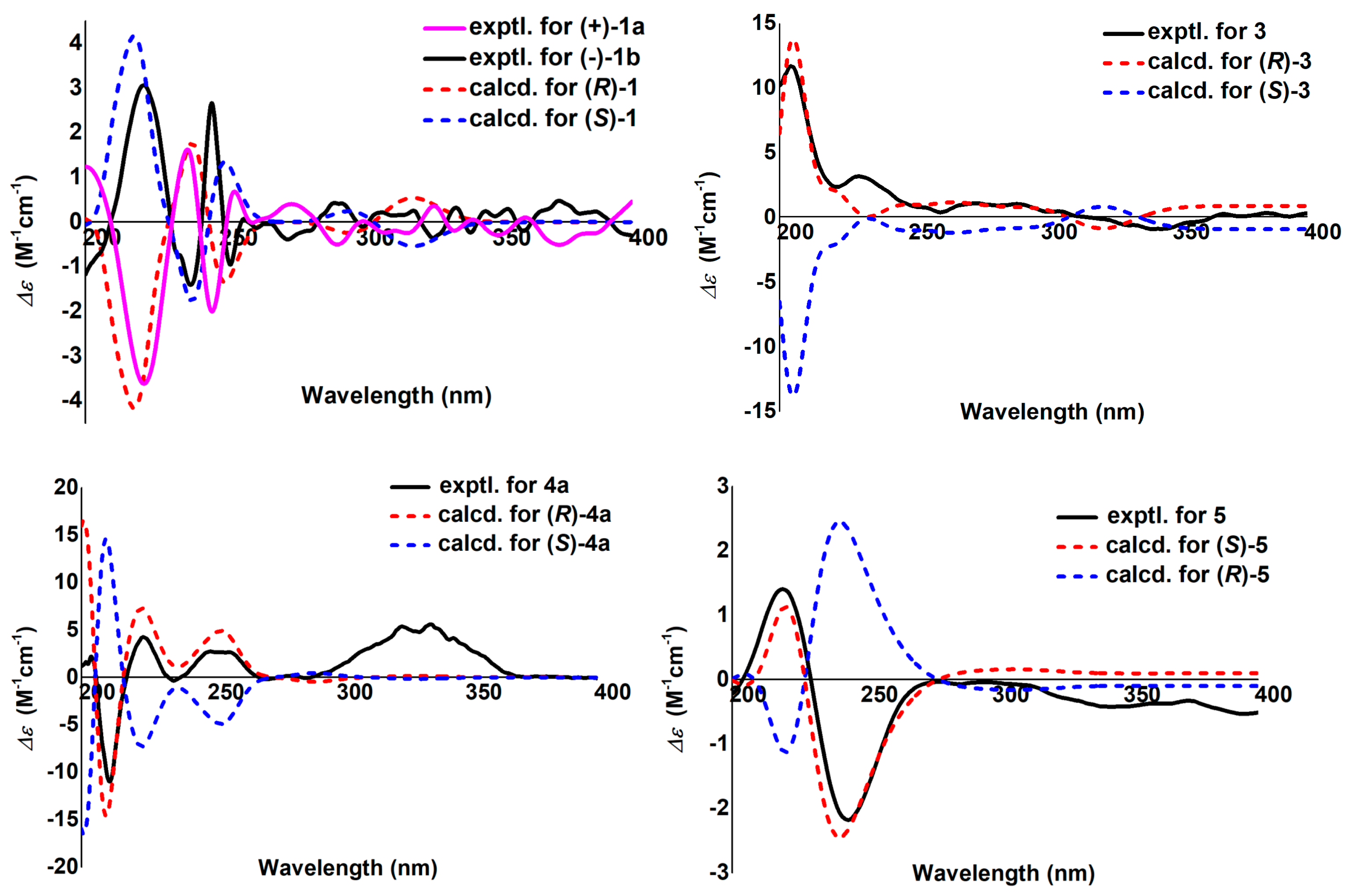
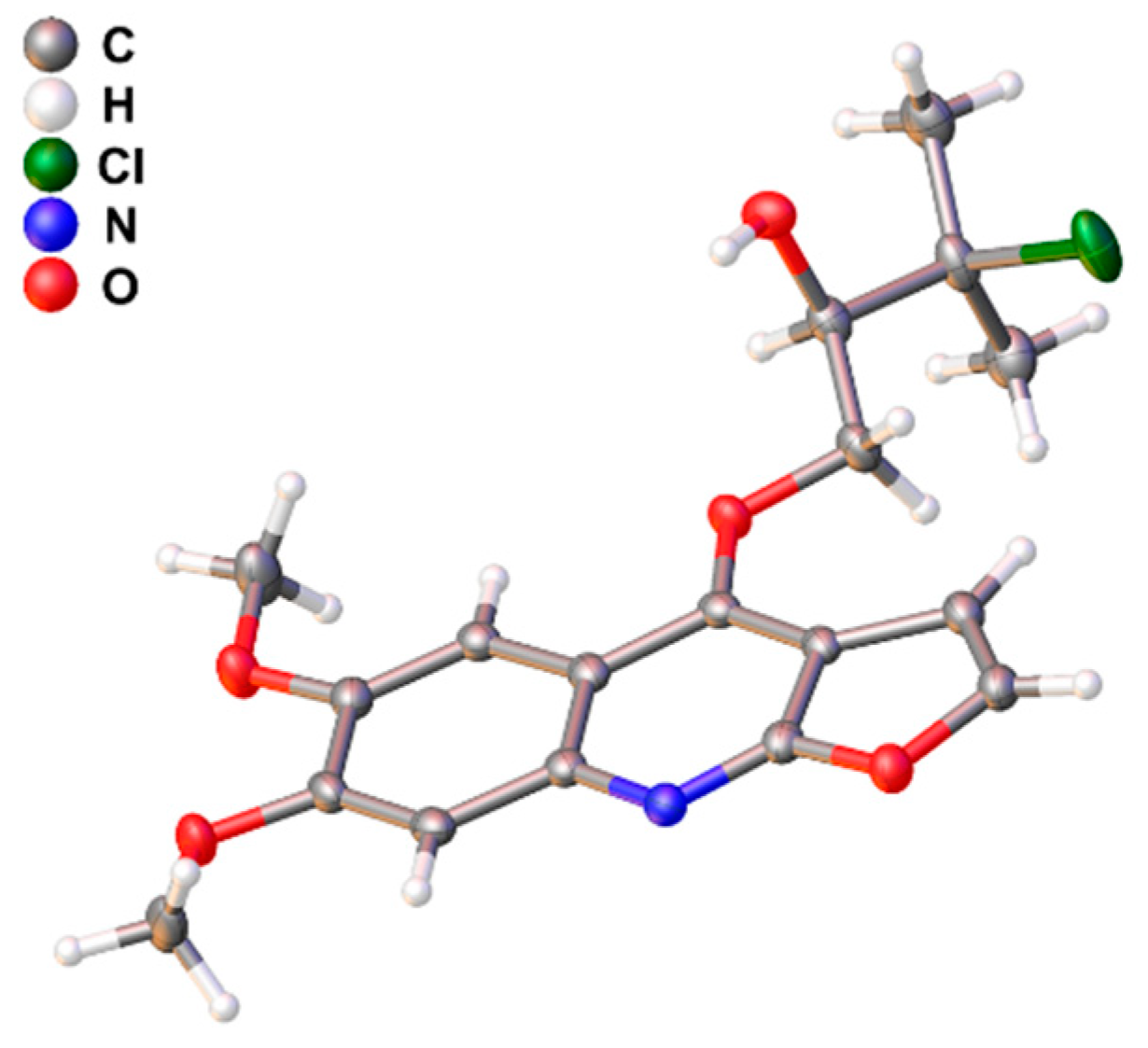
2.1. Structural Elucidation
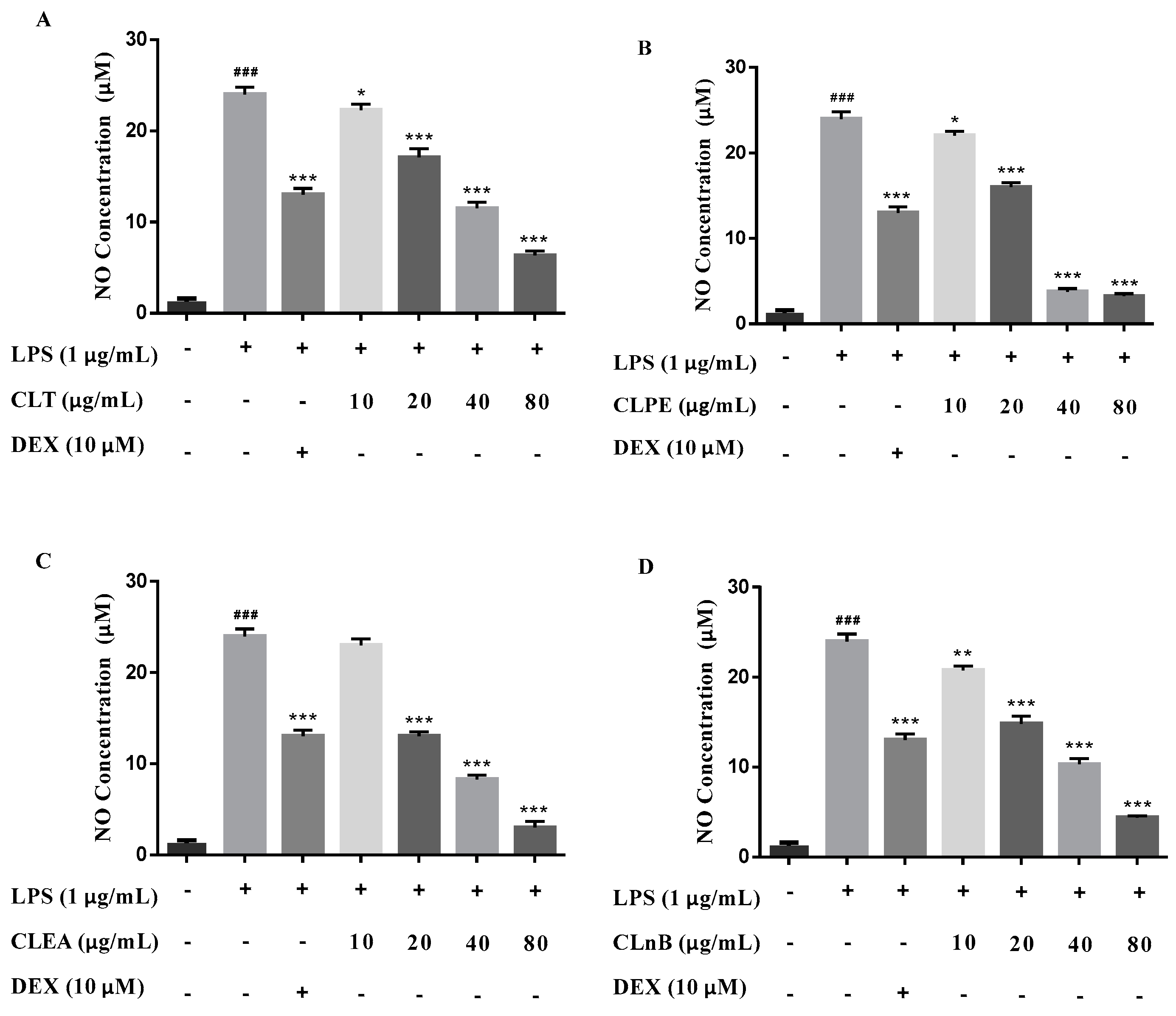
2.2. Anti-Neuroinflammatory Activities
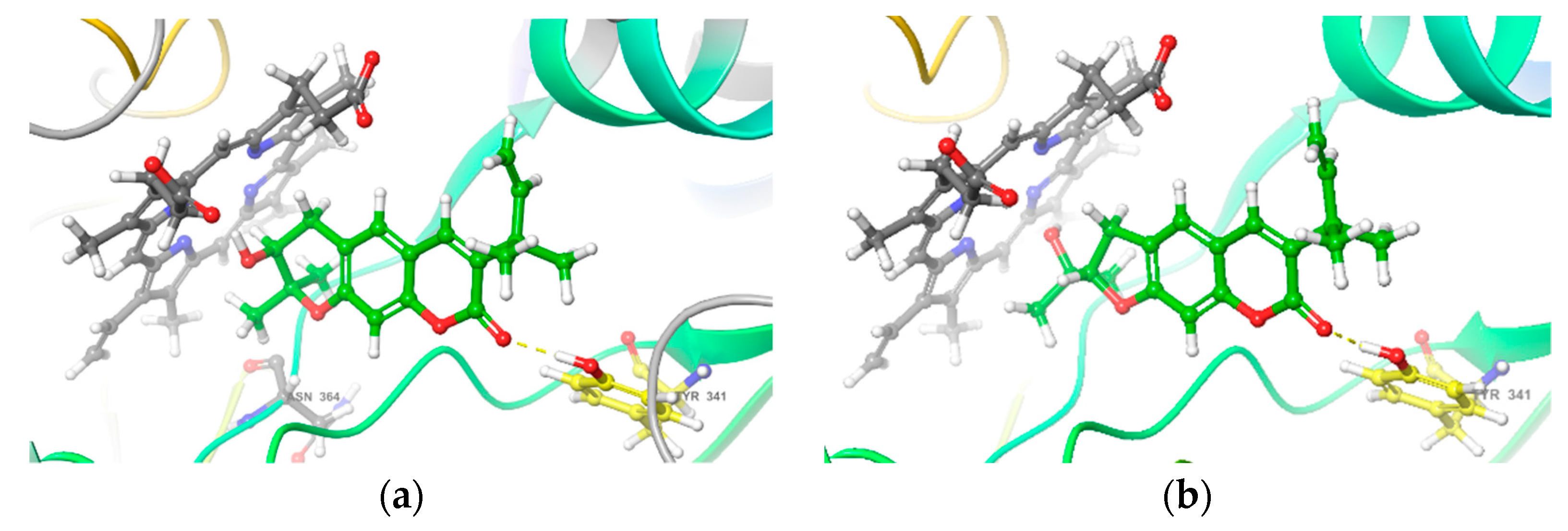
2.3. Interactions of Bioactive Compounds with iNOS
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Plant Material
4.3. Extraction and Isolation
4.3.1. (±)-Claulenine A (1)
4.3.2. Claulenine B (2)
4.3.3. Claulenin A (3)
4.3.4. Clauleside A (4)
4.3.5. Claulenin B (5)
4.3.6. (S)-Swietenocoumarin I (22)
4.4. ECD Calculations of 1, 3, 4a, 5, and 22
4.5. X-ray Crystallography of (+)-1a
4.6. Absolute Configurations Determination of Sugar Moiety for 4
4.7. Cell Culture
4.8. Nitric Oxide (NO) Production Measurement and Cell Viability Assay
4.9. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here. Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, M.; Liang, J.; Wang, C.; Deng, Y.; Chen, Z. Epigenetic regulation of neuroinflammation in Parkinson’s disease. Int. J. Mol. Sci. 2021, 22, 4956. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Wu, J.; Koc, S.; Lu, G. Genetic imaging of neuroinflammation in Parkinson’s disease: Recent advancements. Front. Cell Dev. Biol. 2021, 9, 655819. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, D.C.; Haque, A.; Banik, N.L. Neuroinflammatory responses of microglia in central nervous system trauma. J. Cereb. Blood Flow Metab. 2020, 40, S25–S33. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.A.; Joo, B.J.; Lee, J.S.; Ryu, G.; Han, M.; Kim, W.Y.; Park, H.H.; Lee, J.H.; Lee, C.S. Phytochemicals as anti-inflammatory agents in animal models of prevalent inflammatory diseases. Molecules 2020, 25, 5932. [Google Scholar] [CrossRef] [PubMed]
- Borah, A.; Paul, R.; Mazumder, M.K.; Bhattacharjee, N. Contribution of β-phenethylamine, a component of chocolate and wine, to dopaminergic neurodegeneration: Implications for the pathogenesis of Parkinson’s disease. Neurosci. Bull. 2013, 29, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Kim, B. Anti-cancer natural products and their bioactive compounds inducing ER stress-mediated apoptosis: A review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathaur, P.; SR, K.J. Metabolism and pharmacokinetics of phytochemicals in the human body. Curr. Drug Metab. 2020, 20, 1085–1102. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.M.; Li, C.J.; Yang, J.Z.; Ma, J.; Chen, X.G.; Zhang, D.; Li, L.; Zhang, D.M. A,D-seco-Limonoids from the stems of Clausena emarginata. J. Nat. Prod. 2014, 77, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.M.; Li, C.J.; Yang, J.Z.; Ma, J.; Li, L.; Zhang, D.; Bao, X.Q.; Zhang, D.M. Anti-inflammatory amide alkaloids from the stems of Clausena emarginata. J. Asian Nat. Prod. Res. 2014, 16, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, C.J.; Ni, L.; Yang, J.Z.; Li, L.; Zang, C.X.; Bao, X.Q.; Zhang, D.; Zhang, D.M. Anti-inflammatory alkaloid glycoside and quinoline alkaloid derivates from the stems of Clausena lansium. RSC Adv. 2015, 5, 80553. [Google Scholar] [CrossRef]
- Editorial Committee of Flora of China. Flora of China; Science Press: Beijing, China, 1997; Volume 43, p. 138. [Google Scholar]
- He, H.P.; Chen, S.T.; Shen, Y.M.; Chen, C.X.; Zhao, Y.B.; Hao, X.J. A novel dimeric coumarin from Clausena lenis. Chin. Chem. Lett. 2003, 14, 1150–1153. [Google Scholar]
- He, H.P.; Shen, Y.M.; Chen, S.T.; He, Y.N.; Hao, X.J. Dimeric coumarin and phenylpropanoids from Clausena lenis. Helv. Chim. Acta 2006, 89, 2836–2840. [Google Scholar] [CrossRef]
- Liu, Y.P.; Wen, Q.; Hu, S.; Ma, Y.L.; Jiang, Z.H.; Tang, J.Y.; Fu, Y.H.; Qiu, S.X. Furanocoumarins with potential antiproliferative activities from Clausena lenis. Nat. Prod. Res. 2019, 33, 2631–2637. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Yan, G.; Xie, Y.T.; Lin, T.C.; Zhang, W.; Li, J.; Wu, Y.J.; Zhou, J.Y.; Fu, Y.H. Bioactive prenylated coumarins as potential anti-inflammatory and anti-HIV agents from Clausena lenis. Bioorg. Chem. 2020, 97, 103699. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Li, Y.J.; Zhao, Y.Y.; Guo, J.M.; Zhang, W.H.; Zhang, M.M.; Fu, Y.H.; Liu, Y.P. Neuroprotective carbazole alkaloids from the stems and leaves of Clausena lenis. Nat. Prod. Res. 2021, 35, 2002–2009. [Google Scholar] [CrossRef]
- Wongthet, N.; Sanevas, N.; Schinnerl, J.; Brecker, L.; Santimaleeworagun, W.; Rosenau, T.; Bacher, M.; Vajrodaya, S. Chemical constituents of Clausena lenis. Nat. Prod. Res. 2021, 35, 3873–3879. [Google Scholar] [CrossRef] [Green Version]
- Cao, N.K.; Chen, Y.M.; Ma, X.L.; Zeng, K.W.; Zhao, M.B.; Tu, P.F.; Li, J.; Jiang, Y. Bioactive carbazole and quinoline alkaloids from Clausena dunniana. Phytochemistry 2018, 151, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.K.; Zhu, S.S.; Li, J.; Tu, P.F.; Jiang, Y. Coumarins from the stems and leaves of Clausena dunniana H. Lév. Biochem. Syst. Ecol. 2020, 90, 104048. [Google Scholar] [CrossRef]
- Cao, N.K.; Zhu, S.S.; Chen, Y.M.; Ma, X.L.; Zhao, M.B.; Li, J.; Tu, P.F.; Jiang, Y. A new prenylated coumarin diglycoside with insulin-release promoting activity from Clausena dunniana. J. Asian Nat. Prod. Res. 2021, 23, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Ulubelen, A. Alkaloids from Haplophyllum buxbaumii. Phytochemistry 1985, 24, 372–374. [Google Scholar] [CrossRef]
- He, H.P.; Shen, Y.M.; Zuo, G.Y.; Yang, X.S.; Hao, X.J. Dinorditerpene, diterpenes, alkaloids, and coumarins from Clausena dunniana. Helv. Chim. Acta 2003, 86, 3187–3193. [Google Scholar] [CrossRef]
- Phuwapraisirisan, P.; Puksasook, T.; Jong-aramruang, J.; Kokpol, U. Phenylethyl cinnamides: A new series of a-glucosidase inhibitors from the leaves of Aegle marmelos. Bioorg. Med. Chem. Lett. 2008, 18, 4956–4958. [Google Scholar] [CrossRef] [PubMed]
- Ito, C.; Fujiwara, K.; Kajita, M.; Ju-Ichi, M.; Takemura, Y.; Suzuki, Y.; Tanaka, K.; Omura, M.; Furukawa, H. New coumarins from Citrus plants. Chem. Pharm. Bull. 1991, 39, 2509–2513. [Google Scholar] [CrossRef] [Green Version]
- Suthiwonga, J.; Thongsriband, Y.; Yenjai, C. A new furanocoumarin from the fruits of Scaevola taccada and antifungal activity against Pythium insidiosum. Nat. Prod. Res. 2017, 31, 453–459. [Google Scholar] [CrossRef]
- Chang, H.T.; Okada, T.; Okuyama, T.; Tu, P.F. 1H and 13C NMR assignments for two new angular furanocoumarin glycosides from Peucedanum praeruptorum. Magn. Reson. Chem. 2007, 45, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.D.; Kwon, H.C.; Yang, M.C.; Lee, K.H.; Choi, S.U.; Lee, K.R. Isolation of limonoids and alkaloids from Phellodendron amurense and their multidrug resistance (MDR) reversal activity. Arch. Pharm. Res. 2007, 30, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Boyd, D.R.; Sharma, N.D.; Barr, S.A.; Carroll, J.G.; Mackerracher, D.; Malone, J.F. Synthesis and absolute stereochemistry assignment of enantiopure dihydrofuro- and dihydropyrano-quinoline alkaloids. J. Chem. Soc. Perkin Trans. 2000, 1, 3397–3405. [Google Scholar] [CrossRef]
- Sriphana, U.; Thongsri, Y.; Prariyachatigul, C.; Pakawatchai, C.; Yenjai, C. Clauraila E from the roots of Clausena harmandiana and antifungal activity against Pythium insidiosum. Arch. Pharm. Res. 2013, 36, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Pusset, J.; Lopez, J.L.; Pais, M.; Neirabeyeh, M.A.; Veillon, J.M. Isolation and 2D NMR studies of alkaloids from Comptonella sessilfoliola. Planta Med. 1991, 57, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Calvario, V.; Rios, M.Y. 1H and 13C NMR data, occurrence, biosynthesis, and biological activity of Piper amides. Magn. Reson. Chem. 2019, 57, 994–1070. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.W.; Li, W.; Wang, G.C.; Wu, X.; Ye, W.C.; Li, Y.L. Chemical constituents of Clausena lansium. J. Jinan Univ. (Nat. Sci.) 2012, 33, 506–509. [Google Scholar]
- Ottenbacher, R.V.; Kurganskiy, V.I.; Talsi, E.P.; Bryliakov, K.P. Manganese catalyzed enantioselective epoxidation of α,β-unsaturated amides with H2O2. Adv. Synth. Catal. 2021, 363, 2778–2782. [Google Scholar] [CrossRef]
- Borges-del-Castillo, J.; Vazquez-Bueno, P.; Secundino-Lucas, M.; Martinez-Martir, A.I.; Joseph-Nathan, P. The N-2-phenylethylcinnamamide from Spilanthes ocymifolia. Phytochemistry 1984, 23, 2671–2672. [Google Scholar] [CrossRef]
- Xu, H.Y.; Chen, T.; Huang, L.B.; Shen, Q.J.; Lian, Z.W.; Shi, Y.; Ouyang, M.A.; Song, L.Y. Synthesis and fungicidal activity of lansiumamide A and B and their derivatives. Molecules 2018, 23, 1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, A.; Maier, M.E. Synthesis of enamides from aldenydes and amides. Tetrahedron 2004, 60, 6665–6677. [Google Scholar] [CrossRef]
- Goossen, L.J.; Blanchot, M.; Arndt, M.; Salih, K.S.M. Synthesis of botryllamides and lansiumamides via ruthenium-catalyzed hydroamidation of alkynes. Synlett 2010, 11, 1685–1687. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, X.Y.; Yu, H.B.; Lu, X.L.; Jiao, B.H. Research of the secondary metabolites from Antarctic-derived fungus Penicillium sp. S-2-10. Chin. J. Mar. Drugs 2017, 36, 18–22. [Google Scholar]
- Devi, P.; Wahidullah, S.; Rodrigues, C.; Souza, L.D. The sponge-associated bacterium Bacillus licheniformis SAB1: A source of antimicrobial compounds. Mar. Drugs 2010, 8, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Bigi, F.; Frullanti, B.; Maggi, R.; Sartori, G.; Zambonin, E. Reaction of aliphatic amines with acetoacetanilide in the presence of zeolite catalyst. Solvent-free synthesis of symmetric N,N′-dialkylureas. J. Org. Chem. 1999, 64, 1004–1006. [Google Scholar] [CrossRef] [PubMed]
- Patre, R.E.; Shet, J.B.; Parameswaran, P.S.; Tilve, S.G. Cascade Wittig reaction-double Claisen and Cope rearrangements: One-pot synthesis of diprenylated coumarins gravelliferone, balsamiferone, and 6,8-diprenylumbelliferone. Tetrahedron Lett. 2009, 50, 6488–6490. [Google Scholar] [CrossRef]
- Rao, A.V.R.; Bhide, K.S.; Mujumdar, R.B. Phenolics from the bark of Chloroxylon swietenia DC.: Part II. Isolation of swietenocoumarins G, H and I. Indian J. Chem. 1980, 19B, 1046–1048. [Google Scholar]
- Xiong, Y.T.; Huang, G.; Yao, Z.L.; Zhao, C.; Zhu, X.; Wu, Q.L.; Zhou, X.D.; Li, J.K. Screening effective antifungal substances from the bark and leaves of Zanthoxylum avicennae by the bioactivity-guided isolation method. Molecules 2019, 24, 4207. [Google Scholar] [CrossRef] [Green Version]
- Park, H.Y.; Kwon, S.B.; Heo, N.K.; Chun, W.J.; Kim, M.J.; Kwon, Y.S. Constituents of the stem of Angelica gigas with Rat lens aldose reductase inhibitory activity. J. Korean Soc. Appl. Biol. Chem. 2011, 54, 194–199. [Google Scholar] [CrossRef]
- Malikov, V.M.; Saidkhodzhaev, A.I. Coumarins. Plants, structure, properties. Chem. Nat. Compd. 1998, 34, 345–409. [Google Scholar] [CrossRef]
- Carmo, G.D.; Fernandes, T.S.; Pedroso, M.; Ferraz, A.; Neto, A.T.; Silva, U.F.; Mostardeiro, M.A.; Back, D.F.; Dalcol, I.I.; Morel, A.F. Phytochemical and antimicrobial study of Pilocarpus pennatifolius Lemaire. Fitoterapia 2018, 131, 1–8. [Google Scholar] [CrossRef]
- Macias, F.A.; Massanet, G.M.; Rodriguez-Luis, F.; Salva, J. Carbon-13 NMR of coumarins I: 3-(1,1-dimethylallyl) derivatives. Magn. Reson. Chem. 1989, 27, 705–707. [Google Scholar] [CrossRef]
- De Moura, N.F.; Simionatto, E.; Porto, C.; Hoelzel, S.C.S.; Dessoy, E.C.S.; Zanatta, N.; Morel, A.F. Quinoline alkaloids, coumarins, and volatile constituents of Helietta longifoliata. Planta Med. 2002, 68, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.B.; Li, G.H.; Li, L.; Zheng, L.J.; Huang, R.; Zhang, K.Q. Nematicidal coumarins from Heracleum candicans Wall. Nat. Prod. Res. 2008, 22, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Bergendorff, O.; Dekermendjian, K.; Nielsen, M.; Shan, R.; Witt, R.; Ai, J.; Sterner, O. Furanocoumarins with affinity to brain benzodiazepine receptors in vitro. Phytochemistry 1997, 44, 1121–1124. [Google Scholar] [CrossRef]
- Kviesis, J.; Kļimenkovs, I.; Arbidans, L.; Podjava, A.; Kļaviņš, M.; Liepiņš, E. Evaluation of furanocoumarins from seeds of the wild parsnip (Pastinaca sativa L. s.l.). J. Chromatogr. B 2019, 1105, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Gowri, P.M.; Haribabu, K.; Kishore, H.; Manjusha, O.; Biswas, S.; Murty, U.S.N. Microbial transformation of (+)-heraclenin by Aspergillus niger and evaluation of its antiplasmodial and antimicrobial activities. Curr. Sci. 2011, 100, 1706–1711. [Google Scholar]
- Yamaguchi, S.; Muro, S.; Kobayashi, M.; Miyazawa, M.; Hirai, Y. Absolute structures of some naturally occurring isopropenyldihydrobenzofurans, remirol, remiridiol, angenomalin, and isoangenomalin. J. Org. Chem. 2003, 68, 6274–6278. [Google Scholar] [CrossRef]
- Saeed, M.A.; Sabir, A.W. Irritant and cytotoxic coumarins from Angelica glauca Edgew roots. J. Asian Nat. Prod. Res. 2008, 10, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, F.M.; Sant’ana, A.E.G.; Conserva, L.M.; Maia, J.G.S.; Guilhon, G.M.P. Alkaloids and coumarins from Esenbeckia species. Phytochemistry 1996, 41, 647–649. [Google Scholar] [CrossRef]
- Kamperdick, C.; Phuong, N.M.; Sung, T.V.; Schmidt, J.; Adama, G. Coumarins and dihydrocinnamic acid derivatives from Micromelum falcatum. Phytochemistry 1999, 52, 1671–1676. [Google Scholar] [CrossRef]
- Liu, J.H.; Xu, S.X.; Meng, Z.Y.; Yao, X.S.; Wu, Y.Q. Further isolation of coumarin from Angelica pubescence Maxim f. biserrata Shan et Yuan. J. Chin. Pharm. Sci. 1997, 6, 221–224. [Google Scholar]
- Kassim, N.K.; Lim, P.C.; Ismail, A.; Awang, K. Isolation of antioxidative compounds from Micromelum minutum guided by preparative thin layer chromatography-2,2-diphenyl-1-picrylhydrazyl (PTLC-DPPH) bioautography method. Food Chem. 2019, 272, 185–191. [Google Scholar] [CrossRef]
- Lemmich, J. Monoterpene, chromone and coumarin glucosides of Diplolophium buchananii. Phytochemistry 1995, 38, 427–432. [Google Scholar] [CrossRef]
- Zhang, F.M.; Shi, Z.Z.; Chen, F.; Yuan, Y. An efficient MnCl2-catalyzed tandem acylation-cross-coupling reaction of o-halobenzoyl chloride with diorganyl magnesium compounds. Appl. Organometal. Chem. 2010, 24, 57–63. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Tasleem, M.; Alrehaily, A.; Almeleebia, T.M.; Alshahrani, M.Y.; Ahmad, I.; Asiri, M.; Alabdallah, N.M.; Saeed, M. Investigation of antidepressant properties of yohimbine by employing structure-based computational assessments. Curr. Issues Mol. Biol. 2021, 43, 1805–1827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.Z.; Wang, M.Y.; Sun, C.J.; Li, X.B. Five new compounds from Hosta plantaginea flowers and their anti-inflammatory activities. Bioorg. Chem. 2020, 95, 103494. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Shi, Y.; Zeng, K.; Zhao, M.; Tu, P.; Jiang, Y. Coumarin derivatives from the leaves and twigs of Murraya exotica L. and their anti-inflammatory activities. Phytochemistry 2020, 177, 112416. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.J.; Vanderwal, C.D. Stereoselective halogenation in natural product synthesis. Angew. Chem. Int. Ed. 2016, 55, 4396–4434. [Google Scholar] [CrossRef]
- Li, C.; Shi, D. Structural and bioactive studies of halogenated constituents from sponges. Curr. Med. Chem. 2020, 27, 2335–2360. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Mitchell, A.J.; Glinkerman, C.M.; Li, F.S.; Pluskal, T.; Weng, J.K. The chloroalkaloid (−)-acutumine is biosynthesized via a Fe(II)- and 2-oxoglutarate-dependent halogenase in Menispermaceae plants. Nat. Commun. 2020, 11, 1867. [Google Scholar] [CrossRef] [Green Version]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Pecitelli, G. SpecDis, Version 1.71; Berlin, Germany, 2017. Available online: https:/specdis-software.jimdo.com (accessed on 24 February 2020).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tanaka, T.; Nakashima, T.; Ueda, T.; Tomii, K.; Kouno, I. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 2007, 55, 899–901. [Google Scholar] [CrossRef] [Green Version]
- Garcin, E.D.; Arvai, A.S.; Rosenfeld, R.J.; Kroeger, M.D.; Crane, B.R.; Andersson, G.; Andrews, G.; Hamley, P.J.; Mallinder, P.R.; Nicholls, D.J.; et al. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat. Chem. Biol. 2008, 4, 700–707. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 a | 4 b | 5 a | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | |
| 1 | 135.4, C | 129.5, C | ||||||||
| 2 | 7.59, d (2.7) | 143.0, CH | 7.33, overlap | 128.8, CH | 160.4, C | 163.6, C | 7.52, br s | 131.4, CH | ||
| 3 | 7.00, d (2.7) | 104.5, CH | 7.45, m | 128.1, CH | 131.0, C | 6.13, d (9.4) | 112.4, CH | 127.9, C | ||
| 3a | 103.0, C | |||||||||
| 4 | 154.4, C | 7.33, overlap | 129.8, CH | 7.47, s | 138.2, CH | 7.77, d (9.4) | 146.2, CH | 158.9, C | ||
| 4a | 113.2, C | 113.3, C | 114.3, C | |||||||
| 5 | 7.45, s | 100.1, CH | 7.45, m | 128.1, CH | 7.19, s | 123.4, CH | 7.33, s | 125.3, CH | 126.6, C | |
| 6 | 148.1, C | 7.33, overlap | 128.8, CH | 124.7, C | 126.7, C | 7.52, br s | 130.7, CH | |||
| 7 | 152.8, C | 7.64, d (15.5) | 142.7, CH | 162.4, C | 164.7, C | 9.82, s | 191.4, CH | |||
| 8 | 7.34, s | 106.8, CH | 6.95, d (15.5) | 118.5, CH | 6.70, s | 97.3, CH | 6.67, s | 98.3, CH | ||
| 8a | 142.7, C | 154.8, C | 156.9, C | |||||||
| 9 | 162.9, C | 166.7, C | ||||||||
| 1′ | 4.93, dd (9.8, 3.7) | 72.5, CH2 | 6.38, d (8.6) | 127.1, CH | 40.4, C | 3.43, dd (15.9, 9.5) | 34.7, CH2 | 2.78, d (7.4, 3.8) | 34.7, CH2 | |
| 4.75, dd (9.8, 6.8) | 3.16, overlap | |||||||||
| 2′ | 4.18, dd (6.8, 3.7) | 77.1, CH | 6.20, d (8.6) | 125.6, CH | 6.16, dd (17.4, 10.7) | 145.7, CH | 5.47, t-like (8.7) | 86.3, CH | 2.68, m | 46.4, CH |
| 3′ | 71.9, C | 127.0, C | 5.08, d (10.7) | 112.2, CH2 | 145.6, C | 146.2, C | ||||
| 5.07, d (17.4) | ||||||||||
| 4′ | 1.77, s | 29.5, CH3 | 7.28, d (8.7) | 130.2, CH | 1.46, s | 24.2, CH3 | 4.44, d (12.8) | 70.0, CH2 | 4.78, s | 112.8, CH2 |
| 4.19, d (12.8) | 4.70, s | |||||||||
| 5′ | 1.77, s | 29.0, CH3 | 6.83, d (8.7) | 114.2, CH | 1.35, s | 24.2, CH3 | 5.26, d (13.9) | 114.1, CH2 | 1.71, s | 19.4, CH3 |
| 6′ | 4.00, s | 56.1, CH3 | 159.5, C | 2.44, m | 32.3, CH2 | |||||
| 7′ | 4.03, s | 56.2, CH3 | 6.83, d (8.7) | 114.2, CH | 6.35, t (7.2) | 153.2, CH | ||||
| 8′ | 7.28, d (8.7) | 130.2, CH | 139.7, C | |||||||
| 9′ | 3.78, s | 55.4, CH3 | 9.33, s | 195.4, CH | ||||||
| 10′ | 3.10, s | 34.6, CH3 | 1.70, s | 9.5, CH3 | ||||||
| 1″ | 3.19, m | 29.7, CH2 | 4.23, d (7.8) | 103.6, CH | 3.43, d (7.2) | 30.7, CH2 | ||||
| 2″ | 4.71, t (8.8) | 91.0, CH | 3.16, overlap | 75.0, CH | 5.30, m | 120.5, CH | ||||
| 3″ | 71.8, C | 3.38, m | 78.0, CH | 137.7, C | ||||||
| 4″ | 1.46, s | 26.2, CH3 | 3.21, overlap | 71.6, CH | 1.82, s | 26.0, CH3 | ||||
| 5″ | 1.22, s | 24.4, CH3 | 3.21, overlap | 78.0, CH | 1.84, s | 18.2, CH3 | ||||
| 6″ | 3.81, d (11.9) | 62.7, CH2 | ||||||||
| 3.60, dd (11.9, 4.9) | ||||||||||
| Compound | IC50 (μM) |
|---|---|
| 2 | 32.0 ± 1.4 |
| 23 | 40.9 ± 0.3 |
| 27 | 23.1 ± 0.4 |
| 28 | 38.1 ± 2.0 |
| 33 | 17.6 ± 0.6 |
| 34 | 19.9 ± 0.7 |
| DEX a | 9.6 ± 0.3 |
| Compound | Glide Score |
|---|---|
| 27 | −5.616 |
| 33 | −5.228 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, S.-S.; Zhang, Y.-F.; Ding, M.; Zeng, K.-W.; Tu, P.-F.; Jiang, Y. Anti-Neuroinflammatory Components from Clausena lenis Drake. Molecules 2022, 27, 1971. https://doi.org/10.3390/molecules27061971
Zhu S-S, Zhang Y-F, Ding M, Zeng K-W, Tu P-F, Jiang Y. Anti-Neuroinflammatory Components from Clausena lenis Drake. Molecules. 2022; 27(6):1971. https://doi.org/10.3390/molecules27061971
Chicago/Turabian StyleZhu, Si-Si, Yi-Fan Zhang, Meng Ding, Ke-Wu Zeng, Peng-Fei Tu, and Yong Jiang. 2022. "Anti-Neuroinflammatory Components from Clausena lenis Drake" Molecules 27, no. 6: 1971. https://doi.org/10.3390/molecules27061971
APA StyleZhu, S. -S., Zhang, Y. -F., Ding, M., Zeng, K. -W., Tu, P. -F., & Jiang, Y. (2022). Anti-Neuroinflammatory Components from Clausena lenis Drake. Molecules, 27(6), 1971. https://doi.org/10.3390/molecules27061971