

Formal [3 + 2] Cycloaddition of α-Imino Esters with Azo Compounds: Facile Construction of Pentasubstituted 1,2,4-Triazoline Skeletons

Abstract
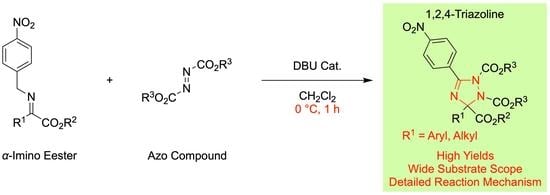
:
1. Introduction

2. Results and Discussion
2.1. Reaction Condition Optimization
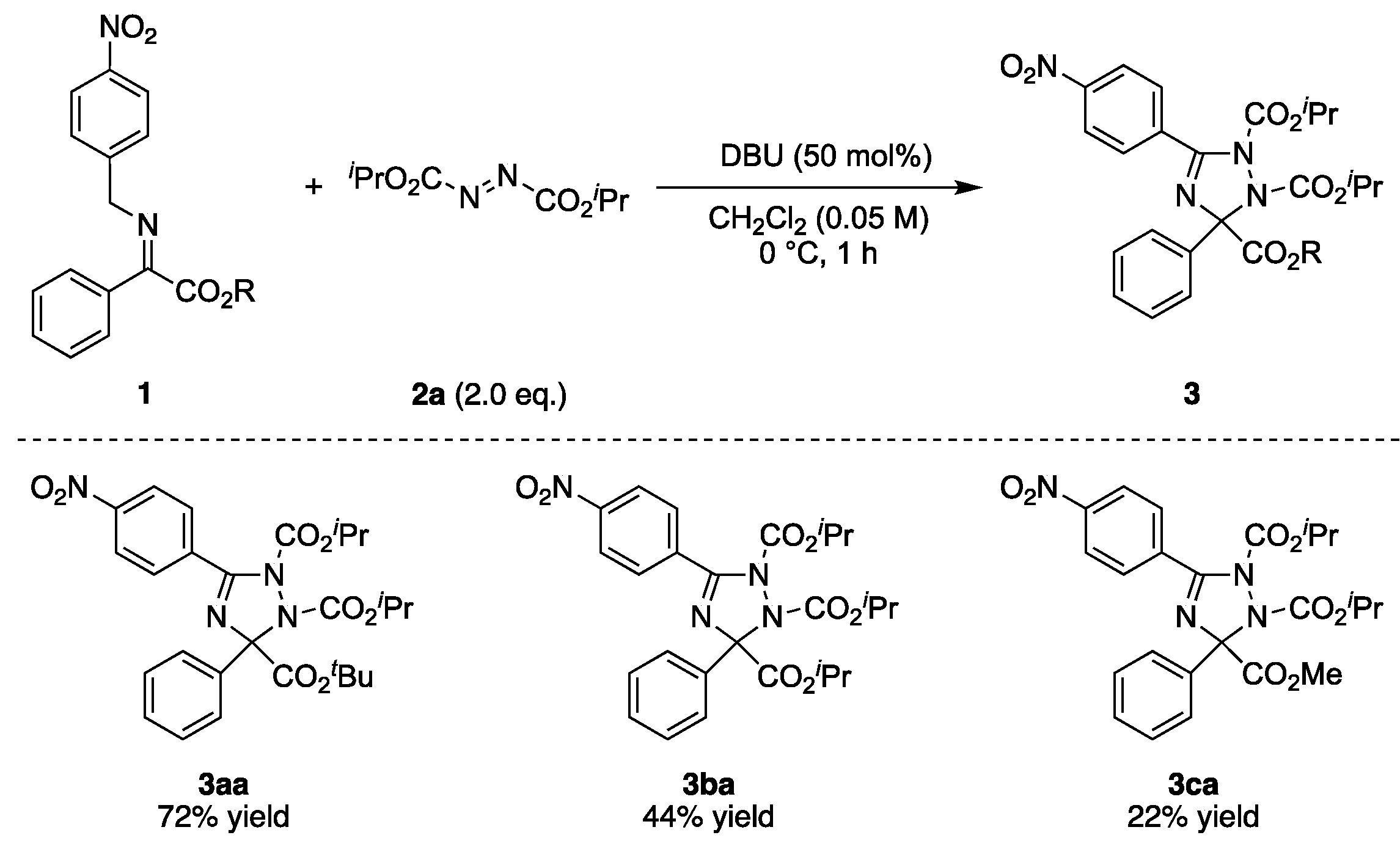
2.2. Substrate Scope
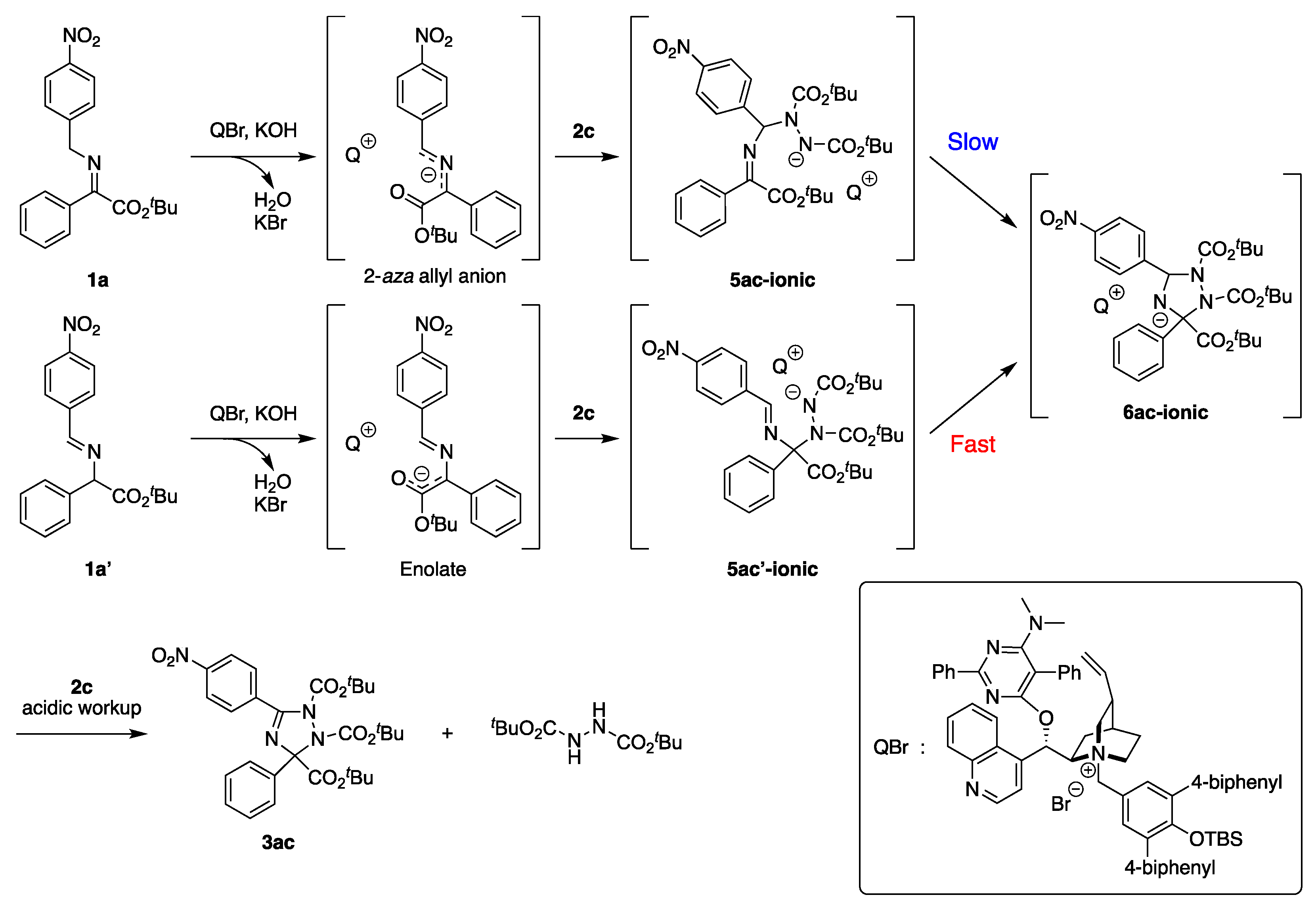
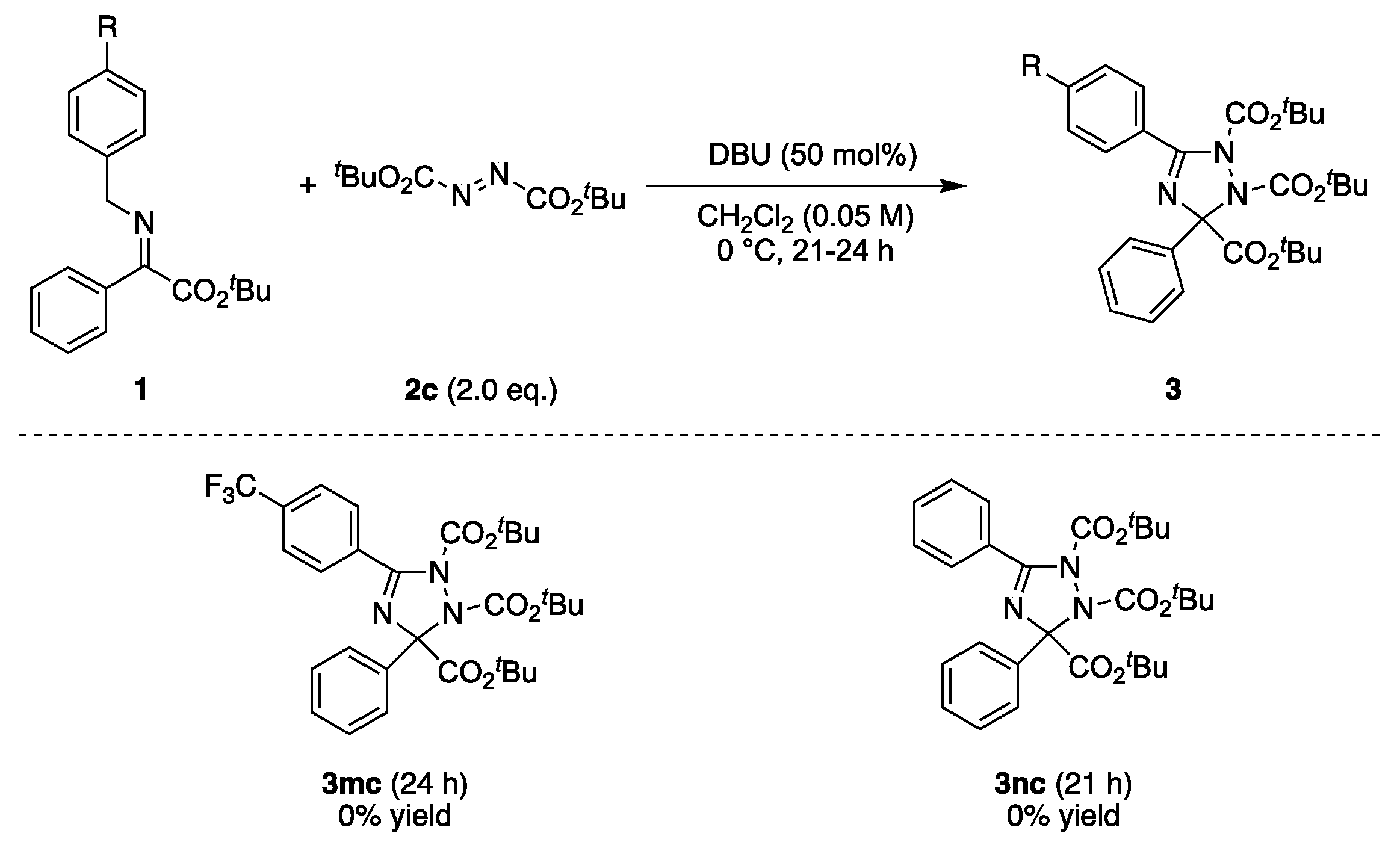
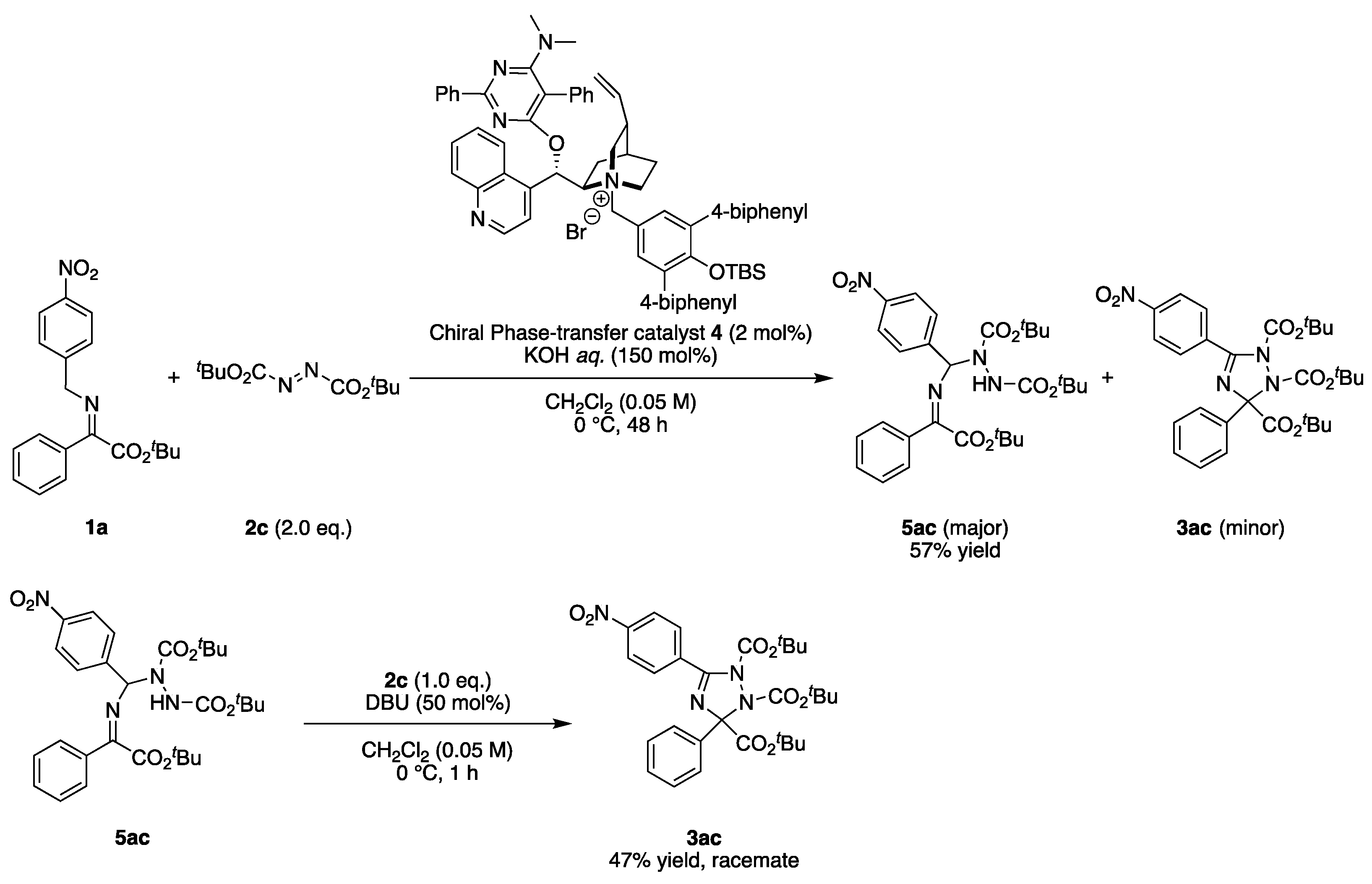
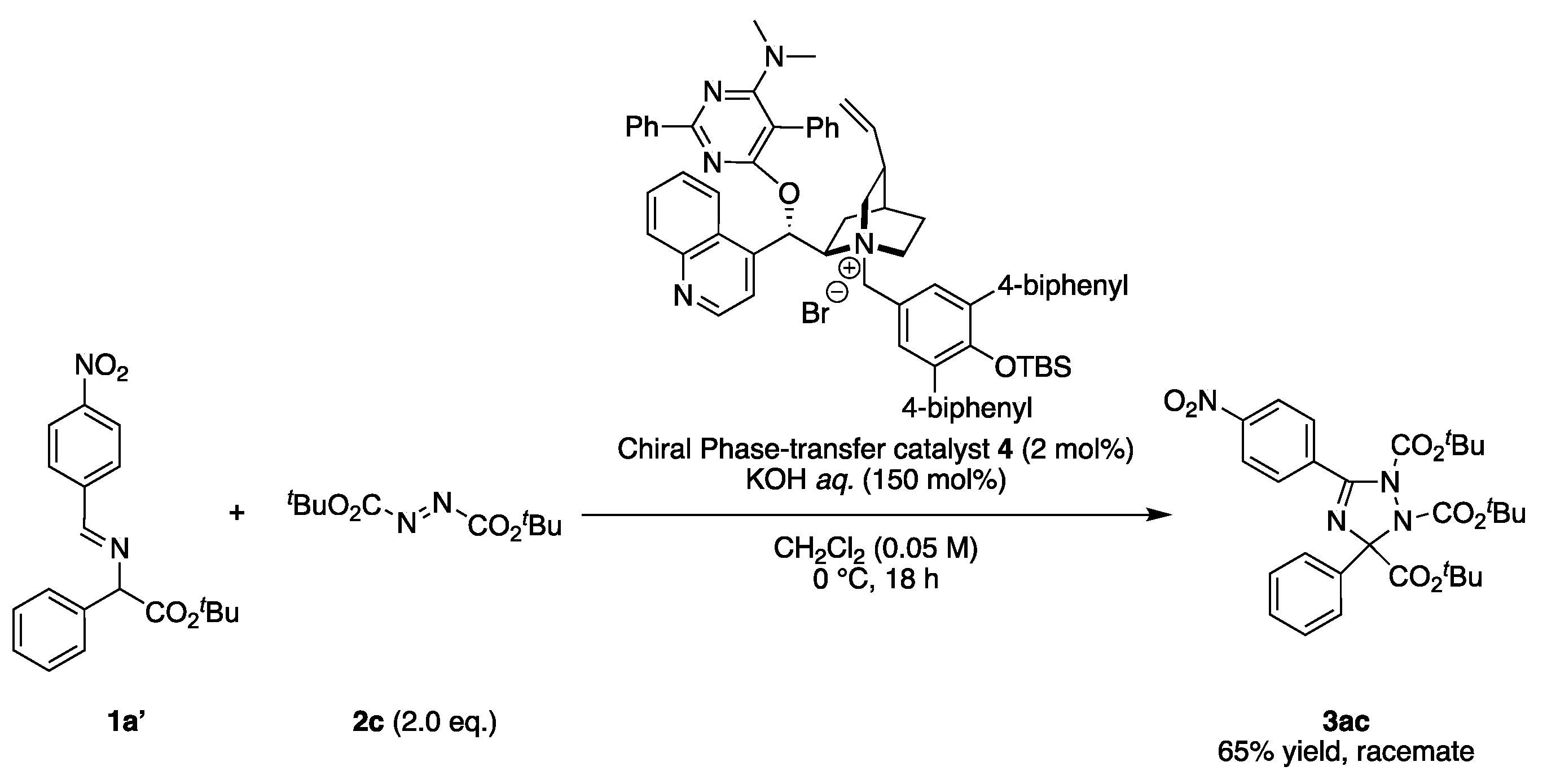
2.3. Asymmetric Synthesis
2.4. Reaction Mechanistic Study
3. Materials and Methods
3.1. Synthesis of Substrates and a Catalyst
3.2. Synthesis of 1,2,4-Triazolines
3.2.1. General Procedure for Table 1
3.2.2. General Procedure for Scheme 1, Scheme 2, Scheme 3 and Scheme 4 (Optimized Protocol)
- 3-(tert-butyl) 1,2-diisopropyl 5-(4-nitrophenyl)-3-phenyl-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3aa), White solid, 19.4 mg, 0.036 mmol, 72% yield (0.050 mmol scale reaction). m.p. 68–70 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 8.00–8.04 (m, 2H), 7.68–7.72 (m, 2H), 7.34–7.43 (m, 3H), 5.10 (sep, J = 6.2 Hz, 1H), 4.81 (sep, J = 6.2 Hz, 1H), 1.40 (s, 9H), 1.39 (d, J = 6.2 Hz, 3H), 1.36 (d, J = 6.2 Hz, 3H), 1.12 (d, J = 6.2 Hz, 3H), 1.11 (d, J = 6.2 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.5, 156.5, 154.5, 152.2, 149.4, 137.0, 135.0, 130.8, 128.5, 127.8, 127.3, 122.9, 95.3, 83.7, 72.5, 71.4, 27.6, 22.2, 21.8, 21.53, 21.46; HRMS (ESI+ in MeCN) calcd. for C27H33O8N4+ (M + H) 541.2293 found 541.2297; IR (KBr) ν 2982, 1752, 1527, 1349, 1260, 1155, 1102, 849 cm−1.
- tri-isopropyl 5-(4-nitrophenyl)-3-phenyl-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3ba), White solid, 12.8 mg, 0.024 mmol, 44% yield (0.055 mmol scale reaction). m.p. 60–62 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 8.02–8.06 (m, 2H), 7.65–7.69 (m, 2H), 7.35–7.44 (m, 3H), 5.08 (sep, J = 6.2 Hz, 1H), 5.03 (sep, J = 6.2 Hz, 1H), 4.84 (sep, J = 6.2 Hz, 1H), 1.37 (d, J = 6.2 Hz, 3H), 1.33 (d, J = 6.2 Hz, 3H), 1.22 (d, J = 6.2 Hz, 3H), 1.18 (d, J = 6.2 Hz, 3H), 1.13 (d, J = 6.2 Hz, 3H), 1.12 (d, J = 6.2 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 166.4, 156.8, 154.2, 152.1, 149.5, 136.8, 134.7, 130.9, 128.7, 127.9, 127.2, 122.9, 94.6, 72.7, 71.4, 71.1, 22.1, 21.7, 21.52, 21.45; HRMS (ESI+ in MeCN) calcd. for C26H31O8N4+ (M + H) 527.2136 found 527.2241; IR (KBr) ν 2983, 1751, 1527, 1349, 1256, 1183, 1099, 849 cm−1.
- 1,2-diisopropyl 3-methyl 5-(4-nitrophenyl)-3-phenyl-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3ca), White solid, 7.6 mg, 0.016 mmol, 23% yield (0.069 mmol scale reaction). m.p. 60–62 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.31 (m, 2H), 8.04–8.08 (m, 2H), 7.66–7.70 (m, 2H), 7.37–7.46 (m, 3H), 5.11 (sep, J = 6.4 Hz, 1H), 4.83 (sep, J = 6.4 Hz, 1H), 3.76 (s, 3H), 1.36 (d, J = 6.4 Hz, 3H), 1.32 (d, J = 6.4 Hz, 3H), 1.14 (d, J = 6.4 Hz, 3H), 1.13 (d, J = 6.4 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 167.5, 157.1, 154.2, 151.9, 149.5, 136.6, 134.4, 131.1, 128.9, 128.1, 127.1, 122.9, 94.3, 72.8, 71.6, 53.6, 22.0, 21.7, 21.54, 21.47; HRMS (ESI+ in MeCN) calcd. for C24H27O8N4+ (M + H) 499.1823 found 499.1828; IR (KBr) ν 2983, 1748, 1526, 1349, 1254, 1184, 1102, 849 cm−1.
- 3-(tert-butyl) 1,2-diisopropyl 5-(4-nitrophenyl)-3-(o-tolyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3da), White solid, 16.6 mg, 0.299 mmol, 60% yield (0.050 mmol scale reaction). m.p. 81–83 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.25–8.29 (m, 2H), 7.96–8.00 (m, 2H), 7.67 (d, J = 7.7 Hz, 1H), 7.25–7.30 (m, 2H), 7.17–7.22 (m, 1H), 5.09 (sep, J = 6.4 Hz, 1H), 4.86 (sep, J = 6.4 Hz, 1H), 2.62 (s, 3H), 1.42 (s, 9H), 1.38 (d, J = 6.4 Hz, 6H), 1.20 (d, J = 6.4 Hz, 3H), 1.19 (d, J = 6.4 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.0, 155.9, 154.5, 152.2, 149.3, 137.5, 135.0, 134.7, 131.6, 130.9, 128.7, 126.7, 125.5, 122.9, 96.9, 83.6, 72.5, 71.4, 27.5, 22.1, 21.99, 21.85, 21.64, 21.58; HRMS (ESI+ in MeCN) calcd. for C28H35O8N4+ (M + H) 555.2449 found 555.2449; IR (KBr) ν 2982, 1744, 1527, 1349, 1257, 1157, 1103, 849 cm−1.
- 3-(tert-butyl) 1,2-diisopropyl 5-(4-nitrophenyl)-3-(m-tolyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3ea), White solid, 14.8 mg, 0.027 mmol, 53% yield (0.050 mmol scale reaction). m.p. 96–98 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 8.04–8.00 (m, 2H), 7.50 (s, 1H), 7.48 (d, J = 6.8 Hz, 1H), 7.28–7.32 (m, 1H), 7.18 (d, J = 7.8 Hz, 1H), 5.11 (sep, J = 6.3 Hz, 1H), 4.81 (sep, J = 6.3 Hz, 1H), 2.40 (s, 3H), 1.40 (s, 9H), 1.39 (d, J = 6.3 Hz, 3H), 1.36 (d, J = 6.3 Hz, 3H), 1.14 (d, J = 6.3 Hz, 6H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.5, 156.3, 154.5, 152.2, 149.4, 137.4, 136.9, 135.0, 130.8, 129.3, 127.93, 127.81, 124.5, 122.9, 95.4, 83.6, 72.6, 71.3, 27.6, 22.2, 21.8, 21.59, 21.56, 21.48; HRMS (ESI+ in MeCN) calcd. for C28H35O8N4+ (M + H) 555.2449 found 555.2454; IR (KBr) ν 2981, 1747, 1526, 1348, 1253, 1155, 1103, 845 cm−1.
- 3-(tert-butyl) 1,2-diisopropyl 5-(4-nitrophenyl)-3-(p-tolyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3fa), White solid, 16.8 mg, 0.030 mmol, 61% yield (0.050 mmol scale reaction). m.p. 76–78 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 7.99–8.03 (m, 2H), 7.55–7.59 (m, 2H), 7.21 (d, J = 8.0 Hz, 2H), 5.09 (sep, J = 6.3 Hz, 1H), 4.81 (sep, J = 6.3 Hz, 1H), 2.37 (s, 3H), 1.40 (s, 9H), 1.39 (d, J = 6.4 Hz, 3H), 1.35 (d, J = 6.4 Hz, 3H), 1.13 (d, J = 6.4 Hz, 3H), 1.20 (d, J = 6.4 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.6, 156.3, 154.5, 152.2, 149.3, 138.4, 135.1, 134.0, 130.7, 128.6, 127.2, 122.9, 95.3, 83.6, 72.6, 71.3, 27.6, 22.2, 21.8, 21.56, 21.45, 21.1; HRMS (ESI+ in MeCN) calcd. for C28H35O8N4+ (M + H) 555.2449 found 555.2455; IR (KBr) ν 2982, 1747, 1526, 1348, 1258, 1155, 1102, 849 cm−1.
- 3-(tert-butyl) 1,2-diisopropyl 3-(4-methoxyphenyl)-5-(4-nitrophenyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3ga), White solid, 18.6 mg, 0.033 mmol, 65% yield (0.050 mmol scale reaction). m.p. 98–100 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 8.00–8.04 (m, 2H), 7.59–7.63 (m, 2H), 6.91–6.95 (m, 2H), 5.09 (sep, J = 6.4 Hz, 1H), 4.81 (sep, J = 6.4 Hz, 1H), 3.83 (s, 3H), 1.40 (s, 9H), 1.39 (d, J = 6.4 Hz, 3H), 1.35 (d, J = 6.4 Hz, 3H), 1.13 (d, J = 6.4 Hz, 3H), 1.10 (d, J = 6.4 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.7, 159.7, 156.3, 154.5, 152.2, 149.3, 135.1, 130.7, 129.1, 128.6, 122.9, 113.2, 95.0, 83.6, 72.6, 71.3, 55.2, 27.6, 22.2, 21.8, 21.56, 21.44; HRMS (ESI+ in MeCN) calcd. for C28H35O9N4+ (M + H) 571.2399 found 571.2404; IR (KBr) ν 2980, 1757, 1526, 1348, 1253, 1155, 1102, 849 cm−1.
- 3-(tert-butyl) 1,2-diisopropyl 3-(4-bromophenyl)-5-(4-nitrophenyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3ha), White solid, 13.3 mg, 0.215 mmol, 43% yield (0.050 mmol scale reaction). m.p. 156–158 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.27–8.31 (m, 2H), 7.99–8.03 (m, 2H), 7.51–7.59 (m, 4H), 5.10 (sep, J = 6.3 Hz, 1H), 4.82 (sep, J = 6.3 Hz, 1H), 1.40 (s, 9H), 1.39 (d, J = 6.3 Hz, 3H), 1.36 (d, J = 6.3 Hz, 3H), 1.13 (d, J = 6.3 Hz, 3H), 1.11 (d, J = 6.3 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.0, 156.7, 154.5, 152.0, 149.5, 136.3, 134.8, 131.0, 130.8, 129.1, 122.99, 122.00, 94.8, 84.1, 72.8, 71.6, 27.6, 22.2, 21.8, 21.56, 21.42; HRMS (ESI+ in MeCN) calcd. for C27H32O8N4Br+ (M + H) 619.1398 found 619.1402; IR (KBr) ν 2981, 1751, 1526, 1348, 1257, 1155, 1102, 849 cm−1.
- 3-(tert-butyl) 1,2-diisopropyl 3-(4-chlorophenyl)-5-(4-nitrophenyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3ia), White solid, 14.4 mg, 0.025 mmol, 50% yield (0.050 mmol scale reaction). m.p. 154–156 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.27–8.31 (m, 2H), 7.99–8.03 (m, 2H), 7.61–7.65 (m, 2H), 7.35–7.40 (m, 2H), 5.10 (sep, J = 6.4 Hz, 1H), 4.82 (sep, J = 6.4 Hz, 1H), 1.40 (s, 9H), 1.39 (d, J = 6.4 Hz, 3H), 1.35 (d, J = 6.4 Hz, 3H), 1.13 (d, J = 6.4 Hz, 3H), 1.11 (d, J = 6.4 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.2, 156.7, 154.5, 152.0, 149.4, 135.7, 134.8, 134.5, 130.8, 128.8, 128.0, 123.0, 94.8, 84.1, 72.8, 71.6, 27.6, 22.2, 21.8, 21.54, 21.42; HRMS (ESI+ in MeCN) calcd. for C27H32O8N4Cl+ (M + H) 575.1903 found 575.1910; IR (KBr) ν 2981, 1751, 1527, 1351, 1259, 1155, 1102, 849 cm−1.
- 3-(tert-butyl) 1,2-diisopropyl 5-(4-nitrophenyl)-3-(4-(trifluoromethyl)phenyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3ja), White solid, 12.7 mg, 0.021mmol, 42% yield (0.050 mmol scale reaction). m.p. 77–79 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.27–8.32 (m, 2H), 8.00–8.04 (m, 2H), 7.83 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.3 Hz, 2H), 5.12 (sep, J = 6.3 Hz, 1H), 4.82 (sep, J = 6.3 Hz, 1H), 1.40 (s, 9H), 1.39 (d, J = 6.3 Hz, 3H), 1.37 (d, J = 6.3 Hz, 3H), 1.13 (d, J = 6.3 Hz, 3H), 1.10 (d, J = 6.3 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.0, 157.0, 154.5, 151.9, 149.5, 141.1, 134.6, 130.86, 130.71 (q, J = 32.3 Hz), 127.8, 124.8 (q, J = 3.9 Hz), 123.9 (q, J = 272.8 Hz), 123.0, 94.8, 84.3, 72.9, 71.7, 27.6, 22.2, 21.8, 21.54, 21.40; 19F-NMR (376 MHz, CHLOROFORM-D) δ -62.5; HRMS (ESI+ in MeCN) calcd. for C28H32O8N4F3+ (M + H) 609.2167 found 609.2172; IR (KBr) ν 2983, 1752,1528,1326, 1257, 1165, 1102, 850 cm−1.
- 3-(tert-butyl) 1,2-diethyl 5-(4-nitrophenyl)-3-phenyl-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3cb), White solid, 13.6 mg, 0.027 mmol, 53% yield (0.050 mmol scale reaction). m.p. 67–69 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 8.01–8.05 (m, 2H), 7.68–7.72 (m, 2H), 7.35–7.45 (m, 3H), 4.38–4.46 (m, 1H), 4.20–4.30 (m, 1H), 4.05–4.18 (m, 2H), 1.40 (s, 9H), 1.36 (t, J = 7.1 Hz, 3H), 1.10 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.4, 156.2, 154.9, 152.4, 149.4, 136.8, 134.8, 130.8, 128.6, 127.9, 127.3, 123.0, 95.5, 83.9, 64.1, 63.1, 27.6, 14.4, 13.8; HRMS (ESI+ in MeCN) calcd. for C25H29O8N4+ (M + H) 513.1980 found 513.1984; IR (KBr) ν 2980, 1752, 1526, 1351, 1258, 1153, 1022, 845 cm−1.
- tri-tert-butyl 5-(4-nitrophenyl)-3-phenyl-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3ac), White solid, 22.4 mg, 0.039 mmol, 78% yield (0.050 mmol scale reaction). Large-scale synthesis was conducted using 1.0 mmol (340.4 mg) of 1a, and 0.79 mmol (447.5 mg, 79% yield) of 3ac was isolated. m.p. 87–89 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 7.99–8.03 (m, 2H), 7.68–7.71 (m, 2H), 7.33–7.44 (m, 3H), 1.58 (s, 9H), 1.41 (s, 9H), 1.29 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.9, 156.6, 153.5, 151.0, 149.3, 137.3, 135.4, 130.7, 128.4, 127.8, 127.3, 122.9, 94.8, 84.8, 83.6, 83.0, 28.2, 27.6 (1 peak is overlapped with the other peak); HRMS (ESI+ in MeCN) calcd. for C29H37O8N4+ (M + H) 569.2606 found 569.2615; IR (KBr) ν 2979, 1744, 1527, 1369, 1349, 1253, 1149, 849 cm−1; HPLC (CHIRALPAK AD-H column, hexane/2-propanol = 95/5, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 5.8 min and second peak: tR = 6.7 min.
- tri-tert-butyl 5-(4-nitrophenyl)-3-(o-tolyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3dc), White solid, 19.0 mg, 0.033 mmol, 65% yield (0.050 mmol scale reaction). m.p. 99–101 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.25–8.29 (m, 2H), 7.94–7.98 (m, 2H), 7.70 (d, J = 7.5 Hz, 1H), 7.19–7.29 (m, 3H), 2.62 (s, 3H), 1.58 (s, 9H), 1.42 (s, 9H), 1.36 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.5, 156.0, 153.7, 151.0, 149.2, 137.5, 135.4, 135.0, 131.6, 130.8, 128.6, 126.6, 125.5, 122.9, 96.4, 84.8, 83.6, 83.0, 28.2, 27.7, 27.5, 22.0; HRMS (ESI+ in MeCN) calcd. for C30H39O8N4+ (M + H) 583.2762 found 583.2767; IR (KBr) ν 2979, 1742, 1527, 1369, 1348, 1254, 1150, 849 cm−1.
- tri-tert-butyl 5-(4-nitrophenyl)-3-(m-tolyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3ec), White solid, 23.0 mg, 0.039 mmol, 79% yield (0.050 mmol scale reaction). m.p. 76–78 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 7.99–8.03 (m, 2H), 7.50 (s, 1H), 7.49 (d, J = 7.7 Hz, 1H), 7.28–7.33 (m, 1H), 7.17 (d, J = 7.8 Hz, 1H), 2.41 (s, 3H), 1.58 (s, 9H), 1.42 (s, 9H), 1.31 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.9, 156.5, 153.5, 151.0, 149.2, 137.3, 137.1, 135.4, 130.7, 129.2, 127.9, 127.7, 124.5, 122.9, 94.9, 84.7, 83.5, 82.9, 28.2, 27.6, 21.6 (1 peak is overlapped with the other peak); HRMS (ESI+ in MeCN) calcd. for C30H39O8N4+ (M + H) 583.2762 found 583.2770; IR (KBr) ν 2979, 1743, 1526, 1369, 1348, 1257, 1149, 851 cm−1.
- tri-tert-butyl 5-(4-nitrophenyl)-3-(p-tolyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3fc), White solid, 21.4 mg, 0.0367 mmol, 73% yield (0.050 mmol scale reaction). m.p. 96–98 °C;1H-NMR (400 MHz, CHLOROFORM-D) δ 8.25–8.30 (m, 2H), 7.98–8.02 (m, 2H), 7.56–7.59 (m, 2H), 7.22 (d, J = 8.0 Hz, 2H), 2.38 (s, 3H), 1.57 (s, 9H), 1.41 (s, 9H), 1.29 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 166.0, 156.5, 153.4, 151.0, 149.2, 138.2, 135.5, 134.3, 130.7, 128.5, 127.2, 122.9, 94.8, 84.7, 83.5, 82.9, 28.2, 27.6, 21.1 (1 peak is overlapped with the other peak); HRMS (ESI+ in MeCN) calcd. for C30H39O8N4+ (M + H) 583.2762 found 583.2767; IR (KBr) ν 2979, 1744, 1527, 1369, 1348, 1254, 1150, 850 cm−1.
- tri-tert-butyl 3-(4-methoxyphenyl)-5-(4-nitrophenyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3gc), White solid, 24.4 mg, 0.041 mmol, 82% yield (0.050 mmol scale reaction). m.p. 87–89 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 7.98–8.02 (m, 2H), 7.59–7.64 (m, 2H), 6.91–6.96 (m, 2H), 3.83 (s, 3H), 1.58 (s, 9H), 1.41 (s, 9H), 1.29 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 166.1, 159.6, 156.5, 153.4, 151.0, 149.2, 135.5, 130.6, 129.4, 128.6, 122.9, 113.2, 94.5, 84.7, 83.5, 82.9, 55.2, 28.2, 27.6 (1 peak is overlapped with the other peak); HRMS (ESI+ in MeCN) calcd. for C30H39O9N4+ (M + H) 599.2712 found 599.2715; IR (KBr) ν 2979, 1744, 1527, 1369, 1348, 1253, 1150, 849 cm−1.
- tri-tert-butyl 3-(4-bromophenyl)-5-(4-nitrophenyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3hc), White solid, 27.0 mg, 0.042 mmol, 83% yield (0.050 mmol scale reaction). m.p. 94–96 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 7.97–8.00 (m, 2H), 7.52–7.60 (m, 4H), 1.58 (s, 9H), 1.41 (s, 9H), 1.29 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.6, 157.0, 153.4, 150.8, 149.3, 136.6, 135.2, 130.9, 130.7, 129.1, 123.0, 122.7, 94.2, 85.1, 84.2, 83.2, 28.2, 27.6 (1 peak is overlapped with the other peak); HRMS (ESI+ in MeCN) calcd. for C29H36O8N4Br+ (M + H) 647.1711 found 647.1721; IR (KBr) ν 2979, 1751, 1527, 1369, 1348, 1253, 1149, 849 cm−1.
- tri-tert-butyl 3-(4-chlorophenyl)-5-(4-nitrophenyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3ic), White solid, 25.4 mg, 0.042 mmol, 84% yield (0.050 mmol scale reaction). m.p. 86–88 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.31 (m, 2H), 7.97–8.02 (m, 2H), 7.62–7.66 (m, 2H), 7.36–7.40 (m, 2H), 1.58 (s, 9H), 1.41 (s, 9H), 1.28 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.6, 156.9, 153.4, 150.8, 149.3, 136.0, 135.2, 134.4, 130.7, 128.8, 128.0, 123.0, 94.3, 85.0, 84.0, 83.2, 28.2, 27.6 (1 peak is overlapped with the other peak); HRMS (ESI+ in MeCN) calcd. for C29H36O8N4Cl+ (M + H) 603.2216 found 603.2227; IR (KBr) ν 2979, 1752, 1527,1369, 1348, 1255, 1149, 848 cm−1.
- tri-tert-butyl 5-(4-nitrophenyl)-3-(4-(trifluoromethyl)phenyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3jc), White solid, 27.7 mg, 0.044 mmol, 87% yield (0.050 mmol scale reaction). m.p. 106–108 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.27–8.31 (m, 2H), 7.98–8.02 (m, 2H), 7.84 (d, J = 8.2 Hz, 2H), 7.67 (d, J = 8.2 Hz, 2H), 1.59 (s, 9H), 1.41 (s, 9H), 1.28 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 165.5, 157.1, 153.5, 150.7, 149.4, 141.4, 135.1, 130.8, 130.5 (q, J = 33.1 Hz), 127.8, 124.8 (q, J = 3.7 Hz), 124.0 (q, J = 273.1 Hz), 123.0, 94.3, 85.1, 84.2, 83.4, 28.2, 27.6 (1 peak is overlapped with the other peak); 19F-NMR (376 MHz, CHLOROFORM-D) δ -62.5; HRMS (ESI+ in MeCN) calcd. for C30H36O8N4F3+ (M + H) 637.2480 found 637.2484; IR (KBr) ν 2980, 1751, 1528, 1370, 1326, 1253, 1149, 850 cm−1.
- tri-tert-butyl 3-(naphthalen-2-yl)-5-(4-nitrophenyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3kc), White solid, 23.6 mg, 0.038 mmol, 76% yield (0.050 mmol scale reaction). m.p. 108–110 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.26–8.30 (m, 2H), 8.13 (s, 1H), 8.00–8.04 (m, 2H), 7.83–7.91 (m, 4H), 7.46–7.53 (m, 2H), 1.61 (s, 9H), 1.43 (s, 9H), 1.31 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 166.0, 156.8, 153.6, 151.0, 149.3, 135.3, 134.83 133.3, 132.7, 130.8, 128.4, 127.6, 127.3, 126.4, 126.02, 125.96, 125.7, 123.0, 95.0, 84.8, 83.9, 83.1, 28.2, 27.7 (1 peak is overlapped with the other peak); HRMS (ESI+ in MeCN) calcd. for C33H39O8N4+ (M + H) 619.2762 found 619.2767; IR (KBr) ν 2979, 1746, 1526, 1369, 1348, 1252, 1149, 851 cm−1.
- tri-tert-butyl 3-(tert-butyl)-5-(4-nitrophenyl)-1H-1,2,4-triazole-1,2,3(3H)-tricarboxylate (3lc), White solid, 7.92 mg, 0.014 mmol, 29% yield (0.050 mmol scale reaction). m.p. 68–70 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.27–8.31 (m, 2H), 7.91–7.95 (m, 2H), 1.56 (s, 9H), 1.40 (s, 9H), 1.30 (s, 9H), 1.19 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 164.8, 155.9, 154.6, 150.7, 149.0, 136.4, 130.0, 123.0, 98.7, 84.3, 82.8, 82.6, 39.1, 28.1, 27.76, 27.62, 25.2; HRMS (ESI+ in MeCN) calcd. For C27H41O8N4+ (M + H) 549.2919 found 549.2920; IR (KBr) ν 2979, 1758, 1528, 1370, 1348, 1255, 1149, 850 cm−1.
3.2.3. General Procedure for Scheme 5 (for the Synthesis of 5ac)
- di-tert-butyl (Z)-1-(((2-(tert-butoxy)-2-oxo-1-phenylethylidene)amino)(4-nitrophenyl)methyl)hydrazine-1,2-dicarboxylate (5ac), White solid, 30.3 mg, 0.053 mmol, 53% yield (0.10 mmol scale reaction). m.p. 85–87 °C: 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.15 (d, J = 8.8 Hz, 2H), 7.84–7.86 (m, 2H), 7.64 (d, J = 8.5 Hz, 2H), 7.49–7.53 (m, 1H), 7.42–7.46 (m, 2H), 6.88 (br, 1H), 6.50 (br, 1H), 1.48 (s, 9H), 1.46 (s, 9H), 1.31 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 163.4, 162.2, 154.4, 147.8, 145.4, 133.9, 131.8, 128.9, 128.6, 127.9, 123.0, 84.9, 82.4, 80.9, 28.2, 28.08, 28.01 (2 peaks are overlapped with the other peaks); HRMS (ESI+ in MeCN) calcd. for C29H39O8N4+ (M + H) 571.2762 found 571.2761; IR (KBr) ν 2979, 1727, 1525, 1368, 1346, 1259, 1153, 854 cm−1.
3.2.4. General Procedure for Scheme 5 (for the Synthesis of 3ac)
3.2.5. General Procedure for Scheme 6
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Murphy, M.; Bernard, E.M.; Ishimaru, T.; Armstrong, D. Activity of voriconazole (UK-109496) against clinical isolates of Aspergillus species and its effectiveness in an experimental model of invasive pulmonary aspergillosis. Antimicrob. Agents Chemother. 1997, 41, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Moosa, M.-Y.S.; Sobel, J.D.; Elhalis, H.; Du, W.; Akins, R.A. Fungicidal Activity of Fluconazole against Candida Albicans in a Synthetic Vagina-Simulative Medium. Antimicrob. Agents Chemother. 2004, 48, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Abdelli, A.; Azzouni, S.; Plais, R.; Gaucher, A.; Efrit, M.L.; Prim, D. Recent advances in the chemistry of 1,2,4-triazoles: Synthesis, reactivity and biological activities. Tetrahedron Lett. 2021, 86, 153518. [Google Scholar] [CrossRef]
- Imberg, L.; Platte, S.; Erbacher, C.; Daniliuc, C.G.; Kalinina, S.A.; Dorner, W.; Poso, A.; Karst, U.; Kalinin, D.V. Amide-functionalized 1,2,4-Triazol-5-amines as Covalent Inhibitors of Blood Coagulation Factor XIIa and Thrombin. ACS Pharmacol. Transl. Sci. 2022, 5, 1318–1347. [Google Scholar] [CrossRef] [PubMed]
- Riederer, S.K.U.; Bechlars, B.; Herrmann, W.A.; Kühn, F.E. Chiral N-heterocyclic biscarbenes based on 1,2,4-triazole as ligands for metal-catalyzed asymmetric synthesis. Dalton Trans. 2011, 40, 41–43. [Google Scholar] [CrossRef]
- Yang, X.; Birman, V.B. Acyl transfer catalysis with 1,2,4-triazole anion. Org. Lett. 2009, 11, 1499–1502. [Google Scholar] [CrossRef]
- Kerr, M.S.; Rovis, T. Enantioselective Synthesis of Quaternary Stereocenters via a Catalytic Asymmetric Stetter Reaction. J. Am. Chem. Soc. 2004, 126, 8876–8877. [Google Scholar] [CrossRef]
- Huang, H.; Guo, W.; Wu, W.; Li, C.-J.; Jiang, H. Copper-Catalyzed Oxidative C(sp3)-H Functionalization for Facile Synthesis of 1,2,4-Triazoles and 1,3,5-Triazines from Amidines. Org. Lett. 2015, 17, 2894–2897. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, J.; Zheng, L.; Liu, Z.-Q. Recent Advances in the Synthesis of Nitrogen Heterocycles Using Arenediazonium Salts as Nitrogen Sources. Adv. Synth. Catal. 2020, 362, 4876–4895. [Google Scholar] [CrossRef]
- Liu, J.Q.; Shen, X.Y.; Wang, Y.H.; Wang, X.S.; Bi, X.H. [3 + 2] Cycloaddition of Isocyanides with Aryl Diazonium Salts: Catalyst-Dependent Regioselective Synthesis of 1,3- and 1,5-Disubstituted 1,2,4-Triazoles. Org. Lett. 2018, 20, 6930–6933. [Google Scholar] [CrossRef]
- Li, Y.; Ye, Z.; Chen, N.; Chen, Z.; Zhang, F. Intramolecular electrochemical dehydrogenative N–N bond formation for the synthesis of 1,2,4-triazolo [1,5-a]pyridines. Green Chem. 2019, 21, 4035–4039. [Google Scholar] [CrossRef]
- Ueda, S.; Nagasawa, H. Facile Synthesis of 1,2,4-Triazoles via a Copper-Catalyzed Tandem Addition–Oxidative Cyclization. J. Am. Chem. Soc. 2009, 131, 15080–15081. [Google Scholar] [CrossRef]
- Shang, E.; Zhang, J.; Bai, J.; Wang, Z.; Li, X.; Zhu, B.; Lei, X. Syntheses of [1,2,4]Triazolo [1,5-a]benzazoles Enabled by the Transition-Metal-Free Oxidative N–N Bond Formation. Chem. Commun. 2016, 52, 7028–7031. [Google Scholar] [CrossRef]
- Eliseeva, A.I.; Nesterenko, O.O.; Slepukhin, P.A.; Benassi, E.; Belskaya, N.P. Synthesis and Fluorescent Behaviour of 2-Aryl-4,5-dihydro-1H-1,2,4-triazoles. J. Org. Chem. 2017, 82, 86–100. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, F.-G.; Ma, J.-A. Cu-Catalysed three-component reaction of aryldiazonium salts with fluorinated diazo reagents and nitriles: Access to difluoro- and trifluoromethylated N1-aryl-1,2,4-triazoles. Adv. Synth. Catal. 2020, 362, 4432–4437. [Google Scholar] [CrossRef]
- Matsuzaki, H.; Takeda, N.; Yasui, M.; Okazaki, M.; Suzuki, S.; Ueda, M. Synthesis of multi-substituted 1,2,4-triazoles utilising the ambiphilic reactivity of hydrazones. Chem. Commun. 2021, 57, 12187–12190. [Google Scholar] [CrossRef]
- Liu, X.; Li, W.; Jiang, W.; Lu, H.; Liu, J.; Lin, Y.; Cao, H. Cu(II)-catalyzed C–H amidation/cyclization of azomethine imines with dioxazolones via acyl nitrenes: A direct access to diverse 1,2,4-triazole derivatives. Org. Lett. 2022, 24, 613–618. [Google Scholar] [CrossRef]
- Hirayama, T.; Watanabe, M.; Akazawa, C.; Ishigami, M.; Fujikawa, F.; Kasahara, T.; Otsuka, M.; Nishida, N.; Mizuno, D. Anti-tumor Activities of Some Heterocyclic and Nitrogen-containing Compounds. Yakugaku Zazzhi 1980, 100, 1225–1234. [Google Scholar] [CrossRef]
- Saleem, R.S.Z.; Tepe, J.J. Synthesis of 1,2,4-Triazolines and Triazoles Utilizing Oxazolones. J. Org. Chem. 2010, 75, 4330–4332. [Google Scholar] [CrossRef]
- Ibata, T.; Suga, H.; Isogami, Y.; Tamura, H.; Shi, X. Abnormal Diels–Alder Reaction of Oxazoles with 4-Phenyl-3H-1,2,4-triazole-3,5(4H)-dione and Diethyl Azodicarboxylate, and X-Ray Crystal Structure of an Adduct. Bull. Chem. Soc. Jpn. 1992, 65, 2998–3007. [Google Scholar] [CrossRef]
- Monge, D.; Jensen, K.L.; Marín, I.; Jørgensen, K.A. Synthesis of 1,2,4-Triazolines: Base-Catalyzed Hydrazination/Cyclization Cascade of α-Isocyano Esters and Amides. Org. Lett. 2011, 13, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, X.-H.; He, P.; Lin, L.-L.; Feng, X.-M. Enantioselective synthesis of 1,2,4-triazolines by chiral iron(ii)-complex catalyzed cyclization of α-isocyano esters and azodicarboxylates. Chem. Commun. 2013, 49, 2572–2574. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-X.; Bi, H.-L.; Zhou, H.; Yang, H.; Shi, M. Cinchona Alkaloid Squaramide Catalyzed Enantioselective Hydrazination/Cyclization Cascade Reaction of α-Isocyanoacetates and Azodicarboxylates. J. Org. Chem. 2013, 78, 9377–9382. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Chen, J.; Tu, M.; Piotrowski, D.W.; Huang, Y. Enantioselective Synthesis of 1,2,4-Triazolines Catalyzed by a Cinchona Alkaloid-derived Organocatalyst. Chem. Commun. 2013, 49, 1098–11100. [Google Scholar] [CrossRef]
- Ma, B.-W.; Luo, W.-W.; Lin, L.-L.; Liu, X.-H.; Feng, X.-M. A chiral cobalt(ii) complex catalyzed asymmetric formal [3 + 2] cycloaddition for the synthesis of 1,2,4-triazoline. Chem. Commun. 2017, 53, 4077–4079. [Google Scholar] [CrossRef]
- Wang, H.; Ren, Y.; Wang, K.; Man, Y.; Xiang, Y.; Li, N.; Tang, B. Visible light-induced cyclization reactions for the synthesis of 1,2,4-triazolines and 1,2,4-triazoles. Chem. Commun. 2017, 53, 9644–9647. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent Development and Application of Chiral Phase-Transfer Catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef]
- Lasch, R.; Heinrich, M.R. Cycloaddition reactions of glycine imine anions to phenylazocarboxylic esters—A new access to 1,3,5-trisubstituted 1,2,4-triazoles. Tetrahedron 2015, 71, 4282–4295. [Google Scholar] [CrossRef]
- Eftekhari-Sis, B.; Zirak, M. α-Imino Esters in Organic Synthesis: Recent Advances. Chem. Rev. 2017, 117, 8326–8419. [Google Scholar] [CrossRef]
- Mazuela, J.; Antonsson, T.; Johansson, M.J.; Knerr, L.; Marsden, S.P. Direct Synthesis of N-Alkyl Arylglycines by Organocatalytic Asymmetric Transfer Hydrogenation of N-Alkyl Aryl Imino Esters. Org. Lett. 2017, 19, 5541–5544. [Google Scholar] [CrossRef]
- Chen, J.; Li, F.; Wang, F.; Hu, Y.; Zhang, Z.; Zhao, M.; Zhang, W. Pd(OAc)2-Catalyzed Asymmetric Hydrogenation of α-Iminoesters. Org. Lett. 2019, 21, 9060–9065. [Google Scholar] [CrossRef]
- Hu, X.H.; Hu, X.P. Ir-Catalyzed Asymmetric Hydrogenation of α-Imino Esters with Chiral Ferrocenylphosphine-Phosphoramidite Ligands. Adv. Synth. Catal. 2019, 361, 5063–5068. [Google Scholar] [CrossRef]
- Kim, J.; Shin, M.; Cho, S.H. Copper-catalyzed diastereoselective and enantioselective addition of 1, 1-diborylalkanes to cyclic ketimines and α-imino esters. ACS Catal. 2019, 9, 8503–8508. [Google Scholar] [CrossRef]
- Liu, D.; Li, B.; Chen, J.; Gridnev, I.D.; Yan, D.; Zhang, W. Ni-catalyzed asymmetric hydrogenation of N-aryl imino esters for the efficient synthesis of chiral α-aryl glycines. Nat. Commun. 2020, 11, 5935. [Google Scholar] [CrossRef]
- Cabré, A.; Verdaguer, X.; Riera, A. Recent Advances in the Enantioselective Synthesis of Chiral Amines via Transition Metal-Catalyzed Asymmetric Hydrogenation. Chem. Rev. 2022, 122, 269–339. [Google Scholar] [CrossRef]
- Wu, Y.; Hu, L.; Li, Z.; Deng, L. Catalytic asymmetric umpolung reactions of imines. Nature 2015, 523, 445–450. [Google Scholar] [CrossRef]
- Mizota, I.; Shimizu, M. Umpolung Reactions of α-Imino Esters: Useful Methods for the Preparation of α-Amino Acid Frameworks. Chem. Rec. 2016, 16, 688–702. [Google Scholar] [CrossRef]
- Chen, P.; Yue, Z.; Zhang, J.; Lv, X.; Wang, L.; Zhang, J. Phosphine-Catalyzed Asymmetric Umpolung Addition of Trifluoromethyl Ketimines to Morita–Baylis–Hillman Carbonates. Angew. Chem. Int. Ed. 2016, 55, 13316–13320. [Google Scholar] [CrossRef]
- Su, Y.-L.; Li, Y.-H.; Chen, Y.-G.; Han, Z.-Y. Ir/PTC Cooperatively Catalyzed Asymmetric Umpolung Allylation of α-Imino Ester Enabled Synthesis of α-Quaternary Amino Acid Derivatives Bearing Two Vicinal Stereocenters. Chem. Commun. 2017, 53, 1985–1988. [Google Scholar] [CrossRef]
- Feng, B.; Lu, L.-Q.; Chen, J.-R.; Feng, G.; He, B.-Q.; Lu, B.; Xiao, W.-J. Umpolung of Imines Enables Catalytic Asymmetric Regio-reversed [3 + 2] Cycloadditions of Iminoesters with Nitroolefins. Angew. Chem. Int. Ed. 2018, 57, 5888–5892. [Google Scholar] [CrossRef]
- Yoshida, Y.; Mino, T.; Sakamoto, M. Organocatalytic Highly Regio- and Enantioselective Umpolung Michael Addition Reaction of α-Imino Esters. Chem. Eur. J. 2017, 23, 12749–12753. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Moriya, Y.; Mino, T.; Sakamoto, M. Regio- and Enantioselective Synthesis of α-Amino-δ-Ketoesters Through Catalytic Umpolung Reaction of α-Iminoesters with Enones. Adv. Synth. Catal. 2018, 360, 4142–4146. [Google Scholar] [CrossRef]
- Yoshida, Y.; Hiroshige, T.; Omori, K.; Mino, T.; Sakamoto, M. Chemo- and Regioselective Asymmetric Synthesis of Cyclic Enamides through the Catalytic Umpolung Organocascade Reaction of α-Imino Amides. J. Org. Chem. 2019, 84, 7362–7371. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Omori, K.; Hiroshige, T.; Mino, T.; Sakamoto, M. Chemoselective Catalytic Asymmetric Synthesis of Functionalized Aminals Through the Umpolung Organocascade Reaction of α-Imino Amides. Chem.-Asian J. 2019, 14, 2737–2743. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Kukita, M.; Omori, K.; Mino, T.; Sakamoto, M. Iminophosphorane-mediated regioselective umpolung alkylation reaction of α-iminoesters, Org. Biomol. Chem. 2021, 19, 4551–4564. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, Y.; Deng, L. Cinchonium Betaines as Efficient Catalysts for Asymmetric Proton Transfer Catalysis: The Development of a Practical Enantioselective Isomerization of Trifluoromethyl Imines. J. Am. Chem. Soc. 2016, 138, 12297–12302. [Google Scholar] [CrossRef]
- Wang, G.; Piva de Silva, G.; Wiebe, N.E.; Fehr, G.M.; Davis, R.L. Development of a Metal–Free Amine Oxidation Method Utilizing DEAD Chemistry. RSC Adv. 2017, 7, 48848–48852. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Solvent | X | Temp. (°C) | Time (h) | Yield (%) b |
| 1 | toluene | 50 | −40 | 18 | 9 |
| 2 | Et2O | 50 | −40 | 18 | 4 |
| 3 | CH2Cl2 | 50 | −40 | 18 | 43 |
| 4 | tetrahydrofuran | 50 | −40 | 18 | 28 |
| 5 | MeOH | 50 | −40 | 18 | 0 |
| 6 | CH2Cl2 | 100 | −40 | 18 | 43 |
| 7 | CH2Cl2 | 150 | −40 | 18 | 42 |
| 8 | CH2Cl2 | 50 | −20 | 18 | 53 |
| 9 | CH2Cl2 | 50 | 0 | 18 | 61 |
| 10 | CH2Cl2 | 50 | r.t | 18 | 51 |
| 11 | CH2Cl2 | 50 | 0 | 1 | 64 |
| 12 | CH2Cl2 | 50 | 0 | 53 | 64 |
| 13 c | CH2Cl2 | 50 | 0 | 1 | 88 (72 d) |
| 14 c,e | CH2Cl2 | 50 | 0 | 1 | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, Y.; Ida, H.; Mino, T.; Sakamoto, M. Formal [3 + 2] Cycloaddition of α-Imino Esters with Azo Compounds: Facile Construction of Pentasubstituted 1,2,4-Triazoline Skeletons. Molecules 2023, 28, 4339. https://doi.org/10.3390/molecules28114339
Yoshida Y, Ida H, Mino T, Sakamoto M. Formal [3 + 2] Cycloaddition of α-Imino Esters with Azo Compounds: Facile Construction of Pentasubstituted 1,2,4-Triazoline Skeletons. Molecules. 2023; 28(11):4339. https://doi.org/10.3390/molecules28114339
Chicago/Turabian StyleYoshida, Yasushi, Hidetoshi Ida, Takashi Mino, and Masami Sakamoto. 2023. "Formal [3 + 2] Cycloaddition of α-Imino Esters with Azo Compounds: Facile Construction of Pentasubstituted 1,2,4-Triazoline Skeletons" Molecules 28, no. 11: 4339. https://doi.org/10.3390/molecules28114339
APA StyleYoshida, Y., Ida, H., Mino, T., & Sakamoto, M. (2023). Formal [3 + 2] Cycloaddition of α-Imino Esters with Azo Compounds: Facile Construction of Pentasubstituted 1,2,4-Triazoline Skeletons. Molecules, 28(11), 4339. https://doi.org/10.3390/molecules28114339