

Engineered Lipidic Nanomaterials Inspired by Sphingomyelin Metabolism for Cancer Therapy


Abstract
:
1. Introduction
2. Synthesis and Metabolism of SM
2.1. Structure and Distribution of SM
2.2. SM Synthesis
2.3. SM Metabolism
2.3.1. Ceramide (Cer)
2.3.2. Sphingosine (Sph)
2.3.3. Sphingosine-1-Phosphate (S1P)
3. Role of SM in the Development of Tumors
3.1. Oncogenesis
3.2. Proliferation and Metastasis
3.3. Multidrug Resistance
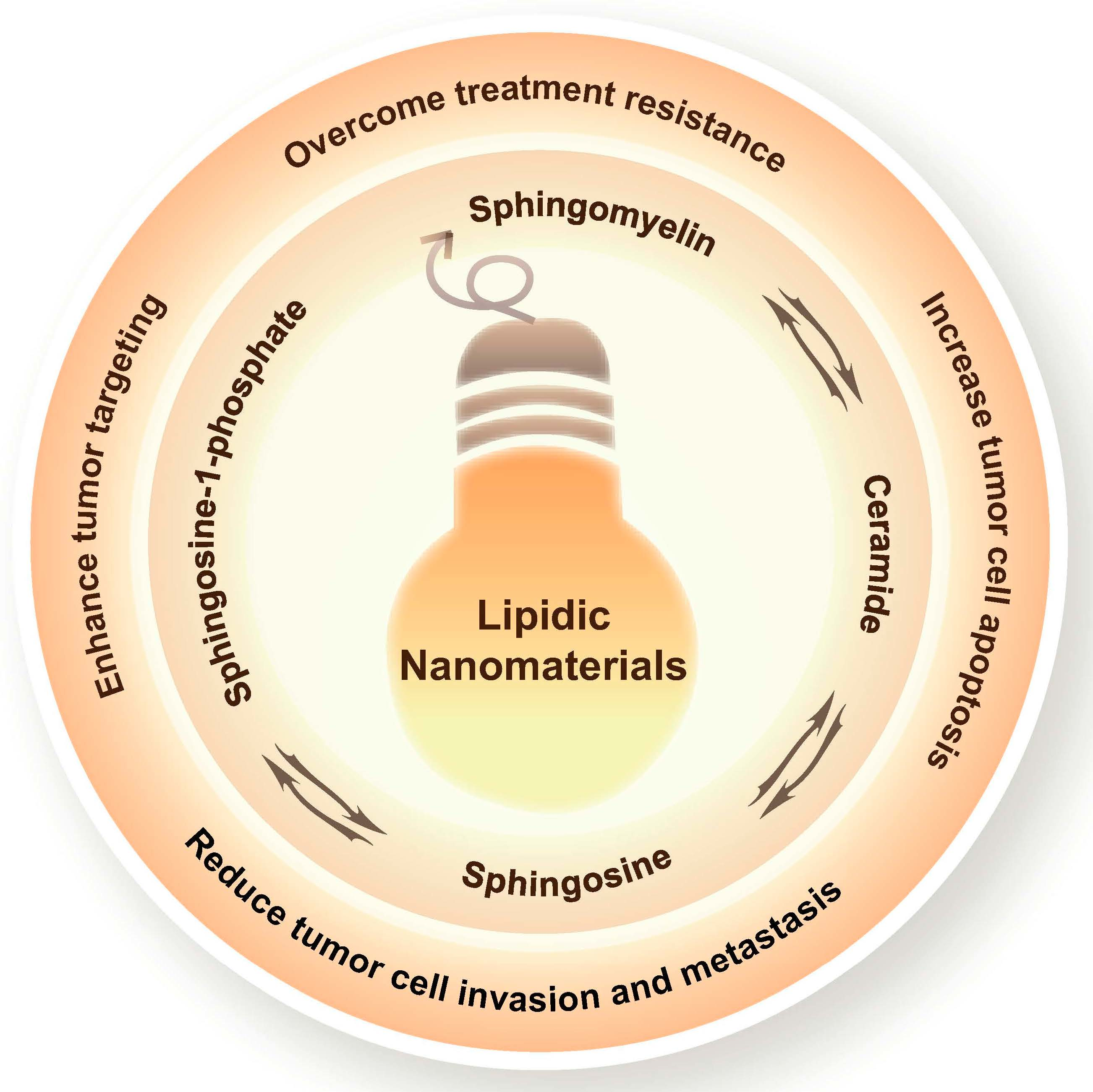
4. SM Metabolism-Based Lipidic Nanomaterials for Cancer Therapy
4.1. SM-Based Lipidic Nanomaterials for Cancer Therapy
4.2. Cer-Based Lipidic Nanomaterials for Cancer Therapy
4.3. S1P-Based Lipidic Nanomaterials for Cancer Therapy
5. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Riehemann, K.; Schneider, S.W.; Luger, T.A.; Godin, B.; Ferrari, M.; Fuchs, H. Nanomedicine--challenge and perspectives. Angew. Chem. Int. Ed. 2009, 48, 872–897. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Zhang, L.; Wu, H. Nanomaterials for cancer therapies. Nanotechnol. Rev. 2017, 6, 473–496. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 12320–12364. [Google Scholar] [CrossRef] [PubMed]
- Dadfar, S.M.; Camozzi, D.; Darguzyte, M.; Roemhild, K.; Varvara, P.; Metselaar, J.; Banala, S.; Straub, M.; Guvener, N.; Engelmann, U.; et al. Size-isolation of superparamagnetic iron oxide nanoparticles improves MRI, MPI and hyperthermia performance. J. Nanobiotechnol. 2020, 18, 22. [Google Scholar] [CrossRef]
- Li, M.; Huang, Y.; Wu, J.; Li, S.; Mei, M.; Chen, H.; Wang, N.; Wu, W.; Zhou, B.; Tan, X.; et al. A PEG-lipid-free COVID-19 mRNA vaccine triggers robust immune responses in mice. Mater. Horiz. 2023, 10, 466–472. [Google Scholar] [CrossRef]
- Chaudhari, V.S.; Murty, U.S.; Banerjee, S. Lipidic nanomaterials to deliver natural compounds against cancer: A review. Environ. Chem. Lett. 2020, 18, 1803–1812. [Google Scholar] [CrossRef]
- Limongi, T.; Susa, F.; Marini, M.; Allione, M.; Torre, B.; Pisano, R.; di Fabrizio, E. Lipid-based nanovesicular drug delivery systems. Nanomaterials 2021, 11, 3391. [Google Scholar] [CrossRef]
- Porter, C.J.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef] [Green Version]
- Wolrab, D.; Jirasko, R.; Cifkova, E.; Horing, M.; Mei, D.; Chocholouskova, M.; Peterka, O.; Idkowiak, J.; Hrnciarova, T.; Kuchar, L.; et al. Lipidomic profiling of human serum enables detection of pancreatic cancer. Nat. Commun. 2022, 13, 124. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, R.Q.; Wang, Z.G.; Liu, S.L. In situ quantification of lipids in live cells by using lipid-binding domain-based biosensors. Bioconjug. Chem. 2022, 33, 2076–2087. [Google Scholar] [CrossRef] [PubMed]
- Radin, N.S. Cancer progression in the kidney and prostate: Vital roles of sphingolipids in chemotherapy. Urology 2002, 60, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Vykoukal, J.; Fahrmann, J.F.; Gregg, J.R.; Tang, Z.; Basourakos, S.; Irajizad, E.; Park, S.; Yang, G.; Creighton, C.J.; Fleury, A.; et al. Caveolin-1-mediated sphingolipid oncometabolism underlies a metabolic vulnerability of prostate cancer. Nat. Commun. 2020, 11, 4279. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, G.; Moorthi, S.; Luberto, C. Role and function of sphingomyelin biosynthesis in the development of cancer. Adv. Cancer Res. 2018, 140, 61–96. [Google Scholar]
- Bienias, K.; Fiedorowicz, A.; Sadowska, A.; Prokopiuk, S.; Car, H. Regulation of sphingomyelin metabolism. Pharmacol. Rep. 2016, 68, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [Green Version]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef]
- Modrak, D.E.; Gold, D.V.; Goldenberg, D.M. Sphingolipid targets in cancer therapy. Mol. Cancer Ther. 2006, 5, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, M.; Okazaki, T. Role of ceramide/sphingomyelin (SM) balance regulated through “SM cycle” in cancer. Cell. Signal. 2021, 87, 110119. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef]
- Taha, T.A.; Mullen, T.D.; Obeid, L.M. A house divided: Ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim. Biophys. Acta 2006, 1758, 2027–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnick, R. The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J. Clin. Investig. 2002, 110, 3–8. [Google Scholar] [CrossRef]
- Huwiler, A.; Pfeilschifter, J. Altering the sphingosine-1-phosphate/ceramide balance: A promising approach for tumor therapy. Curr. Pharm. Des. 2006, 12, 4625–4635. [Google Scholar] [CrossRef]
- Merrill, A.H., Jr.; Schmelz, E.M.; Dillehay, D.L.; Spiegel, S.; Shayman, J.A.; Schroeder, J.J.; Riley, R.T.; Voss, K.A.; Wang, E. Sphingolipids--the enigmatic lipid class: Biochemistry, physiology, and pathophysiology. Toxicol. Appl. Pharmacol. 1997, 142, 208–225. [Google Scholar] [CrossRef]
- Filippov, A.; Oradd, G.; Lindblom, G. Sphingomyelin structure influences the lateral diffusion and raft formation in lipid bilayers. Biophys. J. 2006, 90, 2086–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie; William, W. Lipids: Their structures and occurrence. In Lipid Analysis, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 3–19. [Google Scholar]
- Furland, N.E.; Zanetti, S.R.; Oresti, G.M.; Maldonado, E.N.; Aveldano, M.I. Ceramides and sphingomyelins with high proportions of very long-chain polyunsaturated fatty acids in mammalian germ cells. J. Biol. Chem. 2007, 282, 18141–18150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slotte, J.P. Biological functions of sphingomyelins. Prog. Lipid Res. 2013, 52, 424–437. [Google Scholar] [CrossRef]
- Allan, D.; Quinn, P. Resynthesis of sphingomyelin from plasma-membrane phosphatidylcholine in BHK cells treated with staphylococcus aureus sphingomyelinase. Biochem. J. 1988, 254, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futerman, A.H.; Stieger, B.; Hubbard, A.L.; Pagano, R.E. Sphingomyelin synthesis in rat liver occurs predominantly at the cis and medial cisternae of the Golgi apparatus. J. Biol. Chem. 1990, 265, 8650–8657. [Google Scholar] [CrossRef] [PubMed]
- Huitema, K.; van den Dikkenberg, J.; Brouwers, J.F.; Holthuis, J.C. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004, 23, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef]
- Hanada, K. Regulation of CERT-mediated trafficking of ceramide. Chem. Phys. Lipids 2007, 149, S7. [Google Scholar] [CrossRef]
- Mitsutake, S.; Zama, K.; Yokota, H.; Yoshida, T.; Tanaka, M.; Mitsui, M.; Ikawa, M.; Okabe, M.; Tanaka, Y.; Yamashita, T.; et al. Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. J. Biol. Chem. 2011, 286, 28544–28555. [Google Scholar] [CrossRef] [Green Version]
- Vacaru, A.M.; Tafesse, F.G.; Ternes, P.; Kondylis, V.; Hermansson, M.; Brouwers, J.F.; Somerharju, P.; Rabouille, C.; Holthuis, J.C. Sphingomyelin synthase-related protein SMSr controls ceramide homeostasis in the ER. J. Cell Biol. 2009, 185, 1013–1027. [Google Scholar] [CrossRef] [Green Version]
- Saddoughi, S.A.; Song, P.; Ogretmen, B. Roles of bioactive sphingolipids in cancer biology and therapeutics. Subcell. Biochem. 2008, 49, 413–440. [Google Scholar] [CrossRef] [Green Version]
- Haddadi, N.; Lin, Y.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. “Dicing and splicing” sphingosine kinase and relevance to cancer. Int. J. Mol. Sci. 2017, 18, 1891. [Google Scholar] [CrossRef] [Green Version]
- Turpin-Nolan, S.M.; Bruning, J.C. The role of ceramides in metabolic disorders: When size and localization matters. Nat. Rev. Endocrinol. 2020, 16, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A. Functions of ceramide in coordinating cellular responses to stress. Science 1996, 274, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Testai, F.D.; Landek, M.A.; Dawson, G. Regulation of sphingomyelinases in cells of the oligodendrocyte lineage. J. Neurosci. Res. 2004, 75, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Monastyrskaya, K.; Draeger, A. Fluorescent annexin a1 reveals dynamics of ceramide platforms in living cells. Traffic 2010, 9, 1757–1775. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Beutel, O.; Ebell, K.; Korneev, S.; Holthuis, J.C. Diverting CERT-mediated ceramide transport to mitochondria triggers Bax-dependent apoptosis. J. Cell. Sci. 2017, 130, 360–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalfant, C. Sphingolipids as Signaling and Regulatory Molecules; Springer Science & Business Media: New York, NY, USA, 2010. [Google Scholar]
- Gomez-Munoz, A.; Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Trueba, M.; Ordonez, M. Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 2016, 61, 51–62. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Goi, F.M.; Alonso, A. Sphingomyelinases: Enzymology and membrane activity. FEBS Lett. 2002, 531, 38–46. [Google Scholar]
- Marchesini, N.; Hannun, Y.A. Acid and neutral sphingomyelinases: Roles and mechanisms of regulation. Biochem. Cell Biol. 2004, 82, 27–44. [Google Scholar] [CrossRef]
- Clarke, C.J.; Wu, B.X.; Hannun, Y.A. The neutral sphingomyelinase family: Identifying biochemical connections. Adv. Enzyme Regul. 2011, 51, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, Y.; Tashima, M.; Takahashi, A.; Uchiyama, T.; Okazaki, T. Ceramide generation in nitric oxide-induced apoptosis. Activation of magnesium-dependent neutral sphingomyelinase via caspase-3. J. Biol. Chem. 1999, 274, 10654–10660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, S.; Nakashima, S.; Kiyono, T.; Sawada, M.; Yoshimura, S.; Iwama, T.; Banno, Y.; Shinoda, J.; Sakai, N. p53-independent ceramide formation in human glioma cells during gamma-radiation-induced apoptosis. Cell Death Differ. 2004, 11, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Schütze, S.; Machleidt, T.; Krönke, M. The role of diacylglycerol and ceramide in tumor necrosis factor and interleukin-1 signal transduction. J. Leukoc. Biol. 1994, 56, 533–541. [Google Scholar] [CrossRef]
- Santana, P.; Pena, L.A.; Haimovitz-Friedman, A.; Martin, S.; Green, D.; McLoughlin, M.; Cordon-Cardo, C.; Schuchman, E.H.; Fuks, Z.; Kolesnick, R. Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell 1996, 86, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, A. The presence of spingomyelin- and ceramide-cleaving enzymes in the small intestinal tract. Biochim. Biophys. Acta 1969, 176, 339–347. [Google Scholar] [CrossRef]
- Duan, R.D. Alkaline sphingomyelinase: An old enzyme with novel implications. BBA-Bioenerg. 2006, 1761, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Machala, M.; Prochazkova, J.; Hofmanova, J.; Kralikova, L.; Slavik, J.; Tylichova, Z.; Ovesna, P.; Kozubik, A.; Vondracek, J. Colon Cancer and Perturbations of the Sphingolipid Metabolism. Int. J. Mol. Sci. 2019, 20, 6051. [Google Scholar] [CrossRef] [Green Version]
- Novgorodov, S.A.; Wu, B.X.; Gudz, T.I.; Bielawski, J.; Ovchinnikova, T.V.; Hannun, Y.A.; Obeid, L.M. Novel pathway of ceramide production in mitochondria: Thioesterase and neutral ceramidase produce ceramide from sphingosine and acyl-CoA. J. Biol. Chem. 2011, 286, 25352–25362. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim. Biophys. Acta 2002, 1585, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Spiegel, S. Sphingosine kinase: A mediator of vital cellular functions. Prostag. Other Lipid Mediat. 2001, 64, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase in immunity and cancer: Silencing the siren. Trends Mol. Med. 2007, 13, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Oskeritzian, C.A.; Paugh, S.W.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta 2006, 1758, 2016–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatomi, Y.; Ohmori, T.; Ge, R.L.; Kazama, F.; Okamoto, H.; Sano, T.; Satoh, K.; Kume, S.; Tigyi, G.; Igarashi, Y.; et al. Sphingosine 1-phosphate as a major bioactive lysophospholipid that is released from platelets and interacts with endothelial cells. Blood 2000, 96, 3431–3438. [Google Scholar] [CrossRef] [PubMed]
- Kluk, M.J.; Hla, T. Role of the sphingosine 1-phosphate receptor EDG-1 in vascular smooth muscle cell proliferation and migration. Circ. Res. 2001, 89, 496–502. [Google Scholar] [CrossRef]
- Adada, M.M.; Canals, D.; Jeong, N.; Kelkar, A.D.; Hernandez-Corbacho, M.; Pulkoski-Gross, M.J.; Donaldson, J.C.; Hannun, Y.A.; Obeid, L.M. Intracellular sphingosine kinase 2-derived sphingosine-1-phosphate mediates epidermal growth factor-induced ezrin-radixin-moesin phosphorylation and cancer cell invasion. FASEB J. 2015, 29, 4654–4669. [Google Scholar] [CrossRef] [Green Version]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, S.E.; Milstien, S.; Spiegel, S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol. Metab. 2007, 18, 300–307. [Google Scholar] [CrossRef]
- Zheng, X.; Li, W.; Ren, L.; Liu, J.; Pang, X.; Chen, X.; Kang, D.; Wang, J.; Du, G. The sphingosine kinase-1/sphingosine-1-phosphate axis in cancer: Potential target for anticancer therapy. Pharmacol. Ther. 2019, 195, 85–99. [Google Scholar] [CrossRef]
- Camaré, C.; Trayssac, M.; Garmy-Susini, B.; Mucher, E.; Sabbadini, R.; Salvayre, R.; Negre-Salvayre, A. Oxidized LDL-induced angiogenesis involves sphingosine 1-phosphate: Prevention by anti-S1P antibody. Br. J. Pharmacol. 2014, 172, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, P.; Ramanathan, R.; Takabe, K. S1P promotes breast cancer progression by angiogenesis and lymphangiogenesis. Breast Cancer Manag. 2015, 4, 241–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamekhi, S.; Abdolalizadeh, J.; Ostadrahimi, A.; Mohammadi, S.A.; Barzegari, A.; Lotfi, H.; Bonabi, E.; Zarghami, N. Apoptotic effect of saccharomyces cerevisiae on human colon cancer SW480 cells by regulation of Akt/NF-kB signaling pathway. Probiotics Antimicrob. Proteins 2020, 12, 311–319. [Google Scholar] [CrossRef]
- Nagahashi, M.; Yamada, A.; Katsuta, E.; Aoyagi, T.; Huang, W.C.; Terracina, K.P.; Hait, N.C.; Allegood, J.C.; Tsuchida, J.; Yuza, K.; et al. Targeting the SphK1/S1P/S1PR1 axis that links obesity, chronic inflammation, and breast cancer metastasis. Cancer Res. 2018, 78, 1713–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhuang, T.; Liang, Z.; Li, L.; Xue, M.; Liu, J.; Liang, H. Breast cancer suppression by aplysin is associated with inhibition of PI3K/AKT/FOXO3a pathway. Oncotarget 2017, 8, 63923–63934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Y.; Li, K.; Guo, Y.; Wang, Q.; Li, Z.; Yang, Y.; Chen, Z.; Wang, J.; Zhao, W.; Zhang, H.; et al. Tumor suppressor PRSS8 targets Sphk1/S1P/Stat3/Akt signaling in colorectal cancer. Oncotarget 2016, 7, 26780–26792. [Google Scholar] [CrossRef]
- Zheng, X.D.; Zhang, Y.; Qi, X.W.; Wang, M.H.; Sun, P.; Zhang, Y.; Jiang, J. Role of Sphk1 in the malignant transformation of breast epithelial cells and breast cancer progression. Indian J. Cancer 2014, 51, 524–529. [Google Scholar] [CrossRef]
- Kawahara, S.; Otsuji, Y.; Nakamura, M.; Murakami, M.; Murate, T.; Matsunaga, T.; Kanoh, H.; Seishima, M.; Banno, Y.; Hara, A. Sphingosine kinase 1 plays a role in the upregulation of CD44 expression through extracellular signal-regulated kinase signaling in human colon cancer cells. Anticancer Drugs 2013, 24, 473–483. [Google Scholar] [CrossRef]
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642. [Google Scholar] [CrossRef] [Green Version]
- Pchejetski, D.; Golzio, M.; Bonhoure, E.; Calvet, C.; Doumerc, N.; Garcia, V.; Mazerolles, C.; Rischmann, P.; Teissie, J.; Malavaud, B.; et al. Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 2005, 65, 11667–11675. [Google Scholar] [CrossRef] [Green Version]
- Alshaker, H.; Wang, Q.; Kawano, Y.; Arafat, T.; Bohler, T.; Winkler, M.; Cooper, C.; Pchejetski, D. Everolimus (RAD001) sensitizes prostate cancer cells to docetaxel by down-regulation of HIF-1alpha and sphingosine kinase 1. Oncotarget 2016, 7, 80943–80956. [Google Scholar] [CrossRef] [Green Version]
- Sauer, L.; Nunes, J.; Salunkhe, V.; Skalska, L.; Kohama, T.; Cuvillier, O.; Waxman, J.; Pchejetski, D. Sphingosine kinase 1 inhibition sensitizes hormone-resistant prostate cancer to docetaxel. Int. J. Cancer 2009, 125, 2728–2736. [Google Scholar] [CrossRef]
- Sukocheva, O.; Wang, L.; Verrier, E.; Vadas, M.A.; Xia, P. Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology 2009, 150, 4484–4492. [Google Scholar] [CrossRef] [Green Version]
- Giussani, P.; Bassi, R.; Anelli, V.; Brioschi, L.; De Zen, F.; Riccitelli, E.; Caroli, M.; Campanella, R.; Gaini, S.M.; Viani, P.; et al. Glucosylceramide synthase protects glioblastoma cells against autophagic and apoptotic death induced by temozolomide and Paclitaxel. Cancer Investig. 2012, 30, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Expression of glucosylceramide synthase, converting ceramide to glucosylceramide, confers adriamycin resistance in human breast cancer cells. J. Biol. Chem. 1999, 274, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Y.; Patwardhan, G.A.; Xie, P.; Gu, X.; Giuliano, A.E.; Cabot, M.C. Glucosylceramide synthase, a factor in modulating drug resistance, is overexpressed in metastatic breast carcinoma. Int. J. Oncol. 2011, 39, 425–431. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, F.R.; Pearson, J.M.; Tan, S.F.; Cheon, H.; Xing, J.C.; Dunton, W.; Feith, D.J.; Loughran, T.P., Jr. Sphingosine kinase-2 is overexpressed in large granular lymphocyte leukaemia and promotes survival through Mcl-1. Br. J. Heaematol. 2020, 190, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Oskouian, B.; Sooriyakumaran, P.; Borowsky, A.D.; Crans, A.; Dillard-Telm, L.; Tam, Y.Y.; Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 17384–17389. [Google Scholar] [CrossRef]
- Zheng, K.; Chen, Z.; Feng, H.; Chen, Y.; Zhang, C.; Yu, J.; Luo, Y.; Zhao, L.; Jiang, X.; Shi, F. Sphingomyelin synthase 2 promotes an aggressive breast cancer phenotype by disrupting the homoeostasis of ceramide and sphingomyelin. Cell Death Dis. 2019, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Lafont, E.; Milhas, D.; Carpentier, S.; Garcia, V.; Jin, Z.X.; Umehara, H.; Okazaki, T.; Schulze-Osthoff, K.; Levade, T.; Benoist, H.; et al. Caspase-mediated inhibition of sphingomyelin synthesis is involved in FasL-triggered cell death. Cell Death Differ. 2010, 17, 642–654. [Google Scholar] [CrossRef] [Green Version]
- Grassme, H.; Schwarz, H.; Gulbins, E. Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem. Biophys. Res. Commun. 2001, 284, 1016–1030. [Google Scholar] [CrossRef]
- Eroica, S.; Susan, C.E.; Cynthia, C.; Elroy, F. Characterizing the sphingomyelinase pathway triggered by PRIMA-1 derivatives in lung cancer cells with differing p53 status. Anticancer Res. 2014, 34, 3271. [Google Scholar]
- Kohno, M.; Momoi, M.; Oo, M.L.; Paik, J.H.; Lee, Y.M.; Venkataraman, K.; Ai, Y.; Ristimaki, A.P.; Fyrst, H.; Sano, H.; et al. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol. Cell. Biol. 2006, 26, 7211–7223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senkal, C.E.; Ponnusamy, S.; Manevich, Y.; Meyers-Needham, M.; Saddoughi, S.A.; Mukhopadyay, A.; Dent, P.; Bielawski, J.; Ogretmen, B. Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J. Biol. Chem. 2011, 286, 42446–42458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.T.; Wu, C.Y.; Lin, Y.C.; Wu, M.T.; Su, K.L.; Yuan, S.S.; Wang, H.D.; Fong, Y.; Lin, Y.H.; Chiu, C.C. C(2)-ceramide-induced rb-dominant senescence-like phenotype leads to human breast cancer Mcf-7 escape from p53-dependent cell death. Int. J. Mol. Sci. 2019, 20, 4292. [Google Scholar] [CrossRef] [Green Version]
- Tallima, H.; Azzazy, H.M.E.; El Ridi, R. Cell surface sphingomyelin: Key role in cancer initiation, progression, and immune evasion. Lipids Health Dis. 2021, 20, 150. [Google Scholar] [CrossRef]
- Mombelli, E.; Morris, R.; Taylor, W.; Fraternali, F. Hydrogen-bonding propensities of sphingomyelin in solution and in a bilayer assembly: A molecular dynamics study. Biophys. J. 2003, 84, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Barenholz, Y.; Thompson, T.E. Sphingomyelin: Biophysical aspects. Chem. Phys. Lipids 1999, 102, 29–34. [Google Scholar] [CrossRef]
- Migliardo, F.; Tallima, H.; Ridi, R.E. Is there a sphingomyelin-based hydrogen bond barrier at the mammalian host-schistosome parasite interface? Cell Biochem. Biophys. 2014, 68, 359–367. [Google Scholar] [CrossRef]
- Slotte, J.P. The importance of hydrogen bonding in sphingomyelin’s membrane interactions with co-lipids. Biochim. Biophys. Acta 2016, 1858, 304–310. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, L.; Liu, D.; Wang, C. Ceramide glycosylation and related enzymes in cancer signaling and therapy. Biomed. Pharmacother. 2021, 139, 111565. [Google Scholar] [CrossRef]
- Sheridan, M.; Ogretmen, B. The role of ceramide metabolism and signaling in the regulation of mitophagy and cancer therapy. Cancers 2021, 13, 2475. [Google Scholar] [CrossRef] [PubMed]
- Visentin, B.; Vekich, J.A.; Sibbald, B.J.; Cavalli, A.L.; Moreno, K.M.; Matteo, R.G.; Garland, W.A.; Lu, Y.; Yu, S.; Hall, H.S.; et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 2006, 9, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.K.; Lee, H.M.; Lee, T.H.; Kang, C.; Yong, S.G. Extracellular membrane vesicles from tumor cells promote angiogenesis via sphingomyelin. Cancer Res. 2002, 62, 6312–6317. [Google Scholar]
- Riboni, L.; Viani, P.; Bassi, R.; Giussani, P.; Tettamanti, G. Basic fibroblast growth factor-induced proliferation of primary astrocytes. evidence for the involvement of sphingomyelin biosynthesis. J. Biol. Chem. 2001, 276, 12797–12804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Xiao, J.; Dong, M.; Qiu, Z.; Jin, J. Human alkaline ceramidase 2 promotes the growth, invasion, and migration of hepatocellular carcinoma cells via sphingomyelin phosphodiesterase acid-like 3B. Cancer Sci. 2020, 111, 2259–2274. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef]
- Zama, K.; Mitsutake, S.; Okazaki, T.; Igarashi, Y. Sphingomyelin in microdomains of the plasma membrane regulates amino acid-stimulated mTOR signal activation. Cell Biol. Int. 2018, 42, 823–831. [Google Scholar] [CrossRef]
- Jing, F.; Jing, C.; Dai, X.; Zhou, G.; Hong, L. Sphingomyelin synthase 2 but not sphingomyelin synthase 1 is upregulated in ovarian cancer and involved in migration, growth and survival via different mechanisms. Am. J. Transl. Res. 2021, 13, 4412–4421. [Google Scholar]
- Don, A.S.; Lim, X.Y.; Couttas, T.A. Re-configuration of sphingolipid metabolism by oncogenic transformation. Biomolecules 2014, 4, 315–353. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, M.; Ueda, Y.; Matsushita, M.; Nagaya, S.; Hashizume, C.; Arai, K.; Kabayama, K.; Fukase, K.; Watanabe, K.; Wardhani, L.O.; et al. Deficiency of sphingomyelin synthase 2 prolongs survival by the inhibition of lymphoma infiltration through ICAM-1 reduction. FASEB J. 2020, 34, 3838–3854. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Hu, J.C.; He, S.H.; Lou, B.; Ding, T.B.; Yang, J.T.; Mo, M.G.; Ye, D.Y.; Zhou, L.; Jiang, X.C.; et al. Sphingomyelin synthase 2 facilitates M2-like macrophage polarization and tumor progression in a mouse model of triple-negative breast cancer. Acta Pharmacol. Sin. 2021, 42, 149–159. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Montfort, A.; Bertrand, F.; Rochotte, J.; Gilhodes, J.; Filleron, T.; Milhes, J.; Dufau, C.; Imbert, C.; Riond, J.; Tosolini, M.; et al. Neutral sphingomyelinase 2 heightens anti-melanoma immune responses and anti-pd-1 therapy efficacy. Cancer Immunol. Res. 2021, 9, 568–582. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Gu, J.; Yu, J.; Guo, S.; Zhu, Y.; Ye, D. Abnormal methylation status of FBXW10 and SMPD3, and associations with clinical characteristics in clear cell renal cell carcinoma. Oncol. Lett. 2015, 10, 3073–3080. [Google Scholar] [CrossRef] [Green Version]
- Jabalee, J.; Towle, R.; Lawson, J.; Dickman, C.; Garnis, C. Sphingomyelin phosphodiesterase 3 methylation and silencing in oral squamous cell carcinoma results in increased migration and invasion and altered stress response. Oncotarget 2020, 11, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Tsuruo, T. Mechanism of multidrug resistance and implication for therapy. Pathophysiology 1994, 1, 285–296. [Google Scholar] [CrossRef]
- Peetla, C.; Vijayaraghavalu, S.; Labhasetwar, V. Biophysics of cell membrane lipids in cancer drug resistance: Implications for drug transport and drug delivery with nanoparticles. Adv. Drug Deliv. Rev. 2013, 65, 1686–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emma, B.; Gfn, B.; Fms, B.; Asc, B.; Bs, B.; Rcf, A.; Ono, B. Role of sphingomyelin on the interaction of the anticancer drug gemcitabine hydrochloride with cell membrane models. Colloids Surf. B 2020, 196, 111357. [Google Scholar]
- Xu, J.X.; Morii, E.; Liu, Y.; Nakamichi, N.; Ikeda, J.; Kimura, H.; Aozasa, K. High tolerance to apoptotic stimuli induced by serum depletion and ceramide in side-population cells: High expression of CD55 as a novel character for side-population. Exp. Cell Res. 2007, 313, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Cervia, D.; Assi, E.; De Palma, C.; Giovarelli, M.; Bizzozero, L.; Pambianco, S.; Di Renzo, I.; Zecchini, S.; Moscheni, C.; Vantaggiato, C.; et al. Essential role for acid sphingomyelinase-inhibited autophagy in melanoma response to cisplatin. Oncotarget 2016, 7, 24995–25009. [Google Scholar] [CrossRef] [Green Version]
- Grammatikos, G.; Teichgraber, V.; Carpinteiro, A.; Trarbach, T.; Weller, M.; Hengge, U.R.; Gulbins, E. Overexpression of acid sphingomyelinase sensitizes glioma cells to chemotherapy. Antioxid. Redox. Signal. 2007, 9, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Lacour, S.; Hammann, A.; Grazide, S.; Lagadic-Gossmann, D.; Athias, A.; Sergent, O.; Laurent, G.; Gambert, P.; Solary, E.; Dimanche-Boitrel, M.-T. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res. 2004, 64, 3593–3598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurmann, L.; Belkacemi, L.; Adams, N.R.; Majmudar, P.M.; Moghaddas, S.; Bose, R.N. A novel cisplatin mediated apoptosis pathway is associated with acid sphingomyelinase and FAS proapoptotic protein activation in ovarian cancer. Apoptosis 2015, 20, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Schuchman, E.H. Acid sphingomyelinase overexpression enhances the antineoplastic effects of irradiation in vitro and in vivo. Mol. Ther. 2008, 16, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.A.; Gran, M.L.; Peppas, N.A. Targeted nanodelivery of drugs and diagnostics. Nano Today 2010, 5, 143–159. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Little, N.; Chen, J.; Lambesis, K.T.; Le, K.T.; Han, W.; Scott, A.J.; Lu, J. Immunogenic camptothesome nanovesicles comprising sphingomyelin-derived camptothecin bilayers for safe and synergistic cancer immunochemotherapy. Nat. Nanotechnol. 2021, 16, 1130–1140. [Google Scholar] [CrossRef]
- Bouzo, B.L.; Lores, S.; Jatal, R.; Alijas, S.; Alonso, M.J.; Conejos-Sanchez, I.; de la Fuente, M. Sphingomyelin nanosystems loaded with uroguanylin and etoposide for treating metastatic colorectal cancer. Sci. Rep. 2021, 11, 17213. [Google Scholar] [CrossRef]
- Medina, O.P.; Tower, R.J.; Medina, T.P.; Ashkenani, F.; Appold, L.; Bötcher, M.; Huber, L.; Will, O.; Ling, Q.; Hauser, C.; et al. Multimodal targeted nanoparticle-based delivery system for pancreatic tumor imaging in cellular and animal models. Curr. Pharm. Des. 2020, 28, 313–323. [Google Scholar] [CrossRef]
- Penate Medina, T.; Gerle, M.; Humbert, J.; Chu, H.; Kopnick, A.L.; Barkmann, R.; Garamus, V.M.; Sanz, B.; Purcz, N.; Will, O.; et al. Lipid-iron nanoparticle with a cell stress release mechanism combined with a local alternating magnetic field enables site-activated drug release. Cancers 2020, 12, 3767. [Google Scholar] [CrossRef]
- Masoumi, F.; Saraiva, S.M.; Bouzo, B.L.; Lopez-Lopez, R.; Esteller, M.; Diaz-Lagares, A.; de la Fuente, M. Modulation of colorectal tumor behavior via lncrna tp53tg1-lipidic nanosystem. Pharmaceutics 2021, 13, 1507. [Google Scholar] [CrossRef]
- Nagachinta, S.; Bouzo, B.L.; Vazquez-Rios, A.J.; Lopez, R.; Fuente, M. Sphingomyelin-based nanosystems (sns) for the development of anticancer miRNA therapeutics. Pharmaceutics 2020, 12, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutter, N.L.; Volaric, J.; Szymanski, W.; Feringa, B.L.; Maglia, G. Reversible photocontrolled nanopore assembly. J. Am. Chem. Soc. 2019, 141, 14356–14363. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.H.E.; Park, H.R.; Yoon, Y.M.; Kim, H.I.; Yoo, S.Y.; Lee, K.W.; Lee, Y.S. Antiproliferative 3-deoxysphingomyelin analogs: Design, synthesis, biological evaluation and molecular docking of pyrrolidine-based 3-deoxysphingomyelin analogs as anticancer agents. Bioorganic Chem. 2019, 84, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Messner, M.C.; Levin, J.C.; Abdelmageed, N.; Park, H.; Merrill, A.H., Jr.; Cabot, M.C. Potential role of acid ceramidase in conversion of cytostatic to cytotoxic end-point in pancreatic cancer cells. Cancer Chemoth. Pharm. 2013, 71, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Flowers, M.; Fabrias, G.; Delgado, A.; Casas, J.; Abad, J.L.; Cabot, M.C. C6-ceramide and targeted inhibition of acid ceramidase induce synergistic decreases in breast cancer cell growth. Breast Cancer Res. Treat. 2012, 133, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, Y.; Liu, X.; Li, Z.; Xu, W.; He, S.; Huang, Y.; Zhang, H. Acid sphingomyelinase contributes to evodiamine-induced apoptosis in human gastric cancer SGC-7901 cells. DNA Cell Biol. 2011, 30, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Kester, M.; Heakal, Y.; Fox, T.; Sharma, A.; Robertson, G.P.; Morgan, T.T.; Altinoglu, E.I.; Tabakovic, A.; Parette, M.R.; Rouse, S.M.; et al. Calcium phosphate nanocomposite particles for in vitro imaging and encapsulated chemotherapeutic drug delivery to cancer cells. Nano Lett. 2008, 8, 4116–4121. [Google Scholar] [CrossRef] [Green Version]
- Ganta, S.; Singh, A.; Patel, N.R.; Cacaccio, J.; Rawal, Y.H.; Davis, B.J.; Amiji, M.M.; Coleman, T.P. Development of EGFR-targeted nanoemulsion for imaging and novel platinum therapy of ovarian cancer. Pharm. Res. 2014, 31, 2490–2502. [Google Scholar] [CrossRef] [Green Version]
- Stover, T.; Kester, M. Liposomal delivery enhances short-chain ceramide-induced apoptosis of breast cancer cells. J. Pharmacol. Exp. Ther. 2003, 307, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Alrbyawi, H.; Poudel, I.; Arnold, R.D.; Babu, R.J. Co-delivery of doxorubicin and ceramide in a liposomal formulation enhances cytotoxicity in murine B16Bl6 melanoma cell lines. AAPS PharmSciTech 2019, 20, 99. [Google Scholar] [CrossRef]
- Li, G.; Liu, D.; Kimchi, E.T.; Kaifi, J.T.; Qi, X.; Manjunath, Y.; Liu, X.; Deering, T.; Avella, D.M.; Fox, T.; et al. Nanoliposome C6-ceramide increases the anti-tumor immune response and slows growth of liver tumors in mice. Gastroenterology 2018, 154, 1024–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.; Li, J.; Hui, S. C6 ceramide potentiates curcumin-induced cell death and apoptosis in melanoma cell lines in vitro. Cancer Chemother. Pharmacol. 2010, 66, 999–1003. [Google Scholar]
- Jiang, Y.; DiVittore, N.A.; Kaiser, J.M.; Shanmugavelandy, S.S.; Fritz, J.L.; Heakal, Y.; Tagaram, H.R.; Cheng, H.; Cabot, M.C.; Staveley-O’Carroll, K.F.; et al. Combinatorial therapies improve the therapeutic efficacy of nanoliposomal ceramide for pancreatic cancer. Cancer Biol. Ther. 2011, 12, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.; Medatwal, N.; Kumar, S.; Kar, A.; Komalla, V.; Yavvari, P.S.; Mishra, D.; Rizvi, Z.A.; Nandan, S.; Malakar, D.; et al. A localized chimeric hydrogel therapy combats tumor progression through alteration of sphingolipid metabolism. ACS Cent. Sci. 2019, 5, 1648–1662. [Google Scholar] [CrossRef] [Green Version]
- Medatwal, N.; Ansari, M.N.; Kumar, S.; Pal, S.; Jha, S.K.; Verma, P.; Rana, K.; Dasgupta, U.; Bajaj, A. Hydrogel-mediated delivery of celastrol and doxorubicin induces a synergistic effect on tumor regression via upregulation of ceramides. Nanoscale 2020, 12, 18463–18475. [Google Scholar] [CrossRef]
- Bi, J.; Khan, A.; Tang, J.; Armando, A.M.; Wu, S.; Zhang, W.; Gimple, R.C.; Reed, A.; Jing, H.; Koga, T.; et al. Targeting glioblastoma signaling and metabolism with a re-purposed brain-penetrant drug. Cell. Rep. 2021, 37, 109957. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Li, J.; Tang, J.; Li, B.; Zhang, Y.; Gu, N.; Yang, F. Sphingosine 1-phosphate liposomes for targeted nitric oxide delivery to mediate anticancer effects against brain glioma tumors. Adv. Mater. 2021, 33, e2101701. [Google Scholar] [CrossRef]
- Saddoughi, S.A.; Gencer, S.; Peterson, Y.K.; Ward, K.E.; Mukhopadhyay, A.; Oaks, J.; Bielawski, J.; Szulc, Z.M.; Thomas, R.J.; Selvam, S.P.; et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med. 2013, 5, 105–121. [Google Scholar] [CrossRef]
- Companioni, O.; Mir, C.; Garcia-Mayea, Y.; ME, L.L. Targeting Sphingolipids for Cancer Therapy. Front. Oncol. 2021, 11, 745092. [Google Scholar] [CrossRef]
- Meng, Q.; Zhao, B.; Xu, Q.; Xu, X.; Deng, G.; Li, C.; Luan, L.; Ren, F.; Wang, H.; Xu, H. Indole-propionic acid derivatives as potent, S1P3-sparing and EAE efficacious sphingosine-1-phosphate 1 (S1P1) receptor agonists. Bioorg. Med. Chem. Lett. 2012, 22, 2794–2797. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Guo, Z.; Zhao, X.; Wu, L.; Liu, X.; Zhang, Y.; Zhang, Y.; Deng, Z.; Qu, X.; Cui, S.; et al. Novel 5-fluorouracil sensitizers for colorectal cancer therapy: Design and synthesis of S1P receptor 2 (S1PR2) antagonists. Eur. J. Med. Chem. 2021, 227, 113923. [Google Scholar] [CrossRef] [PubMed]
- Beljanski, V.; Knaak, C.; Smith, C.D. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J. Pharmacol. Exp. Ther. 2010, 333, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Taeb, S.; Jahangiri, S.; Emmenegger, U.; Tran, E.; Bruce, J.; Mesci, A.; Korpela, E.; Vesprini, D.; Wong, C.S.; et al. miRNA-95 mediates radioresistance in tumors by targeting the sphingolipid phosphatase SGPP1. Cancer Res. 2013, 73, 6972–6986. [Google Scholar] [CrossRef] [Green Version]
- Billich, A.; Bornancin, F.; Mechtcheriakova, D.; Natt, F.; Huesken, D.; Baumruker, T. Basal and induced sphingosine kinase 1 activity in A549 carcinoma cells: Function in cell survival and IL-1beta and TNF-alpha induced production of inflammatory mediators. Cell. Signal. 2005, 17, 1203–1217. [Google Scholar] [CrossRef]
- Leroux, M.E.; Auzenne, E.; Evans, R.; Hail, N., Jr.; Spohn, W.; Ghosh, S.C.; Farquhar, D.; McDonnell, T.; Klostergaard, J. Sphingolipids and the sphingosine kinase inhibitor, SKI II, induce BCL-2-independent apoptosis in human prostatic adenocarcinoma cells. Prostate 2007, 67, 1699–1717. [Google Scholar] [CrossRef]
- Liu, H.; Kong, Y.; Liu, Z.; Guo, X.; Yang, B.; Yin, T.; He, H.; Gou, J.; Zhang, Y.; Tang, X. Sphingomyelin-based PEGylation Cu (DDC)(2) liposomes prepared via the dual function of Cu(2+) for cancer therapy: Facilitating DDC loading and exerting synergistic antitumor effects. Int. J. Pharm. 2022, 621, 121788. [Google Scholar] [CrossRef]
- Bidan, N.; Lores, S.; Vanhecke, A.; Nicolas, V.; Domenichini, S.; Lopez, R.; de la Fuente, M.; Mura, S. Before in vivo studies: In vitro screening of sphingomyelin nanosystems using a relevant 3D multicellular pancreatic tumor spheroid model. Int. J. Pharm. 2022, 617, 121577. [Google Scholar] [CrossRef]
- Massiot, J.; Rosilio, V.; Ibrahim, N.; Yamamoto, A.; Nicolas, V.; Konovalov, O.; Tanaka, M.; Makky, A. Newly synthesized lipid-porphyrin conjugates: Evaluation of their self-assembling properties, their miscibility with phospholipids and their photodynamic activity in vitro. Chemistry 2018, 24, 19179–19194. [Google Scholar] [CrossRef]
- Lim, E.B.; Haam, S.; Lee, S.W. Sphingomyelin-based liposomes with different cholesterol contents and polydopamine coating as a controlled delivery system. Colloid Surface A 2021, 618, 126447. [Google Scholar] [CrossRef]
- Zembruski, N.C.; Nguyen, C.D.; Theile, D.; Ali, R.M.; Herzog, M.; Hofhaus, G.; Heintz, U.; Burhenne, J.; Haefeli, W.E.; Weiss, J. Liposomal sphingomyelin influences the cellular lipid profile of human lymphoblastic leukemia cells without effect on P-glycoprotein activity. Mol. Pharm. 2013, 10, 1020–1034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kitatani, K.; Toyoshima, M.; Ishibashi, M.; Usui, T.; Minato, J.; Egiz, M.; Shigeta, S.; Fox, T.; Deering, T.; et al. Ceramide nanoliposomes as a MLKL-dependent, necroptosis-inducing, chemotherapeutic reagent in ovarian cancer. Mol. Cancer Ther. 2018, 17, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Molecule | Pathway | Function or Activity | Cancer | Refs. |
|---|---|---|---|---|
| Sphingosine kinase | Akt/NF-κB | Cancer progression and chemoresistance | Colon | [74] |
| S1P/S1PR1 | Inflammation and angiogenesis | Breast | [75] | |
| PI3k/Akt/FOXO3a | Apoptosis resistance | Breast | [76] | |
| S1P/Stat3/AKT | Proliferation | Colon | [77] | |
| E-cad | Tumorigenesis and metastasis | Breast | [78] | |
| S1P/ERK/CD44 | Chemoresistance | Colon | [79] | |
| S1P/AKT/GSK-3β | HIF-1α stabilization | Glioblastoma | [80] | |
| Sphingomyelinase | ↑ Sensitivity | Glioblastoma | [81] | |
| ↑ Resistance | Melanoma | [82] | ||
| Induce apoptosis | Colon | [83] | ||
| Induce apoptosis and resistance | Ovarian | [84] | ||
| Acid ceramidase | ↑ Proliferative ↓ Sensitivity | Melanoma | [85,86] | |
| ↓ Sensitivity | Prostate | [87] | ||
| ↑ Radioresistant | Glioblastoma | [87] | ||
| Sphingosine kinase 2 | Mcl-1 | ↑ Cell survival | Leukemia | [88] |
| Sphingosine-1-phosphate lyase | p53 and p38 | ↑ Apoptosis | Colon | [89] |
| Sphingomyelin synthase | TGF- b1 | ↑ Migratory, invasion | Breast | [90] |
| Overexpression of SMS1 | ↓ Cell death | Lymphatic | [91] | |
| Sphingomyelinase | CD95 | ↑ Apoptosis | Lymphatic | [92] |
| p53 | ↑ Apoptosis | Lung | [93] | |
| Sphingosine | Cdk4 | ↓ Cell proliferation | Intestinal adenoma | [94] |
| Ceramide | CerS6/C16-ceramide activated | ↑ Apoptosis | Lung | [95] |
| High cytotoxicity in p53 | ↑ Apoptosis | Breast | [96] |
| Materials | Size (d. nm) | Therapeutics | Cancer Cell Type | Refs. |
|---|---|---|---|---|
| SM-CSS-CPT | 93.1 ± 7.63 | Intravenous injection | CT26, b16, mc38 | [129] |
| UroGm-SNs | 131 ± 12 | SW620 | [130] | |
| DOTAP (DSN) | 142 ± 2 | HCT-116 | [133] | |
| PEGylated Cu (DDC)2 liposomes | 121.5 ± 0.57 | Intravenous injection | 4T1 | [159] |
| SNs–ST | 131 ± 8 | SW480 | [134] | |
| SNs_PEG | 77 ± 3 | PANC-1, | [160] | |
| Lipid–porphyrin conjugates | 180 ± 10 | Kyse-30 | [161] | |
| SMLs@PDA | 229.5 ± 26.3 | [162] | ||
| C6-NBD-SM Liposomes | 71 ± 3 | CCRF-CEM | [163] | |
| CPNPs | 20 | MCF-7 | [142] | |
| C6 ceramide | ~80 | B16, WM-115 | [145] | |
| TRI-Gel | Subcutaneous injection | Lewis lung carcinoma | [147] | |
| Celastrol | Subcutaneous injection | CT26, HCT-8, HCT-116, DLD-1 | [148] | |
| LipC6 | 90 | Intra-splenic injections | liver tumors | [144] |
| Thirty molar percent C6-ceramide in a twelve molar percent pegylated | 80 | Intraperitoneal injection | SKOV3, TOV112D, A2780, A2780CP, PE01, PE04 | [164] |
| S1P/JS-K/Lipo | 189 | Intravenous injection | U87MG | [150] |
| PP2A | A549 | [151] | ||
| 3-[4-(5-aryl-1, 2, 4-oxadiazol-3-yl)-1H-indol-1-yl]propanoic acid series | Intravenous injection | peripheral lymphocyte | [153] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Chen, H.-J.; Wen, H.-Y.; Wang, Z.-G.; Liu, S.-L. Engineered Lipidic Nanomaterials Inspired by Sphingomyelin Metabolism for Cancer Therapy. Molecules 2023, 28, 5366. https://doi.org/10.3390/molecules28145366
Zhu H, Chen H-J, Wen H-Y, Wang Z-G, Liu S-L. Engineered Lipidic Nanomaterials Inspired by Sphingomyelin Metabolism for Cancer Therapy. Molecules. 2023; 28(14):5366. https://doi.org/10.3390/molecules28145366
Chicago/Turabian StyleZhu, Han, Hua-Jie Chen, Hai-Yan Wen, Zhi-Gang Wang, and Shu-Lin Liu. 2023. "Engineered Lipidic Nanomaterials Inspired by Sphingomyelin Metabolism for Cancer Therapy" Molecules 28, no. 14: 5366. https://doi.org/10.3390/molecules28145366
APA StyleZhu, H., Chen, H. -J., Wen, H. -Y., Wang, Z. -G., & Liu, S. -L. (2023). Engineered Lipidic Nanomaterials Inspired by Sphingomyelin Metabolism for Cancer Therapy. Molecules, 28(14), 5366. https://doi.org/10.3390/molecules28145366