4. Materials and Methods
4.1. General Experiments
A Thermo Scientific Q-Exactive mass spectrometer with electrospray ionization (ESI) was utilized to obtain the HRMS. A Nicolet Nexus 470 FTIR spectrophotometer (Waltham, MA, USA) was used to gather the IR spectra. A Bruker Fourier 300 spectrometer was employed to acquire NMR spectra, with CDCl
3 as the solvent. The chemical shifts of the NMR spectra are reported in ppm, with reference to the corresponding solvent peak, while the coupling constants are expressed in Hz. The PureSolv MD 7 Solvent Purification System from Innovative Technologies (MB-SPS-800) (Herndon, VA, USA) or activated molecular sieves (heating at 180–200 °C for 6 h under vacuum) were used to remove the trace amount of water from acetone and dichloromethane. All remaining reagents and solvents were directly used as received from commercial sources. All column chromatography was carried out on silica gel with a particle size of 32–63 μm. Preparative thin-layer chromatography (PTLC) purifications were conducted on silica gel 60 GF254-loaded plates (EMD Millipore Corporation, Burlington, MA, USA). Abietic acid (80% purity) and dehydroabietylamine (55% purity) were purchased from Fisher Scientific (Portland, OR, USA). All NMR spectra and high-resolution mass spectra were included in
Supplementary Materials.
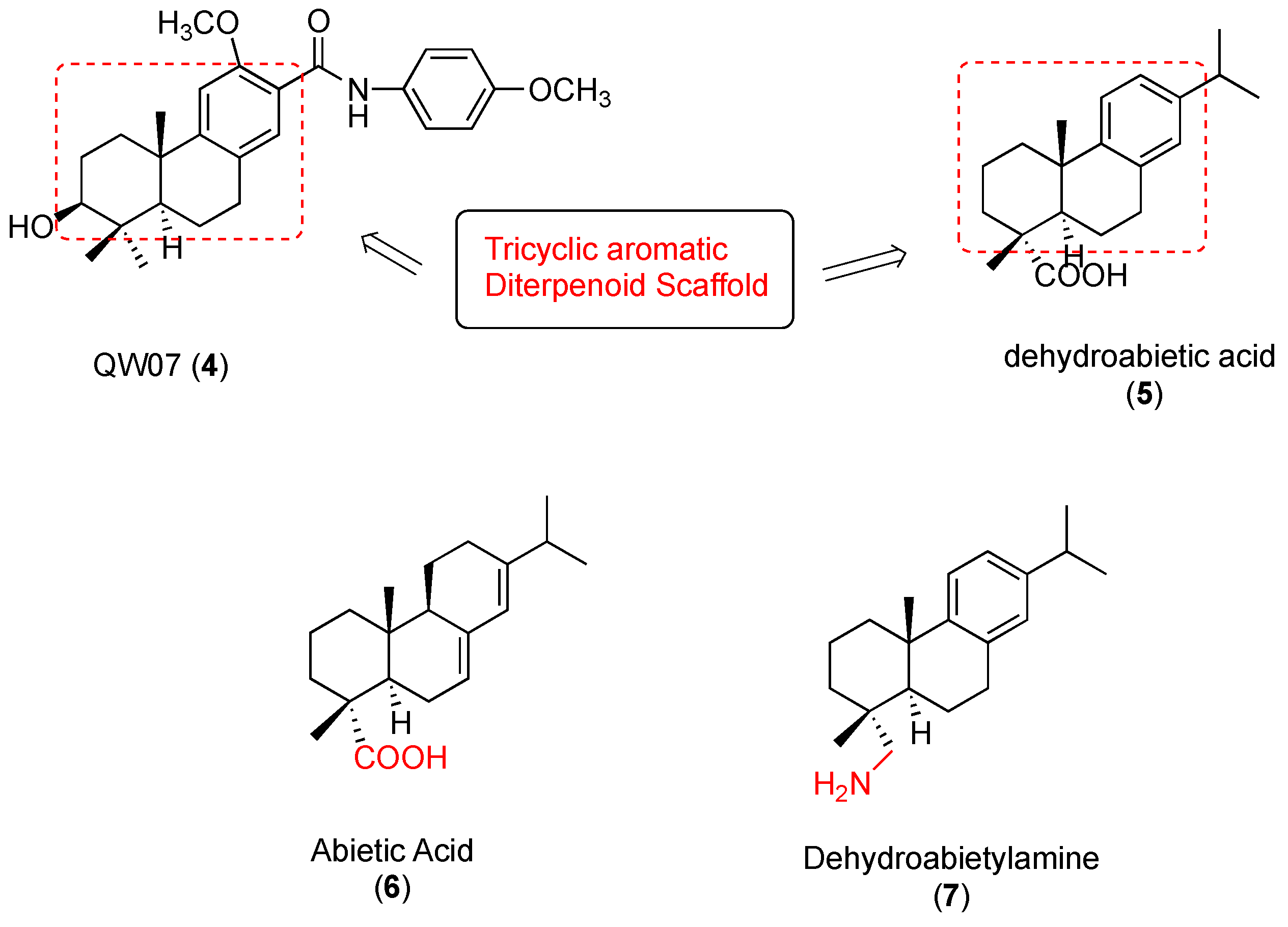
4.2. Purification of Abietic Acid (6)
The purchased abietic acid (6) has only 80% purity, which was purified via PTLC eluting twice with hexane-ethyl acetate (2:1, v/v) to give the pure abietic acid as a yellow oil. The recovery rate is 66%. 1H NMR (300 MHz, CDCl3) δ 5.77 (s, 1H), 5.37 (s, 1H), 2.26–2.18 (m, 1H), 2.10–2.03 (m, 4H), 1.97–1.76 (m, 5H), 1.62–1.55 (m, 2H), 1.28–1.09 (m, 3H), 1.25 (s, 3H), 1.01 (d, J = 6.9 Hz, 3H), 1.00 (d, J = 6.9 Hz, 3H), 0.83 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 184.69, 145.00, 135.28, 122.12, 120.24, 50.65, 46.05, 44.61, 37.98, 36.89, 34.60, 34.18, 27.17, 25.32, 22.19, 21.13, 20.59, 17.77, 16.43, 13.75. HRMS (ESI): m/z calculated for C20H31O2 [M + H]+: 303.2324. Found: 303.2321. IR (film) vmax: 3395–2600, 2951, 1686, 1277, 1154, 993, 891,789 cm−1.
4.3. Synthesis of 8
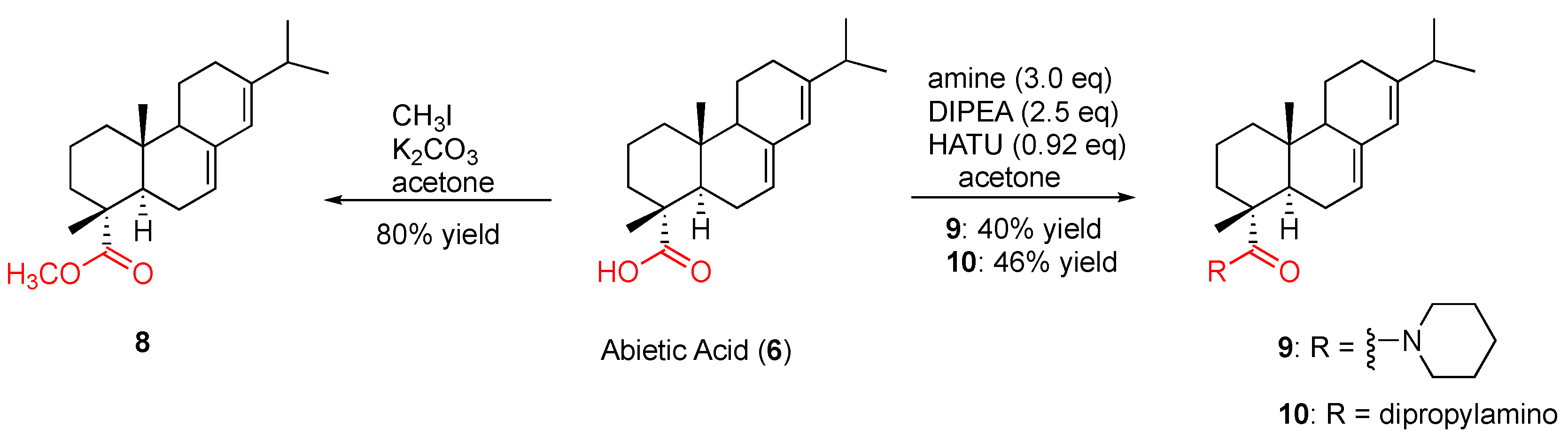
To a solution of abietic acid (5.00 g, 80%, 13.2 mmol) in acetone (25 mL) at room temperature were sequentially added K2CO3 (2.51 g, 18.2 mmol) and methyl iodide (1.51 mL, 24.3 mmol) dropwise. The reaction was then stirred at room temperature for two days and monitored with TLC (hexane-ethyl acetate, 3:1, v/v) for completeness. The solution was diluted with ethyl acetate (300 mL) and rinsed with brine (30 mL × 5). The organic fraction was dried over anhydrous Na2SO4 and concentrated. The crude product was purified through column chromatography, eluting with hexane-ethyl acetate (3:1, v/v) to give the desired product an 80% yield as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 5.77 (s, 1H), 5.36 (s, 1H), 3.63 (s, 3H), 2.26–2.17 (m, 1H), 2.13–2.03 (m, 4H), 1.91–1.70 (m, 5H), 1.62–1.57 (m, 2H), 1.27–1.19 (m, 3H), 1.25 (s, 3H), 1.01 (d, J = 6.9 Hz, 3H), 1.00 (d, J = 6.9 Hz, 3H), 0.82 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 178.66, 144.95, 135.18, 122.00, 120.25, 51.52, 50.60, 46.25, 44.76, 37.98, 36.77, 34.54, 34.19, 27.13, 25.33, 22.12, 21.51, 17.79, 16.67, 13.68. HRMS (ESI): m/z calculated for C21H33O2 [M + H]+: 317.2480. Found: 317.2478. IR (film) vmax: 2928, 2868, 1724, 1459, 1385, 1243, 1185, 1106 cm−1.
4.4. Synthesis of 9
Piperidine (89 μL, 0.9 mmol) was added to a solution of abietic acid (113 mg, 80% purity, 0.3 mmol) in acetone (5 mL) under argon at 0 °C, to which was added a solution of DIPEA (0.13 mL, 0.75 mmol) and HATU (105 mg, 0.28 mmol) in acetone (5 mL). The resulting reaction mixture was stirred at 0 °C for 15 to 20 min, when it turned to a yellow color. The reaction was then allowed to proceed at room temperature overnight prior to removing the solvent. The residue was diluted with EtOAc (75 mL), which was rinsed with brine (25 mL × 3). The EtOAc layer was dried over anhydrous Na2SO4 and concentrated to afford a yellow oil, which was subjected to PTLC purification eluting with hexane/EtOAc (2:1, v/v) to give the desired product in 40% yield as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 5.74 (s, 1H), 5.35 (s, 1H), 3.58 (t, J = 5.6 Hz, 4H), 2.24–2.14 (m, 2H), 2.07–2.02 (m, 2H), 1.89–1.75 (m, 5H), 1.67–1.45 (m, 10H), 1.30 (s, 3H), 1.26–1.17 (s, 2H), 0.99 (d, J = 6.9 Hz, 3H), 0.98 (d, J = 6.9 Hz, 3H), 0.84 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 176.04, 144.41, 134.83, 122.27, 120.97, 51.01, 46.92, 46.12, 44.01, 37.48, 35.04, 34.44, 27.03, 25.89, 25.54, 24.43, 22.25, 21.03, 20.46, 19.96, 18.17, 13.79. HRMS (ESI): m/z calculated for C25H40NO [M + H]+: 370.3110. Found: 370.3106. IR (film) vmax: 2931, 2853, 1681, 1416, 1247, 1040 cm−1.
4.5. Synthesis of 10
Dipropylamine (123 μL, 0.9 mmol) was added to a solution of abietic acid (113 mg, 80% purity, 0.3 mmol) in acetone (5 mL) under argon at 0 °C, to which was added a solution of DIPEA (0.13 mL, 0.75 mmol) and HATU (105 mg, 0.28 mmol) in acetone (5 mL). The resulting reaction mixture was stirred at 0 °C for 15 to 20 min, when it turned to a yellow color. The reaction was then allowed to proceed at room temperature overnight prior to removing the solvent. The residue was diluted with EtOAc (75 mL), which was rinsed with brine (25 mL × 3). The EtOAc layer was dried over anhydrous Na2SO4 and concentrated to afford a crude product, which was purified with PTLC eluting with hexane/EtOAc (6:1, v/v) to give 10 as a red oil in 46% yield. 1H NMR (300 MHz, CDCl3) δ 5.75 (s, 1H), 5.36 (s, 1H), 3.46–3.13 (m, 4H), 2.85–2.74 (m, 1H), 2.33–1.99 (m, 6H), 1.86–1.73 (m, 6H), 1.62–1.44 (m, 8H), 1.30 (s, 3H), 1.25–1.17 (m, 4H), 1.00 (d, J = 6.9 Hz, 3H), 0.99 (d, J = 6.9 Hz, 3H), 0.92–0.82 (overlapped, 9H). 13C NMR (75 MHz, CDCl3) δ 177.12, 144.89, 135.35, 122.70, 121.41, 51.65, 50.63, 46.96, 44.63, 38.17, 35.70, 34.96, 27.51, 25.97, 24.06, 22.74, 21.50, 20.95, 20.25, 18.72, 14.31, 11.44. HRMS (ESI): m/z calculated for C26H44NO [M + H]+: 386.3423. Found: 386.3423. IR (film) vmax: 2931, 1693, 1506, 1471, 1385, 1100 cm−1.
4.6. Synthesis of Dehydroabietic Acid (5)
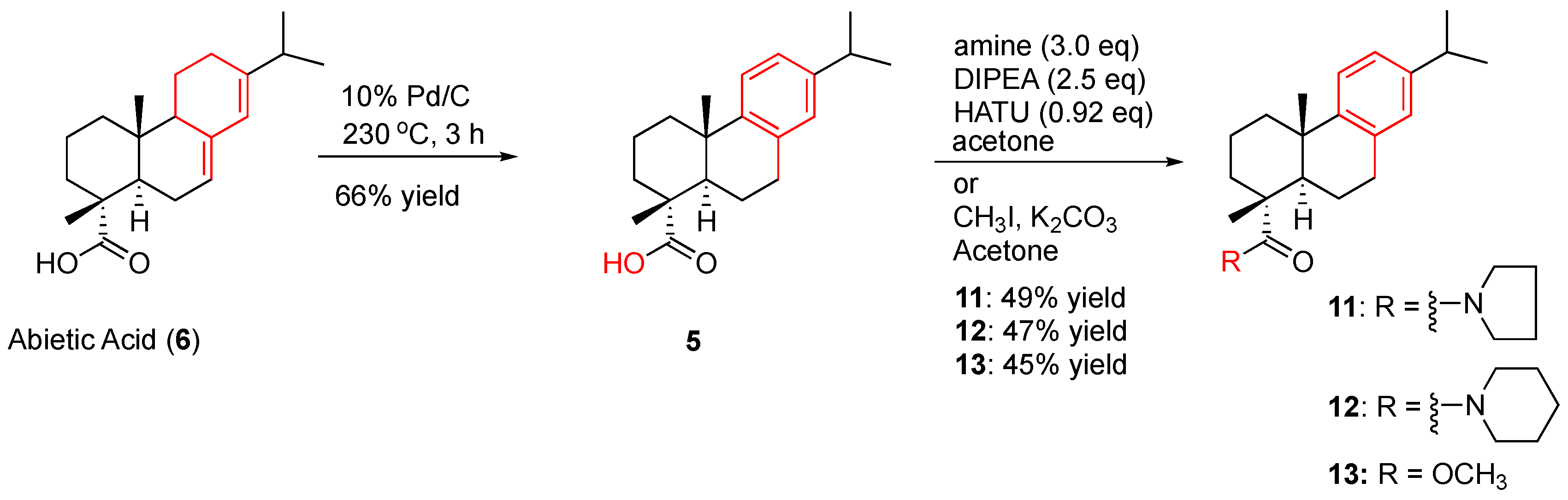
Abietic acid (502 mg, 80%, 1.33 mmol) and 10% Pd/C (12.6 mg) were added to a 5 mL conical vial with a triangular spin vane. The reaction mixture was heated under argon using aluminum beads to 220–230 °C (the melting point of abietic acid is 250 °C) for four hours. TLC (hexane/EtOAc, 4:1) was used to check the completeness of the reaction. The reaction mixture was then cooled down to room temperature and washed with ethyl acetate (10 mL) before it completely solidified. The black solids were placed in a celite pad and rinsed with ethyl acetate (10 mL). The combined ethyl acetate fractions were concentrated to give a yellow solid, which was purified with column chromatography eluting with hexane/EtOAC (4:1, v/v) to give dehydroabietic acid in 66% yield as a clear crystal solid. 1H NMR (300 MHz, CDCl3) δ 7.19 (d, J = 8.1 Hz, 1H), 7.02 (d, J = 8.1 Hz, 1H), 6.92 (s, 1H), 2.98–2.80 (m, 3H), 2.35–2.26 (m, 2H), 1.97–1.72 (m, 5H), 1.61–1.52 (m, 2H), 1.31 (s, 3H), 1.25 (d, J = 6.9 Hz, 6H), 1.25 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 185.60, 146.89, 145.86, 134.83, 127.04, 124.25, 124.03, 47.58, 44.70, 38.04, 36.99, 36.88, 33.59, 30.14, 25.26, 24.12, 21.91, 18.67, 16.33. HRMS (ESI): m/z calculated for C20H29O2 [M + H]+: 301.2167. Found: 301.2164. IR (film) vmax: 3047, 2954, 2928, 2867, 1690, 1612, 1458, 1276, 1134 cm−1.
4.7. General Synthesis Procedures of Amides (11 and 12)
The corresponding amine (3 equv.) was added to a solution of dehydroabietic acid in half the volume of acetone (the concentration of the limiting reagent in acetone is 0.03 M) under argon at 0 °C. A solution of DIPEA (2.5 equiv.) and HATU (0.92 equiv.) in the remaining acetone was then added at 0 °C. The resulting reaction mixture was stirred for 15 to 20 min when it turned a yellow color. Then the reaction was allowed to proceed at room temperature overnight. After the removal of the organic solvent, the residue was diluted with 75 mL of ethyl acetate, which was rinsed with brine (25 mL × 3). The ethyl acetate layer was dried over anhydrous Na2SO4 and concentrated to yield a crude product as a yellow oil.
4.7.1. Amide 11
The crude product was subjected to PTLC purification eluting with hexane/EtOAC (4:1, v/v) to give amide 11 in 49% yield as a colorless solid. 1H NMR (300 MHz, CDCl3) δ 7.17 (d, J = 8.1 Hz, 1H), 6.99 (d, J = 8.1 Hz, 1H), 6.89 (s, 1H), 3.57 (t, J = 6.9 Hz, 4H), 3.01– 2.78 (m, 3H), 2.39 (dd, J = 12.0, 2.1 Hz, 1H), 2.29 (d, J = 13.2 Hz, 1H), 1.86–1.69 (m, 9H), 1.60–1.40 (m, 2H), 1.35 (s, 3H), 1.25 (s, 3H), 1.22 (d, J = 6.9 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 176.84, 147.37, 145.63, 135.15, 127.10, 124.21, 123.85, 48.84, 47.52, 44.29, 37.91, 37.45, 34.59, 33.53, 30.47, 25.64, 24.11, 24.08, 21.86, 18.93, 18.18. HRMS (ESI): m/z calculated for C24H36NO [M + H]+: 354.2797. Found: 354.2796. IR (film) vmax: 2953, 2867, 1711, 1609, 1497, 1279, 1036 cm−1. HRMS (ESI): m/z calculated for C24H36NO [M + H]+: 354.2797. Found: 354.2796.
4.7.2. Amide 12
The crude product was subjected to PTLC purification eluting with hexane-EtOAc (4:1, v/v) to afford amide 12 as a clear oil in 47% yield. 1H NMR (300 MHz, CDCl3) δ 7.16 (d, J = 8.3 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 6.90 (s, 1H), 3.65–3.51 (m, 4H), 3.06–2.76 (m, 1H), 2.90–2.76 (m, 2H), 2.37–2.25 (m, 2H), 1.84–1.70 (m, 5H), 1.65–1.58 (m, 3H), 1.55–1.41 (m, 5H), 1.34 (s, 3H), 1.25 (s, 3H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 177.02, 147.25, 145.62, 135.36, 127.19, 124.24, 123.80, 47.20, 46.88, 45.40, 37.81, 37.60, 35.33, 33.55, 30.81, 26.37, 25.67, 24.89, 24.11, 24.08, 22.26, 19.05. HRMS (ESI): m/z calculated for C25H38NO [M + H]+: 368.2953. Found: 368.2948. IR (film) vmax: 2935, 1768, 1615, 1464, 1262, 1007 cm−1.
4.8. Methylation of Dehydroabietic Acid (Synthesis of 13)
To a solution of dehydroabietic acid (97 mg, 0.32 mmol) in acetone (3.2 mL, 0.1 M) was added K2CO3 (133 mg, 0.96 mmol), and the reaction mixture was stirred for 5 min before adding methyl iodide (0.09 mL, 1.45 mmol). The reaction was allowed to proceed with stirring under argon at room temperature overnight when the reaction was completed as monitored by TLC (hexane/EtOAc, 5:1). After removing the acetone; the crude product was diluted with 50 mL of ethyl acetate and rinsed with brine (10 mL × 5). The organic layer was dried over anhydrous Na2SO4 and concentrated to give a crude product, which was subjected to PTLC purification eluting with hexane/EtOAc (5:1) to give the desired product as a white solid in 45% yield. 1H NMR (300 MHz, CDCl3) δ 7.19 (d, J = 8.3 Hz, 1H), 7.02 (dd, J = 8.3, 2.4 Hz, 1H), 6.91 (d, J = 2.4 Hz, 1H), 3.68 (s, 3H), 2.94–2.80 (m, 3H), 2.35–2.29 (m, 1H), 2.27 (dd, J = 12.6, 2.4 Hz, 1H), 1.93–1.64 (m, 5H), 1.57–1.40 (m, 2H), 1.30 (s, 3H), 1.25 (d, J = 6.9 Hz, 6H), 1.23 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 179.24, 147.03, 145.82, 134.80, 126.99, 124.25, 124.02, 52.05, 47.77, 44.97, 38.11, 37.05, 36.75, 33.58, 30.12, 25.22, 24.11, 21.83, 18.69, 16.62. HRMS (ESI): m/z calculated for C21H31O2 [M + H]+: 315.2324. Found: 315.2321. IR (film) vmax: 2928, 2867, 1725, 1611, 1432, 1243, 1057 cm−1.
4.9. Purification of Dehydroabietylamine (7)
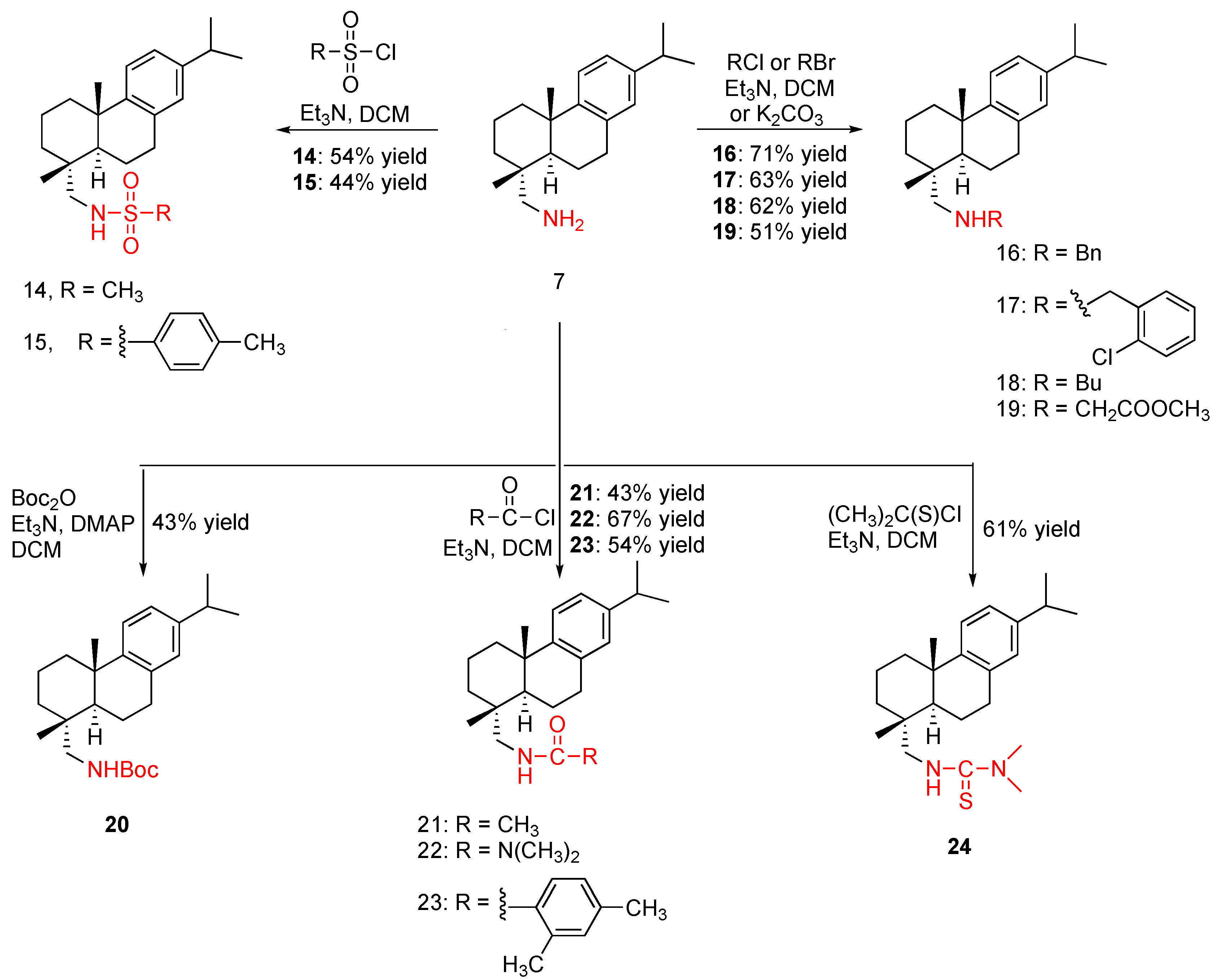
The purchased dehydroabietylamine (204.5 mg, 55% purity) was purified via PTLC developing with hexane-EtOAc (1:3 with 3% Et3N, v/v) three times to remove the impurity that is on top of the desired product. Pure dehydroabietylamine (7) was obtained in 31% yield as a clear oil. 1H NMR (300 MHz, CDCl3) δ 7.19 (d, J = 8.1, 1H), 7.00 (d, J = 8.1, 1H), 6.90 (d, J = 2.1 Hz, 1H), 2.92–2.78 (m, 3H), 2.62 (d, J = 13.2 Hz, 1H), 2.47 (d, J = 13.2 Hz, 1H), 2.38 (br.s, 2H), 2.30 (br.d, J = 14.1 Hz, 1H), 1.79–1.64 (m, 4H), 1.53–1.34 (m, 4H), 1.23 (s, 3H), 1.23 (d, J = 6.9 Hz, 6H), 0.92 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 147.49, 145.58, 134.73, 126.87, 124.31, 123.90, 53.81, 45.01, 38.59, 37.45, 37.22, 35.30, 33.51, 30.23, 25.33, 24.08, 24.05, 18.79, 18.72. HRMS (ESI): m/z calculated for C20H32N [M + H]+: 286.2535. Found: 286.2530. IR (film) vmax: 3305, 2922, 2865, 2085, 1611, 1555, 1497, 1237, 1173, 1058, 908 cm−1.
4.10. Synthesis of 14
To a solution of dehydroabietylamine (156 mg, 55% purity, 0.3 mmo) in DCM (3 mL) was added triethylamine (104 μL, 0.75 mmol) at 0 °C. The solution was stirred for 30 min before adding mesyl chloride (23 μL, 0.3 mmol). The reaction was allowed to proceed at room temperature under argon overnight before diluting with EtOAc (75 mL). The resulting mixture was rinsed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated. The yellow crude oil was subjected to PTLC purification developing four times with hexane/EtOAc (3:1, v/v) to give the desired product as a white crystal in 54% yield. 1H NMR (300 MHz, CDCl3) δ 7.17 (d, J = 8.4 Hz, 1H), 6.99 (dd, J = 8.4, 2.4 Hz, 1H), 6.89 (d, J = 2.1 Hz, 1H), 4.69 (t, J = 6.9 Hz, 1H), 3.04–2.78 (m, 5H), 2.89 (s, 3H, SO2CH3), 2.29 (d, J = 12.6 Hz, 1H), 1.7–1.65 (m, 4H), 1.52 (dd, J = 10.8, 3.9 Hz, 1H), 1.44–1.30 (m, 3H), 1.23 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.22 (s, 3H, CH3), 0.95 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 147.04, 145.82, 134.66, 126.96, 124.26, 123.98, 45.02, 40.17, 38.35, 37.49, 37.10, 35.92, 33.55, 29.98, 25.31, 24.12, 24.09, 18.89, 18.61, 18.56. HRMS (ESI): m/z calculated for C21H34NO2S [M + H]+: 364.2310. Found: 364.2303. IR (film) vmax: 3255, 3000, 2959, 2927, 2871, 1496, 1433, 1375, 1232, 1134, 1050 cm−1.
4.11. Synthesis of 15
To a solution of dehydroabietylamine (156 mg, 55% purity, 0.3 mmo) in DCM (3 mL) was added triethylamine (104 μL, 0.75 mmol) at 0 °C. The solution was stirred for 30 min before adding 4-tolenesulfonyl chloride (57 mg, 0.3 mmol). The reaction was allowed to proceed at room temperature under argon for 5 h before diluting with EtOAc (75 mL). The resulting mixture was rinsed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated. The crude product is purified with column chromatography eluting with hexane-EtOAc (5:1, v/v) followed by further PTLC purification developing four times with hexane-EtOAc (8:1, v/v) to give 15 as a clear oil in 44% yield. 1H NMR (300 MHz, CDCl3) δ 7.74 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.1 Hz, 1H), 6.98 (dd, J = 8.1, 2.4 Hz, 1H), 6.87 (d, J = 2.4 Hz,1H), 4.87 (t, J = 7.2 Hz, 1H, NH), 2.87–2.77 (m, 4H), 2.64 (dd, J = 12.9, 7.5 Hz, 1H), 2.40 (s, 3H, Ar-CH3), 2.24 (br.d, J = 12.9 Hz, 1H), 1.74–1.60 (m, 4H), 1.53–1.48 (m, 1H), 1.34–1.26 (m, 3H), 1.23 (d, J = 6.9 Hz, 6H), 1.18 (s, 3H), 0.88 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 147.07, 145.66, 143.33, 137.19, 134.76, 129.81, 127.08, 126.93, 124.22, 123.88, 53.89, 44.92, 38.29, 37.47, 37.04, 35.83, 33.55, 29.95, 25.31, 24.15, 21.61, 18.80, 18.66, 18.60. HRMS (ESI): m/z calculated for C27H38NO2S [M + H]+: 440.2623. Found: 440.2616. IR (film) vmax: 3273, 3273, 2929, 2851, 2448, 2216, 2182, 1952, 1459, 1093 cm−1.
4.12. Synthesis of 16
To a solution of dehydroabietylamine (156 mg, 55% purity, 0.3 mmo) in DCM (3 mL) was added triethylamine (104 μL, 0.75 mmol) at 0 °C. The solution was stirred for 30 min before adding benzyl bromide (36 μL, 0.3 mmol). The reaction was allowed to proceed at room temperature under argon for 5 h before diluting with EtOAc (75 mL). The resulting mixture was rinsed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated. The clear crude oil is subjected to PTLC purification by developing twice with hexane/EtOAc (4:1, v/v) to give the desired product as wax in 71% yield. 1H NMR (300 MHz, CDCl3) δ 7.40–7.27 (m, 5H), 7.17 (d, J = 8.1 Hz, 1H), 6.97 (dd, J = 8.1, 2.4 Hz, 1H), 6.88 (d, J = 2.4 Hz, 1H), 3.84 (s, 2H), 2.88–2.77 (m, 3H), 2.54 (d, J = 12.0 Hz, 1H), 2.37 (d, J = 12.0 Hz, 1H), 2.27 (br.d, J = 12.6 Hz, 1H), 1.75–1.63 (m, 4H), 1.50–1.36 (m, 4H), 1.22 (d, J = 6.9 Hz, 6H), 1.21 (s, 3H), 0.95 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 147.53, 145.55, 134.87, 128.52, 127.31, 126.89, 124.34, 123.90, 60.33, 54.26, 45.52, 38.48, 37.55, 37.06, 33.53, 30.27, 25.49, 24.11, 19.31, 18.93. HRMS (ESI): m/z calculated for C27H38N [M + H]+: 376.3004. Found: 376.3001. IR (film) vmax: 2923, 2866, 1538, 1495, 1361, 1264, 1173, 1075, 971 cm−1.
4.13. Synthesis of 17
To a solution of dehydroabietylamine (156 mg, 55% purity, 0.3 mmo) in DCM (3 mL) was added triethylamine (104 μL, 0.75 mmol) at 0 °C. The solution was stirred for 30 min before adding 2-chlorobenzyl bromide (62 mg, 0.3 mmol). The reaction was allowed to proceed at room temperature under argon for 4 h before diluting with EtOAc (75 mL). The resulting mixture was rinsed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated. The clear crude oil was purified via PTLC, developing twice with hexane/EtOAc (7:1, v/v) to yield the desired product as a clear oil in 63% yield. 1H NMR (300 MHz, CDCl3) δ 7.45 (dd, J = 7.2, 2.1 Hz, 1H), 7.40 (dd, J = 7.2, 1.8 Hz, 1H), 7.31–7.21 (overlapped, 3H), 7.04 (dd, J = 8.1, 2.1 Hz, 1H), 6.94 (d, J = 2.1 Hz, 1H), 3.95 (d, J = 14.1 Hz, 1H, benzylic H), 3.88 (d, J = 14.1 Hz, 1H, benzylic H), 2.95–2.83 (m, 3H), 2.58 (d, J = 11.7 Hz, 1H), 2.28 (d, J = 11.7 Hz, 1H), 2.34–2.30 (overlapped, 1H), 1.87–1.66 (m, 5H), 1.60–1.41 (m, 3H), 1.28 (d, J = 6.9 Hz, 6H), 1.27 (s, 3H), 0.98 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 147.67, 145.55, 138.32, 134.96, 133.76, 130.15, 129.51, 128.19, 126.92, 126.79, 124.45, 123.92, 61.02, 52.30, 45.34, 38.66, 37.58, 37.19, 36.33, 33.57, 30.43, 25.54, 24.13, 19.56, 19.03, 18.88. HRMS (ESI): m/z calculated for C27H37ClN [M + H]+: 410.2614 and 412.2585. Found: 410.2611 and 412.2575. IR (film) vmax: 2924, 2866, 1572, 1497, 1442, 1381, 1264, 1196, 1109 cm−1.
4.14. Synthesis of 18
To a solution of dehydroabietylamine (156 mg, 55% purity, 0.3 mmol) in anhydrous acetonitrile (3 mL) was added potassium carbonate (124 mg, 0.9 mmol) followed by 1-bromobutane (96 μL, 0.9 mmol) at room temperature. The reaction was allowed to proceed at room temperature under argon overnight before diluting with EtOAc (75 mL). The resulting mixture was rinsed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated. The reaction mixture was stirred under argon at room temperature for six hours. The clear crude oil was subjected to PTLC purification, developing twice with hexane/EtOAc (5:1, v/v) to give the desired product as a colorless oil in 62% yield. 1H NMR (300 MHz, CDCl3) δ 7.19 (d, J = 8.1 Hz, 1H), 7.00 (dd, J = 8.1, 2.4 Hz, 1H), 6.90 (d, J = 2.4 Hz, 1H), 2.91–2.79 (m, 3H), 2.60 (d, J = 6.9 Hz, 2H), 2.52 (d, J = 11.7 Hz, 1H), 2.34 (d, J = 11.7 Hz, 1H), 2.28 (br.d, J = 12.7 Hz, 1H), 1.81–1.57 (m, 5H), 1.51–1.30 (m, 7H), 1.24 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.22 (s, 3H, CH3), 0.95 (s, 3H, CH3), 0.92 (t, J = 7.2 Hz, 3H, CH2CH3). 13C NMR (75 MHz, CDCl3) δ 147.77, 145.55, 135.00, 126.92, 124.46, 123.92, 61.99, 50.90, 45.68, 38.68, 37.59, 37.10, 36.40, 33.59, 32.41, 30.50, 25.51, 24.14, 20.64, 19.40, 19.05, 18.95, 14.18. HRMS (ESI): m/z calculated for C24H40N [M + H]+: 342.3161. Found: 342.3155. IR (film) vmax: 2955, 2868, 1458, 1379, 1121, 974.8 cm−1.
4.15. Synthesis of 19
To a solution of dehydroabietylamine (156 mg, 55% purity, 0.3 mmol) in DCM (3 mL) was added triethylamine (104 μL, 0.75 mmol) at 0 °C. The solution was stirred for 30 min before adding methyl chloroacetate (32 mg, 0.3 mmol). The reaction was allowed to proceed at room temperature under argon for 5 h before diluting with EtOAc (75 mL). The resulting mixture was rinsed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated. The crude oil was subjected to PTLC purification by developing twice with hexane/EtOAc (6:1, v/v) to give the desired product as a clear oil in 51% yield. 1H NMR (300 MHz, CDCl3) δ 7.19 (d, J= 8.3 Hz, 1H), 6.99 (dd, J = 8.3, 2.4 Hz, 1H), 6.90 (d, J = 2.4 Hz, 1H), 4.19 (q, J = 7.2 Hz, 2H, CH2CH3), 3.43 (d, J = 17.3 Hz, 1H), 3.33 (d, J = 17.3 Hz, 1H), 2.93–2.79 (m, 3H), 2.56 (d, J = 11.7 Hz, 1H), 2.28 (d, J = 11.5 Hz, 1H), 1.82–1.69 (m, 5H), 1.51–1.40 (m, 4H), 1.29 (t, J = 7.2 Hz, 3H), 1.24 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.23 (s, 3H, CH3), 0.94 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 172.88, 147.54, 145.46, 134.84, 126.82, 124.30, 123.81, 61.44, 60.67, 52.07, 45.35, 38.55, 37.47, 37.15, 36.12, 33.50, 30.30, 25.35, 24.08, 19.18, 18.90, 18.83, 14.32. HRMS (ESI): m/z calculated for C24H38NO2 [M + H]+: 372.2902. Found: 372.2896. IR (film) vmax: 2926,1736,1497,1192, 1036 cm−1.
4.16. Synthesis of 20
To a solution of dehydroabietylamine (379 mg, 55% purity, 0.73 mmol) in DCM (3.5 mL) were added triethylamine (0.195 mL, 1.4 mmol) and 4-dimethylaminopyridine (21 mg, 0.17 mmol) under argon at 0 °C. A solution of di-tert-butyl dicarbonate (135 mg, 0.62 mmol) in DCM (3.8 mL) was then added to the reaction mixture. The reaction was allowed to proceed with stirring overnight at room temperature until the reaction turned pinkish and the reaction was complete as monitored by TLC (hexane/EtOAc, 10:1). The reaction mixture was diluted with DCM (30 mL), and the resulting mixture was rinsed with HCl solution (1 M, 10 mL) and saturated NaHCO3 solution (10 mL), respectively. The organic layer was dried over anhydrous Na2SO4 and concentrated. The crude product was purified by PTLC developing with hexane/EtOAC (10:1, v/v) to give the desired product as a colorless oil in 43% yield. 1H NMR (300 MHz, CDCl3) δ 7.18 (d, J = 8.1, 1H), 7.00 (dd, J = 8.1, 2.4 Hz, 1H), 6.90 (d, J = 2.4 Hz, 1H), 4.53 (br.s, 1H), 3.11 (dd, J = 13.8, 6.3 Hz, 1H), 2.97–2.78 (m, 5H), 2.28 (d, J = 12.9 Hz, 1H), 1.89–1.58 (m, 7H), 1.42 (s, 9H, C(CH3)3), 1.23 (d, J = 6.9 Hz, 6H, CH(CH3)2)), 1.22 (s, 3H, CH3), 0.91 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 150.30, 147.39, 145.75, 134.97, 127.02, 124.40, 123.98, 79.22, 51.21, 44.99, 38.50, 37.56, 37.49, 36.12, 33.57, 30.38, 28.53, 25.45, 24.13, 24.08, 18.98, 18.76. HRMS (ESI): m/z calculated for C25H40NO2 [M + H]+: 386.3059. Found: 386.3051. IR (film) vmax: 3350, 2959, 1694, 1389, 1165, 1039 cm−1.
4.17. Synthesis of 21
To a solution of dehydroabietylamine (156 mg, 55% purity, 0.3 mmo) in DCM (3 mL) was added triethylamine (104 μL, 0.75 mmol) at 0 °C. The solution was stirred for 30 min before adding acetyl chloride (21 μL, 0.3 mmol). The reaction was allowed to proceed at 0 °C under argon for 1 h before adding 10 M HCl (0.03 mL, 0.3 mmol). The mixture was then diluted with EtOAc (75 mL). The resulting mixture was rinsed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated. The yellow crude oil was purified via PTLC, developing twice with hexane/EtOAc (2:1, v/v) to furnish the desired product as a clear oil in 43% yield. 1H NMR (300 MHz, CDCl3) δ 7.17 (d, J = 8.2 Hz, 1H), 6.99 (dd, J = 8.1, 2.4 Hz, 1H), 6.89 (d, J = 2.4 Hz, 1H), 5.87 (br.s, 1H), 3.24 (dd, J = 13.5, 6.3 Hz, 1H), 3.08 (dd, J = 13.8, 6.6 Hz, 1H), 2.96–2.75 (m, 3H), 2.29 (br.d, J = 14.4 Hz, 1H), 1.98 (s, 3H), 1.92–1.85 (m, 2H), 1.79–1.59 (m, 3H), 1.44–1.36 (m, 3H), 1.22 (d, J = 6.9 Hz, 6H), 1.21 (s, 3H), 0.94 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 170.50, 147.23, 145.72, 134.85, 127.01, 124.21, 123.92, 50.00, 45.22, 38.39, 37.49, 37.39, 36.22, 33.49, 30.23, 25.35, 24.07, 24.03, 23.48, 23.41, 19.02, 18.81, 18.66. HRMS (ESI): m/z calculated for C22H34NO [M + H]+: 328.2640. Found: 328.2639. IR (film) vmax: 2924, 2866, 1693, 1440, 1286, 1039 cm−1.
4.18. Synthesis of 22
To a solution of dehydroabietylamine (156 mg, 55% purity, 0.3 mmo) in DCM (3 mL) was added triethylamine (104 μL, 0.75 mmol) at 0 °C. The solution was stirred for 30 min before adding dimethylcarbamoyl chloride (32 mg, 0.3 mmol). The reaction was allowed to proceed at room temperature under argon for 6 h before diluting with EtOAc (75 mL). The resulting mixture was rinsed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated. The clear crude oil was subjected to PTLC purification eluting with hexane/EtOAc (2:1, v/v) to give the desired product as a clear oil in 67% yield. 1H NMR (300 MHz, CDCl3) δ 7.16 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 2.4 Hz, 1H), 4.49 (br,s, 1H, NH), 3.20 (dd, J = 13.8, 5.7 Hz, 1H), 3.08 (dd, J = 13.8, 5.7 Hz, 1H), 2.87 (s, 6H), 2.27 (d, J = 14.4 Hz, 1H), 1.77–1.58 (m, 6H), 1.45–1.29 (m, 5H), 1.21 (d, J = 6.9 Hz, 6H), 1.20 (s, 3H), 0.91 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 158.52, 147.34, 145.59, 135.03, 127.02, 124.31, 123.87, 60.45, 51.31, 45.43, 38.45, 37.57, 37.44, 36.42, 36.27, 33.48, 30.57, 25.56, 24.06, 24.03, 19.03, 18.75. HRMS (ESI): m/z calculated for C23H37N2O [M + H]+: 357.2906. Found: 357.2902. IR (film) vmax: 3349, 1636, 1529, 1219, 908 cm−1.
4.19. Synthesis of 23
To a solution of dehydroabietylamine (156 mg, 55% purity, 0.3 mmo) in DCM (3 mL) was added triethylamine (104 μL, 0.75 mmol) at 0 °C. The solution was stirred for 30 min before adding 2,4-dimethylbenzoyl chloride (51 mg, 0.3 mmol). The reaction was allowed to proceed at room temperature under argon for 4 h before being diluted with EtOAc (75 mL). The resulting mixture was rinsed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated. The light-yellow crude product was subjected to PTLC purification by developing three times with hexane/EtOAc (8:1, v/v) to give the desired product as a clear wax in 54% yield. 1H NMR (300 MHz, CDCl3) δ 7.33 (s, 2H), 7.17 (d, J = 8.4 Hz, 1H), 7.11 (s, 1H), 6.99 (dd, J = 8.1, 2.4 Hz, 1H), 6.89 (d, J = 2.4 Hz, 1H), 6.12 (t, J = 5.7 Hz, 1H, NH), 3.43 (dd, J = 13.8, 6.6 Hz, 1H), 3.32 (dd, J = 13.8, 6.6 Hz, 1H), 2.97–2.78 (m, 3H), 2.41–2.29 (m, 2H), 2.34 (s, 6H, 2 × CH3), 2.02–1.96 (m, 1H), 1.84–1.66 (m, 3H), 1.55–1.36 (m, 3H), 1.24 (s, 3H, CH3), 1.22 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.01 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 168.14, 147.19, 145.73, 138.38, 134.97, 133.11, 127.07, 124.72, 124.65, 124.33, 123.96, 60.52, 50.48, 38.46, 37.80, 37.67, 36.52, 33.53, 30.53, 25.57, 24.08, 21.34, 19.23, 18.86, 18.75, 14.32. HRMS (ESI): m/z calculated for C29H40NO [M + H]+: 418.3110. Found: 418.3109. IR (film) vmax: 3292, 2916, 2865, 1685, 1497, 1245, 1038 cm−1.
4.20. Synthesis of 24
To a solution of dehydroabietylamine (202 mg, 55% purity, 0.39 mmol) in DCM (4 mL) was added triethylamine (0.14 mL, 1.0 mmol) at 0 °C under argon, and the mixture was stirred for 15–20 min before adding dimethyl thiocarbonyl chloride (73 mg, 0.59 mmol). The reaction was then allowed to proceed with stirring at room temperature for two days prior to being diluted with ethyl acetate (50 mL). The resulting mixture was rinsed with brine (10 mL × 3), dried over anhydrous Na2SO4, and concentrated. The obtained crude product was purified by PTLC developing twice with hexane/EtOAc (1:2, v/v) to give the desired product as a yellow oil in 61% yield. 1H NMR (300 MHz, CDCl3) δ 7.16 (d, J = 8.2 Hz, 1H), 6.99 (dd, J = 8.2, 2.1 Hz, 1H), 6.89 (d, J = 2.1 Hz, 1H), 5.49 (br.s, 1H), 3.72 (dd, J = 13.5, 5.1 Hz, 1H), 3.59 (dd, J = 13.2, 4.5 Hz, 1H), 3.25 (s, 6H, N(CH3)2), 2.91–2.77 (m, 3H), 2.30 (d, J = 13.5 Hz, 1H), 1.98 (dd, J = 13.5, 6.6 Hz, 1H), 1.82–1.65 (m, 3H), 1.51–1.29 (m, 4H), 1.22 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.22 (s, 3H, CH3), 0.99 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 182.23, 147.04, 145.70, 134.83, 127.02, 124.24, 123.93, 56.86, 46.21, 40.58, 38.39, 37.67, 37.63, 36.82, 33.46, 30.45, 25.54, 24.05, 24.00, 19.24, 18.87, 18.67. HRMS (ESI): m/z calculated for C23H37N2S [M + H]+: 373.2677. Found: 373.2675. IR (film) vmax: 3332, 2923,1733, 1533, 1408, 1125, 909 cm−1.
4.21. Cell Culture
The four prostate cancer cell lines (LNCaP, 22Rv1, DU145, and PC-3) were initially procured from the ATCC (American Type Culture Collection, Manassas, VA, USA). The three cell lines (PC-3, LNCaP, and 22Rv1) were cultured on a regular basis in RPMI-1640 medium, supplemented with 10% FBS and 1% penicillin/streptomycin. The cultures were sustained at 37 °C in a humid environment with 5% CO2 supplementation. Eagle’s Minimum Essential Medium (EMEM), supplemented with 10% FBS and 1% penicillin/streptomycin, was employed to regularly culture the DU145 cells.
4.22. WST-1 Cell Proliferation Assay
The PC-3, DU145, and LNCaP prostate cancer cells were placed in 96-well plates at a density of 3200 cells per well in 200 µL of culture medium. A density of 6400 22Rv1 cells per well was used for seeding in 96-well plates, with each well containing 200 µL of culture medium. Subsequently, the cells were treated separately with enzalutamide as a positive control, or tricyclic diterpenoids at varying doses for 72 h. The vehicle control group was treated with equal volumes of DMSO. The cell culture was incubated at 37 °C in a CO2 incubator throughout this period. For cell proliferation assessment, 10 µL of the premixed WST-1 cell proliferation reagent (Clontech, Mountain View, CA, USA) was added to each well. After gently mixing on an orbital shaker for 1 min to ensure even color distribution, the cells were further incubated at 37 °C for 3 h. A microplate reader (Synergy HT, BioTek, Winooski, VT, USA) was utilized to measure the absorbance of each well at a wavelength of 430 nm. The IC50 value represented the concentration of each test compound that suppresses cell proliferation by 50% under the experimental conditions, which was determined by averaging triplicate determinations that were both reproducible and statistically significant. To calculate the IC50 values, a linear or logarithmic proliferative suppression curve was generated based on at least five dosages for each test compound.
4.23. Statistical Analysis
The mean ± SD (standard derivation) was used to represent all the data gathered from the indicated number of experiments. The differences between the treatment and control groups were analyzed using the student’s t-test, with statistical significance defined as a p-value < 0.05.


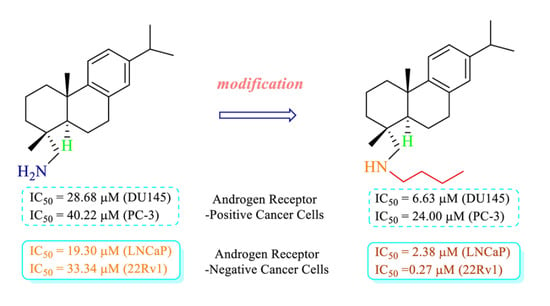
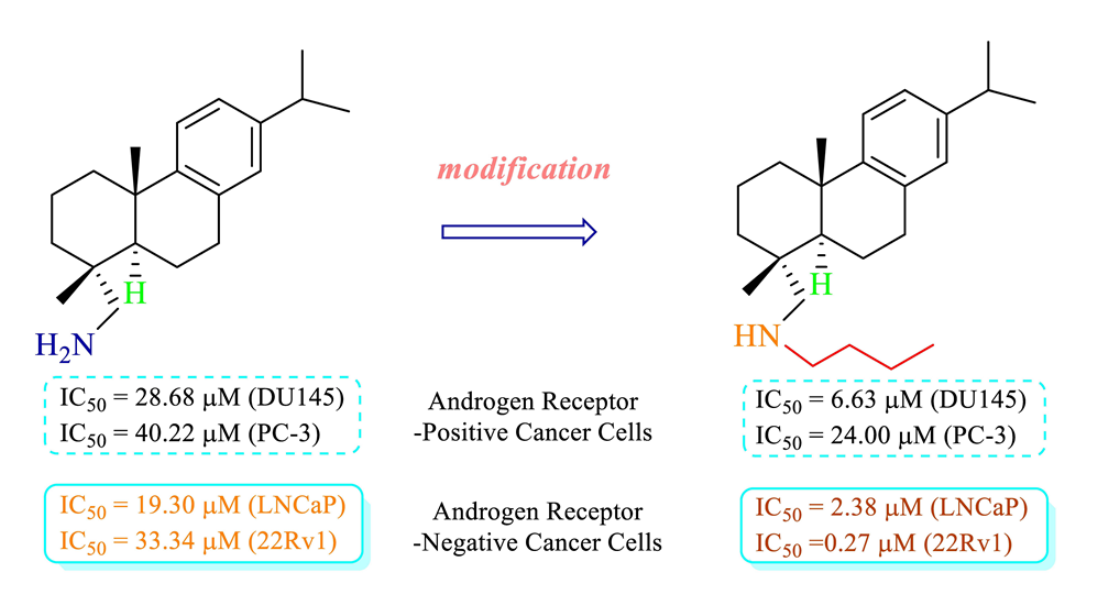
 ,
, 







{kind=link}
{kind=link}
{kind=link}
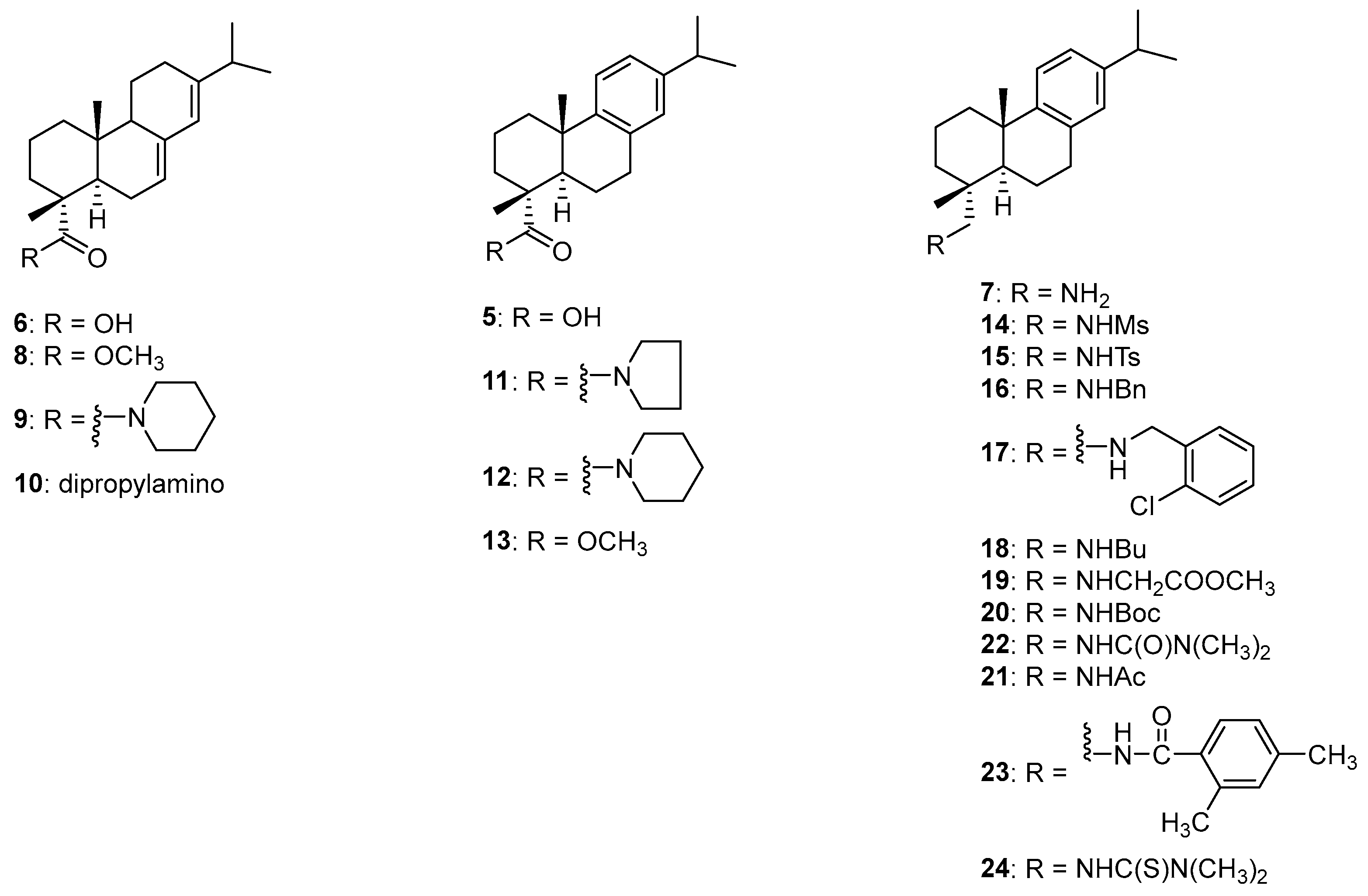{kind=link}
{kind=link}
{kind=link}
{kind=link}