
In Silico Drug Design of Anti-Breast Cancer Agents

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
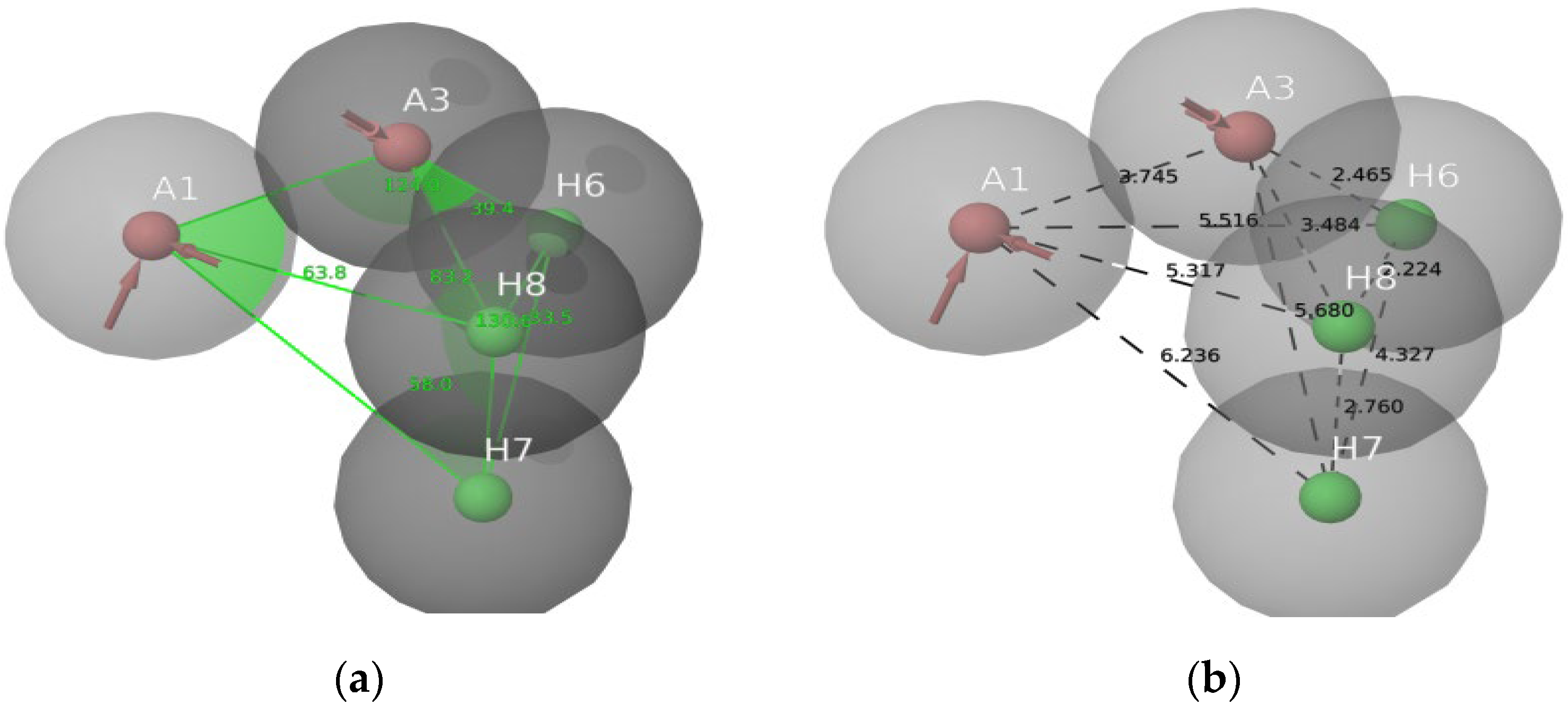
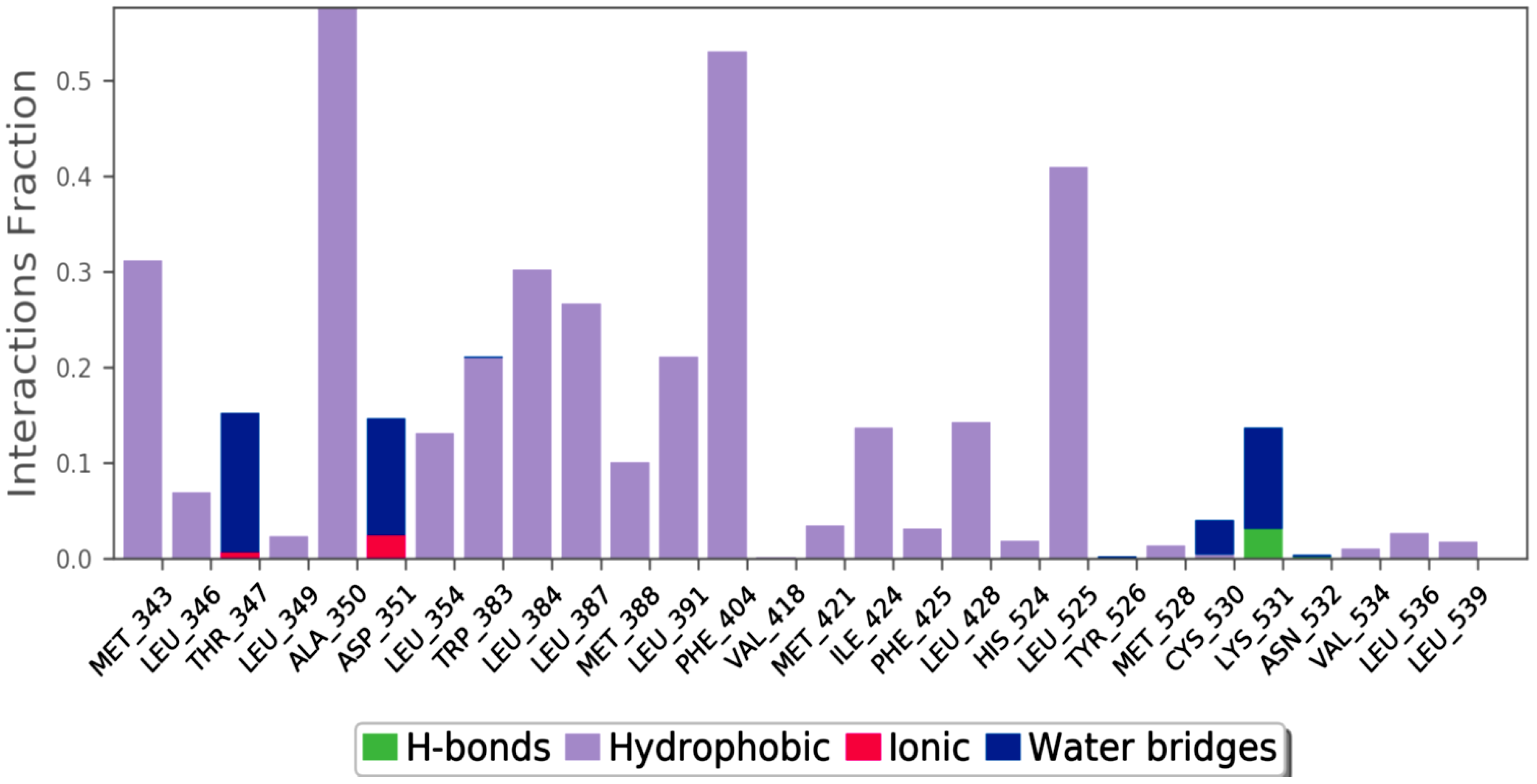
2.1. Molecular Docking Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No | Compound Code | Glide ENERGY | Docking Score | Glide-Gscore | XP H-Bond |
|---|---|---|---|---|---|
| 1 | BT_ER_15f | 53.441 | −15.922 | −16.14 | −1.546 |
| 2 | BT_ER_Tf | 59.574 | −13.560 | −13.563 | −0.9 |
| 3 | BT_ER_21b | 65.999 | −12.577 | −12.964 | −0.385 |
| 4 | BT_ER_15e | 51.353 | −12.155 | −12.825 | −1.164 |
| 5 | BT_ER_15b | 57.5 | −12.007 | −12.776 | −0.688 |
| 6 | BT_ER_23c | 44.431 | −12.394 | −12.404 | −0.599 |
| 7 | BT_ER_15d | 60.801 | −11.524 | −12.321 | −0.9 |
| 8 | BT_ER_15c | 52.788 | −11.459 | −12.32 | −0.7 |
| 9 | BT_ER_23b | 49.559 | −11.622 | −11.632 | −0.35 |
| 10 | BT_ER_Rf | 46.16 | −8.852 | −11.314 | −0.627 |
| 11 | BT_ER_21d | 69.206 | −10.469 | −11.035 | −0.335 |
| 12 | SL_TN_55 | 27.163 | −10.962 | −10.962 | 0 |
| 13 | SL_TN_56 | 27.163 | −10.962 | −10.962 | 0 |
| 14 | SL_TN_34 | 15.699 | −10.856 | −10.856 | 0 |
| 15 | BT_ER_23a | 45.782 | −10.773 | −10.782 | 0 |
| 16 | MS_ER_8b | 18.796 | −10.726 | −10.726 | 0 |
| 17 | SL_TN_51 | 26.449 | −10.553 | −10.553 | 0 |
| 18 | SL_TN_63 | 26.449 | −10.535 | −10.535 | 0 |
| 19 | BT_ER_21c | 62.876 | −9.931 | −10.519 | −0.605 |
| 20 | BT_ER_15a | 48.368 | −7.952 | −10.495 | 0 |
| 21 | SL_TN_32 | 13.11 | −10.492 | −10.492 | 0 |
| 22 | SL_TN_53 | 28.726 | −10.462 | −10.465 | 0 |
| 23 | SL_TN_38 | 17.353 | −10.447 | −10.447 | 0 |
| 24 | MS_ER_8a | 24.947 | −10.418 | −10.418 | −0.027 |
| 25 | HC_TI_CT | 52.763 | −10.333 | −10.333 | −0.854 |
| 26 | SL_TN_35 | 20.901 | −10.255 | −10.255 | 0 |
| 27 | SL_TN_21 | 2.415 | −10.214 | −10.214 | 0 |
| 28 | SL_TN_37 | 17.418 | −10.146 | −10.146 | 0 |
| 29 | BT_ER_21e | 61.13 | −9.576 | −10.101 | −0.7 |
| 30 | SL_TN_60 | 18.294 | −10.065 | −10.065 | 0 |
| 31 | SL_TN_47 | 17.206 | −10.043 | −10.043 | 0 |
| 32 | SL_TN_33_DETD_39 | 18.699 | −10.04 | −10.04 | 0 |
| 33 | BT_ER_21a | 56.951 | −9.415 | −10.027 | −0.7 |
| 34 | SL_TN_40 | 5.994 | −9.892 | −9.892 | 0 |
| 35 | SL_TN_52 | 27.449 | −9.861 | −9.861 | 0 |
| 36 | MS_ER_6a | 24.224 | −9.836 | −9.836 | 0 |
| 37 | SL_TN_39 | 24.888 | −9.765 | −9.765 | 0 |
| 38 | SL_TN_59 | 35.939 | −9.75 | −9.75 | 0 |
| 39 | SL_TN_57 | 29.053 | −9.719 | −9.719 | 0 |
| 40 | SL_TN_58 | 29.053 | −9.719 | −9.719 | 0 |
| 41 | BT_ER_25a | 39.481 | −7.193 | −9.666 | 0 |
| 42 | MS_ER_5b | 19.258 | −9.627 | −9.627 | 0 |
| 43 | MS_ER_4b | 17.706 | −9.603 | −9.603 | −0.178 |
| 44 | SL_TN_44 | 12.471 | −9.559 | −9.559 | 0 |
| 45 | SL_TN_42 | 10.596 | −9.34 | −9.34 | 0 |
| 46 | SL_TN_31 | 11.769 | −9.329 | −9.329 | 0 |
| 47 | SL_TN_41 | 8.182 | −9.286 | −9.286 | 0 |
| 48 | SL_TN_27 | 5.437 | −9.273 | −9.273 | 0 |
| 49 | SL_TN_16 | 9.358 | −9.256 | −9.256 | 0 |
| 50 | SL_TN_19 | 4.803 | −9.249 | −9.249 | 0 |
| 51 | SL_TN_12 | 4.923 | −9.13 | −9.13 | 0 |
| 52 | SL_TN_46 | 8.311 | −8.998 | −8.998 | 0 |
| 53 | SL_TN_20 | 3.842 | −8.994 | −8.994 | 0 |
| 54 | BT_ER_25b | 42.416 | −8.96 | −8.969 | 0 |
| 55 | SL_TN_26 | 12.472 | −8.959 | −8.959 | 0 |
| 56 | SL_TN_25 | 15.772 | −8.941 | −8.941 | 0 |
| 57 | SL_TN_17 | 3.063 | −8.894 | −8.894 | 0 |
| 58 | SL_TN_50 | 19.536 | −8.723 | −8.723 | 0 |
| 59 | HC_TI_14 | 20.434 | −8.713 | −8.713 | 0 |
| 60 | SL_TN_18 | 3.524 | −8.71 | −8.71 | 0 |

2.2. Binding Free Energy Calculation Using MM/GBSA
| Compound | MMGBA dG Bind | MMGBSA dG Bind Coulomb | MMGBA dG Bind Covalent | MMGBA dG Bind H-bond | MMGBA dG Bind Lipo | MMGBA dG Bind vdW |
|---|---|---|---|---|---|---|
| BT_ER_15f | −70.59 | −31.39 | 25.77 | 0.29 | −47.46 | −66.83 |
| BT_ER_Tf | −73.77 | −37.36 | 13.73 | 1.85 | −50.23 | −51.61 |
| BT_ER_21b | −67.84 | 6.28 | 3.91 | 3.72 | −44.3 | −58.54 |
| BT_ER_15e | −58.72 | −7.14 | 11.68 | 0.65 | −43.31 | −50.6 |
| BT_ER_15b | −84.12 | 2.23 | 12.68 | 1.55 | −49.81 | −67.59 |
| BT_ER_23c | −35.85 | 35.14 | 4.56 | 2.43 | −38.46 | −38.31 |
| BT_ER_15d | −69.35 | 15.23 | 16.5 | 1.6 | −50.67 | −76.44 |
| BT_ER_15c | −77.87 | −8.52 | 4.85 | −0.1 | −43.55 | −51.96 |
| BT_ER_23b | −42.39 | 4.45 | 15.46 | 2.18 | −35.55 | −43.15 |
| BT_ER_Rf | −39.6 | 9.74 | 5.84 | 2.32 | −40.64 | −54.61 |
| BT_ER_21d | −83.6 | −23.01 | 9.8 | −0.26 | −46.56 | −61.19 |
| SL_TN_55 | −47.68 | 19.48 | 13.24 | 1.19 | −37.05 | −63.86 |
| SL_TN_56 | −47.68 | 19.48 | 13.24 | 1.19 | −37.05 | −63.86 |
| SL_TN_34 | −47.89 | 2.46 | 17.92 | 0.06 | −34.68 | −54.25 |
| BT_ER_23a | −22.06 | 43.87 | 4.15 | 3.73 | −33.83 | −37.62 |
| MS_ER_8b | −51.07 | 23.16 | 16.5 | 1.72 | −38.3 | −53.5 |
| SL_TN_51 | −23.06 | 19.21 | 16.72 | 0.71 | −27.94 | −44.25 |
| SL_TN_63 | −32.26 | 7.75 | 28.36 | 0.71 | −33.29 | −60.91 |
| BT_ER_21c | −62.02 | −11.89 | 10.67 | −0.17 | −41.19 | −40.9 |
| BT_ER_15a | −48.48 | −28.74 | 20.87 | 0.87 | −40.95 | −69.26 |
| SL_TN_32 | −51.78 | 21.56 | 10.52 | 2.17 | −37.46 | −60.5 |
| SL_TN_53 | −26.15 | 26.63 | 11.07 | 1.24 | −26.98 | −42.96 |
| SL_TN_38 | −43.82 | 36.19 | 3.87 | 4.46 | −32.63 | −52.89 |
| MS_ER_8a | −32.26 | 13.04 | 8.48 | 3.91 | −35.7 | −31.91 |
| HC_TI_CT | −37.49 | 33.67 | 9.55 | 4.97 | −35.4 | −59.22 |
| SL_TN_35 | −70.61 | 8.94 | 17.37 | 0.14 | −35.16 | −71.95 |
| SL_TN_21 | −40.68 | 33.51 | 17.41 | 1.79 | −33.22 | −58.98 |
| SL_TN_37 | −44.62 | 10.16 | 16.17 | 2.79 | −35.66 | −54.67 |
| BT_ER_21e | −77.57 | −12.32 | 8.39 | −0.4 | −42.39 | −57.26 |
| SL_TN_60 | −15.66 | 30.43 | 11.01 | 2.91 | −22.07 | −33.97 |
| SL_TN_47 | −14.45 | 36.34 | 14.47 | 1.62 | −26.13 | −42.54 |
| SL_TN_33 | −44.77 | −4.51 | 23.03 | 1.05 | −37.47 | −54 |
| BT_ER_21a | −74.17 | −25.99 | 19.61 | 0.51 | −42.98 | −64.65 |
| SL_TN_40 | −46.78 | 3.72 | 14.69 | 1.93 | −34.26 | −45.35 |
| SL_TN_52 | −61.66 | 51.17 | 6.73 | 2 | −36.49 | −64.87 |
| MS_ER_6a | −80.51 | 45.68 | 7.44 | 3.79 | −42.18 | −69.73 |
| SL_TN_39 | −32.17 | 27.4 | 5.89 | 4.14 | −29.4 | −38.86 |
| SL_TN_59 | −23.46 | 16.16 | −4.63 | 2.44 | −23.08 | −20.15 |
| SL_TN_57 | −45.01 | 24.7 | 13.93 | 3.5 | −38.92 | −47.17 |
| SL_TN_58 | −45.01 | 24.7 | 13.93 | 3.5 | −38.92 | −47.17 |
| BT_ER_25a | −20.73 | 20.05 | 8.4 | 1.86 | −32.66 | −40.55 |
| MS_ER_5b | −44.69 | 1.97 | 27.56 | 0.39 | −37.99 | −43.9 |
| MS_ER_4b | −45.14 | −4.23 | 26.93 | −0.77 | −36.71 | −43.04 |
| SL_TN_44 | −51.74 | 29.57 | 3.53 | 1.71 | −31.97 | −53.17 |
| SL_TN_42 | −25.68 | 7.34 | 14.67 | 1.59 | −25.3 | −45.84 |
| SL_TN_31 | −59.09 | 26.08 | 34.57 | 0.18 | −44.44 | −62.36 |
| SL_TN_41 | −32.35 | 27.42 | 8.56 | 4.49 | −31.66 | −39.33 |
| SL_TN_27 | −36.14 | 19.28 | 8.97 | 3.86 | −31.29 | −44.2 |
| SL_TN_16 | −26.89 | 32.38 | 15.44 | 3.74 | −32.99 | −45.44 |
| SL_TN_19 | −28.82 | 41.14 | 10.29 | 3.07 | −32.82 | −41.2 |
| SL_TN_12 | −36.46 | 16.03 | 3.56 | 3.35 | −29.29 | −42.28 |
| SL_TN_46 | −17.05 | 31.24 | 14 | 1.77 | −25.1 | −43.97 |
| SL_TN_20 | −45.87 | 33.37 | 6.75 | 4.13 | −31.83 | −52.63 |
| BT_ER_25b | −63.44 | −23.53 | 13.99 | 0.3 | −43.08 | −57.07 |
| SL_TN_26 | −31.13 | −2.41 | 17.3 | 0.95 | −28.38 | −56.03 |
| SL_TN_25 | −20.67 | 24.88 | 9.38 | 1.91 | −21.97 | −37.14 |
| SL_TN_17 | −24.26 | 41.87 | −1.61 | 4.43 | −25.99 | −34.99 |
| SL_TN_50 | −40.32 | 3.61 | 8.68 | 1.26 | −29.99 | −35.15 |
| HC_TI_14 | −36.29 | 47.93 | −4.62 | 5.56 | −29.23 | −33.83 |
| SL_TN_18 | −15.33 | 45.87 | 3.18 | 4.13 | −25.08 | −29.85 |
2.3. ADMET Studies
| Compound | Mol MW | Dipole | Donor HB | Accpt HB | QP Log Po/w | Rule of Five |
|---|---|---|---|---|---|---|
| BT_ER_15f | 561.67 | 5.605 | 3 | 7.5 | 5.995 | 2 |
| BT_ER_Tf | 355.522 | 0.865 | 0 | 2 | 6.682 | 1 |
| BT_ER_21b | 518.602 | 6.426 | 1 | 5 | 6.874 | 2 |
| BT_ER_15e | 563.642 | 4.866 | 3 | 9.2 | 5.013 | 2 |
| BT_ER_15b | 506.591 | 6.325 | 2 | 6.5 | 5.827 | 2 |
| BT_ER_23c | 463.522 | 7.699 | 2 | 4.5 | 6.319 | 1 |
| BT_ER_15d | 547.643 | 3.649 | 3 | 7.5 | 6 | 2 |
| BT_ER_15c | 533.616 | 4.877 | 3 | 6 | 6.059 | 2 |
| BT_ER_23b | 463.522 | 10.476 | 2 | 4.5 | 6.354 | 1 |
| BT_ER_Rf | 473.586 | 3.216 | 2 | 6.25 | 4.686 | 0 |
| BT_ER_21d | 559.654 | 6.407 | 2 | 6 | 7.16 | 2 |
| SL_TN_55 | 488.536 | 6.502 | 0 | 8.75 | 3.567 | 0 |
| SL_TN_56 | 488.536 | 6.502 | 0 | 8.75 | 3.567 | 0 |
| SL_TN_34 | 490.432 | 8.923 | 0 | 8 | 3.939 | 0 |
| BT_ER_23a | 449.496 | 6.238 | 2 | 4.5 | 5.939 | 1 |
| MS_ER_8b | 454.648 | 4.201 | 0 | 6 | 5.814 | 1 |
| SL_TN_51 | 444.483 | 5.698 | 0 | 8 | 3.007 | 0 |
| SL_TN_63 | 444.483 | 5.698 | 0 | 8 | 3.002 | 0 |
| BT_ER_21c | 545.627 | 8.062 | 2 | 4.5 | 7.754 | 2 |
| BT_ER_15a | 492.564 | 7.054 | 3 | 6 | 5.127 | 1 |
| SL_TN_32 | 440.879 | 6.971 | 0 | 8 | 3.291 | 0 |
| SL_TN_53 | 445.471 | 7.567 | 0 | 9 | 2.305 | 0 |
| SL_TN_38 | 450.487 | 7.079 | 0 | 8.75 | 3.325 | 0 |
| MS_ER_8a | 480.686 | 7.171 | 0 | 6 | 6.628 | 1 |
| HC_TI_CT | 348.357 | 9.984 | 1 | 7.75 | 1.742 | 0 |
| SL_TN_35 | 472.46 | 6.031 | 0 | 6.75 | 4.818 | 0 |
| SL_TN_21 | 426.508 | 7.617 | 0 | 8 | 3.504 | 0 |
| SL_TN_37 | 450.487 | 6.55 | 0 | 8.75 | 3.323 | 0 |
| BT_ER_21e | 575.653 | 5.019 | 2 | 7.7 | 6.795 | 2 |
| SL_TN_60 | 450.505 | 6.824 | 0 | 8 | 2.934 | 0 |
| SL_TN_47 | 420.418 | 6.224 | 0 | 8.5 | 2.307 | 0 |
| SL_TN_33 | 436.46 | 6.732 | 0 | 8.75 | 2.848 | 0 |
| BT_ER_21a | 504.575 | 7.879 | 2 | 4.5 | 6.882 | 2 |
| SL_TN_40 | 396.396 | 7.295 | 0 | 8.5 | 2.042 | 0 |
| SL_TN_52 | 474.509 | 5.824 | 0 | 8.75 | 3.042 | 0 |
| MS_ER_6a | 452.633 | 6.505 | 0 | 6 | 5.499 | 1 |
| SL_TN_39 | 450.444 | 7.35 | 0 | 9.5 | 2.209 | 0 |
| SL_TN_59 | 488.536 | 6.657 | 0 | 8.75 | 3.815 | 0 |
| SL_TN_57 | 474.509 | 6.071 | 0 | 9.7 | 2.768 | 0 |
| SL_TN_58 | 474.509 | 6.071 | 0 | 9.7 | 2.768 | 0 |
| BT_ER_25a | 461.507 | 7.977 | 1 | 4 | 6.11 | 1 |
| MS_ER_5b | 412.568 | 8.87 | 0 | 6 | 4.603 | 0 |
| MS_ER_4b | 398.541 | 3.782 | 0 | 6 | 4.268 | 0 |
| SL_TN_44 | 410.423 | 6.063 | 0 | 8.75 | 2.101 | 0 |
| SL_TN_42 | 386.419 | 6.579 | 0 | 8 | 1.975 | 0 |
| SL_TN_31 | 420.461 | 7.979 | 0 | 8 | 3.112 | 0 |
| SL_TN_41 | 412.456 | 7.171 | 0 | 8 | 2.707 | 0 |
| SL_TN_27 | 386.444 | 6.974 | 0 | 8 | 2.236 | 0 |
| SL_TN_16 | 386.444 | 8.057 | 0 | 8 | 2.481 | 0 |
| SL_TN_19 | 402.486 | 6.561 | 0 | 8 | 2.848 | 0 |
| SL_TN_12 | 358.39 | 8.016 | 0 | 8 | 1.783 | 0 |
| SL_TN_46 | 459.292 | 6.371 | 0 | 8 | 2.614 | 0 |
| SL_TN_20 | 388.46 | 6.587 | 0 | 8 | 2.382 | 0 |
| BT_ER_25b | 475.533 | 7.097 | 1 | 4 | 6.266 | 1 |
| SL_TN_26 | 372.417 | 6.793 | 0 | 8 | 1.878 | 0 |
| SL_TN_25 | 344.363 | 6.336 | 0 | 8 | 1.196 | 0 |
| SL_TN_17 | 374.433 | 6.114 | 0 | 8 | 1.856 | 0 |
| SL_TN_50 | 424.449 | 5.364 | 0 | 8.75 | 2.215 | 0 |
| HC_TI_14 | 302.405 | 8.12 | 0 | 3.25 | 4.681 | 0 |
| SL_TN_18 | 374.433 | 6.621 | 0 | 8 | 2.159 | 0 |
2.4. Pharmacophore Modeling


2.5. 3D-QSAR Results

2.6. MD Simulation







3. Materials and Method
3.1. Docking Studies
3.2. MM/GBSA Binding Free Energy Calculation
3.3. In Silico Predicted ADMET Properties
3.4. Pharmacophore Modeling
3.5. QSAR-Quantitative Structure Activity Relationship
3.6. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef] [PubMed]
- Lemaine, V.; Simmons, P.S. The adolescent female: Breast and reproductive embryology and anatomy. Clin. Anat. 2013, 26, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Bisoyi, P. Malignant Tumors—As Cancer. In Understanding Cancer; Elsevier: Amsterdam, The Netherlands, 2022; pp. 21–36. ISBN 978-0-323-99883-3. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780323998833000111 (accessed on 13 March 2023).
- Follain, G.; Herrmann, D.; Harlepp, S.; Hyenne, V.; Osmani, N.; Warren, S.C.; Timpson, P.; Goetz, J.G. Fluids and their mechanics in tumour transit: Shaping metastasis. Nat. Rev. Cancer 2020, 20, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Šarenac, T.; Mikov, M. Cervical Cancer, Different Treatments and Importance of Bile Acids as Therapeutic Agents in This Disease. Front. Pharmacol. 2019, 10, 484. [Google Scholar] [CrossRef] [PubMed]
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef]
- Mathur, P.; Sathishkumar, K.; Chaturvedi, M.; Das, P.; Sudarshan, K.L.; Santhappan, S.; Nallasamy, V.; John, A.; Narasimhan, S.; Roselind, F.S.; et al. Cancer Statistics, 2020: Report From National Cancer Registry Programme, India. JCO Glob. Oncol. 2020, 6, 1063–1075. [Google Scholar] [CrossRef]
- Kumar, N.; Gulati, H.K.; Sharma, A.; Heer, S.; Jassal, A.K.; Arora, L.; Kaur, S.; Singh, A.; Bhagat, K.; Kaur, A.; et al. Most recent strategies targeting estrogen receptor alpha for the treatment of breast cancer. Mol. Divers. 2021, 25, 603–624. [Google Scholar] [CrossRef]
- Nilsson, S.; Gustafsson, J.-Å. Estrogen Receptors: Therapies Targeted to Receptor Subtypes. Clin. Pharmacol. Ther. 2011, 89, 44–55. [Google Scholar] [CrossRef]
- Santen, R.J. Inhibition of aromatase: Insights from recent studies. Steroids 2003, 68, 559–567. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Li, X.; Wu, J.; Zhao, L.; Li, W.; Liu, J. Dynamics-Based Discovery of Novel, Potent Benzoic Acid Derivatives as Orally Bioavailable Selective Estrogen Receptor Degraders for ERα+ Breast Cancer. J. Med. Chem. 2021, 64, 7575–7595. [Google Scholar] [CrossRef]
- McDonnell, D.P.; Wardell, S.E.; Norris, J.D. Oral Selective Estrogen Receptor Downregulators (SERDs), a Breakthrough Endocrine Therapy for Breast Cancer. J. Med. Chem. 2015, 58, 4883–4887. [Google Scholar] [CrossRef]
- Baselga, J.; Swain, S.M. CLEOPATRA: A Phase III Evaluation of Pertuzumab and Trastuzumab for HER2-Positive Metastatic Breast Cancer. Clin. Breast Cancer 2010, 10, 489–491. [Google Scholar] [CrossRef]
- Nathan, M.R.; Schmid, P. A Review of Fulvestrant in Breast Cancer. Oncol. Ther. 2017, 5, 17–29. [Google Scholar] [CrossRef]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERα) and beta (ERβ): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef]
- Dahlman-Wright, K.; Cavailles, V.; Fuqua, S.A.; Jordan, V.C.; Katzenellenbogen, J.A.; Korach, K.S.; Maggi, A.; Muramatsu, M.; Parker, M.G.; Gustafsson, J.-Å. International Union of Pharmacology. LXIV. Estrogen Receptors. Pharmacol. Rev. 2006, 58, 773–781. [Google Scholar] [CrossRef]
- Rajagopal, K.; Varakumar, P.; Aparna, B.; Byran, G.; Jupudi, S. Identification of some novel oxazine substituted 9-anilinoacridines as SARS-CoV-2 inhibitors for COVID-19 by molecular docking, free energy calculation and molecular dynamics studies. J. Biomol. Struct. Dyn. 2021, 39, 5551–5562. [Google Scholar] [CrossRef]
- Horvath, D. Pharmacophore-Based Virtual Screening. In Chemoinformatics and Computational Chemical Biology; Bajorath, J., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 672, pp. 261–298. ISBN 978-1-60761-838-6. [Google Scholar] [CrossRef]
- Chaudhaery, S.S.; Roy, K.K.; Saxena, A.K. Consensus Superiority of the Pharmacophore-Based Alignment, Over Maximum Common Substructure (MCS): 3D-QSAR Studies on Carbamates as Acetylcholinesterase Inhibitors. J. Chem. Inf. Model. 2009, 49, 1590–1601. [Google Scholar] [CrossRef]
- Alnajjar, R.; Mostafa, A.; Kandeil, A.; Al-Karmalawy, A.A. Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease. Heliyon 2020, 6, e05641. [Google Scholar] [CrossRef]
- Liang, J.-J.; Yu, W.-L.; Yang, L.; Xie, B.-H.; Qin, K.-M.; Yin, Y.-P.; Yan, J.-J.; Gong, S.; Liu, T.-Y.; Zhou, H.-B.; et al. Design and synthesis of marine sesterterpene analogues as novel estrogen receptor α degraders for breast cancer treatment. Eur. J. Med. Chem. 2022, 229, 114081. [Google Scholar] [CrossRef]
- Nakagawa-Goto, K.; Chen, J.-Y.; Cheng, Y.-T.; Lee, W.-L.; Takeya, M.; Saito, Y.; Lee, K.-H.; Shyur, L.-F. Novel sesquiterpene lactone analogues as potent anti-breast cancer agents. Mol. Oncol. 2016, 10, 921–937. [Google Scholar] [CrossRef]
- Zhang, S.; Won, Y.-K.; Ong, C.-N.; Shen, H.-M. Anti-Cancer Potential of Sesquiterpene Lactones: Bioactivity and Molecular Mechanisms. Curr. Med. Chem.-Anti-Cancer Agents 2005, 5, 239–249. [Google Scholar] [CrossRef]
- Kalirajan, R.; Sivakumar, S.U.; Jubie, S.; Gowramma, B.; Suresh, B. Synthesis and biological evaluation of some heterocyclic derivatives of chalcones. Int. J. Chem. Tech. Res. 2009, 1, 27–34. [Google Scholar]
- Jeon, K.-H.; Yu, H.-B.; Kwak, S.Y.; Kwon, Y.; Na, Y. Synthesis and topoisomerases inhibitory activity of heteroaromatic chalcones. Bioorg. Med. Chem. 2016, 24, 5921–5928. [Google Scholar] [CrossRef]
- Bai, C.; Wu, S.; Ren, S.; Zhu, M.; Luo, G.; Xiang, H. Benzothiophene derivatives as selective estrogen receptor covalent antagonists: Design, synthesis and anti-ERα activities. Bioorg. Med. Chem. 2021, 47, 116395. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Anand, A.; Garg, S.; Khan, M.S.; Bhasin, S.; Asghar, M.N.; Emran, T.B. Structure-Based In Silico Investigation of Agonists for Proteins Involved in Breast Cancer. Evid.-Based Complement. Alternat. Med. 2022, 2022, 7278731. [Google Scholar] [CrossRef] [PubMed]
- Pattar, S.V.; Adhoni, S.A.; Kamanavalli, C.M.; Kumbar, S.S. In silico molecular docking studies and MM/GBSA analysis of coumarin-carbonodithioate hybrid derivatives divulge the anticancer potential against breast cancer. Beni-Suef Univ. J. Basic Appl. Sci. 2020, 9, 36. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins Struct. Funct. Bioinforma. 2006, 65, 15–26. [Google Scholar] [CrossRef]
- Bissantz, C.; Folkers, G.; Rognan, D. Protein-Based Virtual Screening of Chemical Databases. 1. Evaluation of Different Docking/Scoring Combinations. J. Med. Chem. 2000, 43, 4759–4767. [Google Scholar] [CrossRef]
- Hussein, K.; Shihab, N.; Saeed, B. Anti-Cancer, Anti-Osteoporosis, and Molecular Docking Studies of Novel Chalcone and Epoxy Chalcone. Biointerface Res. Appl. Chem. 2021, 12, 6668–6685. [Google Scholar] [CrossRef]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1233–1237. [Google Scholar] [CrossRef]
- Wang, W.; Kollman, P.A. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J. Mol. Biol. 2000, 303, 567–582. [Google Scholar] [CrossRef]
- Rajagopal, K.; Kannan, R.; Aparna, B.; Varakumar, P.; Pandiselvi, A.; Gowramma, B. COVID-19 Activity of Some 9-Anilinoacridines substituted with Pyrazole against SARS CoV2 Main Protease: An In-silico Approach. Res. J. Pharm. Technol. 2023, 16, 529–534. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Etti, I.; Abdullah, R.; Hashim, N.; Kadir, A.; Abdul, A.; Etti, C.; Malami, I.; Waziri, P.; How, C. Artonin E and Structural Analogs from Artocarpus Species Abrogates Estrogen Receptor Signaling in Breast Cancer. Molecules 2016, 21, 839. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S. In Silico Models for Ecotoxicity of Pharmaceuticals. In In Silico Methods for Predicting Drug Toxicity; Benfenati, E., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 237–304. ISBN 978-1-4939-3609-0. [Google Scholar] [CrossRef]
- Vuorinen, A.; Odermatt, A.; Schuster, D. In silico methods in the discovery of endocrine disrupting chemicals. J. Steroid Biochem. Mol. Biol. 2013, 137, 18–26. [Google Scholar] [CrossRef]
- Yang, S.-Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Seidel, T.; Ibis, G.; Bendix, F.; Wolber, G. Strategies for 3D pharmacophore-based virtual screening. Drug Discov. Today Technol. 2010, 7, e221–e228. [Google Scholar] [CrossRef]
- Veeramachaneni, G.K.; Raj, K.K.; Chalasani, L.M.; Bondili, J.S.; Talluri, V.R. High-throughput virtual screening with e-pharmacophore and molecular simulations study in the designing of pancreatic lipase inhibitors. Drug Des. Devel. Ther. 2015, 9, 4397–4412. [Google Scholar] [CrossRef]
- Patel, H.M.; Noolvi, M.N.; Sharma, P.; Jaiswal, V.; Bansal, S.; Lohan, S.; Kumar, S.S.; Abbot, V.; Dhiman, S.; Bhardwaj, V. Quantitative structure–activity relationship (QSAR) studies as strategic approach in drug discovery. Med. Chem. Res. 2014, 23, 4991–5007. [Google Scholar] [CrossRef]
- Kalirajan, R.; Kulshrestha, V.; Sankar, S. Synthesis, Characterization and Antitumour Activity of Some Novel Oxazine Substituted 9-Anilinoacridines and their 3D-QSAR Studies. Indian J. Pharm. Sci. 2018, 80, 921–929. [Google Scholar] [CrossRef]
- Adcock, S.A.; McCammon, J.A. Molecular Dynamics: Survey of Methods for Simulating the Activity of Proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajagopal, K.; Kalusalingam, A.; Bharathidasan, A.R.; Sivaprakash, A.; Shanmugam, K.; Sundaramoorthy, M.; Byran, G. In Silico Drug Design of Anti-Breast Cancer Agents. Molecules 2023, 28, 4175. https://doi.org/10.3390/molecules28104175
Rajagopal K, Kalusalingam A, Bharathidasan AR, Sivaprakash A, Shanmugam K, Sundaramoorthy M, Byran G. In Silico Drug Design of Anti-Breast Cancer Agents. Molecules. 2023; 28(10):4175. https://doi.org/10.3390/molecules28104175
Chicago/Turabian StyleRajagopal, Kalirajan, Anandarajagopal Kalusalingam, Anubhav Raj Bharathidasan, Aadarsh Sivaprakash, Krutheesh Shanmugam, Monall Sundaramoorthy, and Gowramma Byran. 2023. "In Silico Drug Design of Anti-Breast Cancer Agents" Molecules 28, no. 10: 4175. https://doi.org/10.3390/molecules28104175
APA StyleRajagopal, K., Kalusalingam, A., Bharathidasan, A. R., Sivaprakash, A., Shanmugam, K., Sundaramoorthy, M., & Byran, G. (2023). In Silico Drug Design of Anti-Breast Cancer Agents. Molecules, 28(10), 4175. https://doi.org/10.3390/molecules28104175