3. Experimental Section
3.1. General Methods
All NMR experiments were performed with a Varian Mercury 300 MHz, Varian VNMR-S 400 MHz, Bruker Ascend™ 400 MHz NANOBA and Bruker Avance 600 MHz spectrometers. Full assignment of all NMR signals and determination of the stereochemistry were possible with the use of various NMR techniques, including 1H, 1H{/19F}, 1H{/31P}, 1H-1H COSY, 1H-1H NOE, 1H-19F HOE, 1H-13C HSQC, 1H-13C HMBC, 13C, 19F, and 31P{/1H} experiments. The NMR shifts were determined in relation to the residual solvent proton signal (for CDCl3: 7.26 ppm–1H NMR, 77.16 ppm–13C NMR) and are expressed in parts per million (ppm) in CDCl3. Coupling constants (J) were reported in hertz (Hz). The following abbreviations were used to express the multiplicities: s–singlet, d–doublet, t–triplet, q–quartet, quint–quintet, dd–doublet of doublets, dt–doublet of triplets, dq–doublet of quartets, td–triplet of doublets, ddd–doublet of doublet of doublets, m–multiplet, br d–broad doublet, br s–broad singlet. 19F NMR spectra were measured with trichlorofluoromethane (CFCl3) as the internal standard, while for 31P NMR spectroscopy, 85% H3PO4 was used as the external standard. High-resolution mass spectra (HRMS) for the final compounds were performed on an Agilent 6210 ESI using electrospray ionization. Electron ionization mass spectroscopy (EI-MS; low-resolution, direct injection) was performed on a Bruker 320MS/420GC spectrometer.
The obtained compounds were purified by column chromatography using silica gel Merck Kieselgel 60 (230–400 mesh) as the stationary phase, and ethyl acetate/hexane or ethyl acetate/petroleum ether as developing systems. Thin Layer Chromatography (TLC) was performed on commercially available Merck Kieselgel 60-F254 with ethyl acetate/hexane as the mobile phase. Visualization of the TLC plates was done using UV light and/or permanganate solution.
Solvents were dried by commonly used methods: toluene was freshly distilled over sodium hydride (NaH2) and acetonitrile was distilled over calcium hydride (CaH2) prior to use. Anhydrous MeOH and DMF were stored over 4Å molecular sieves. All of the reagents were purchased from Fluorochem®, Acros®, Alfa Aesar® or Sigma-Aldrich®, and used as received.
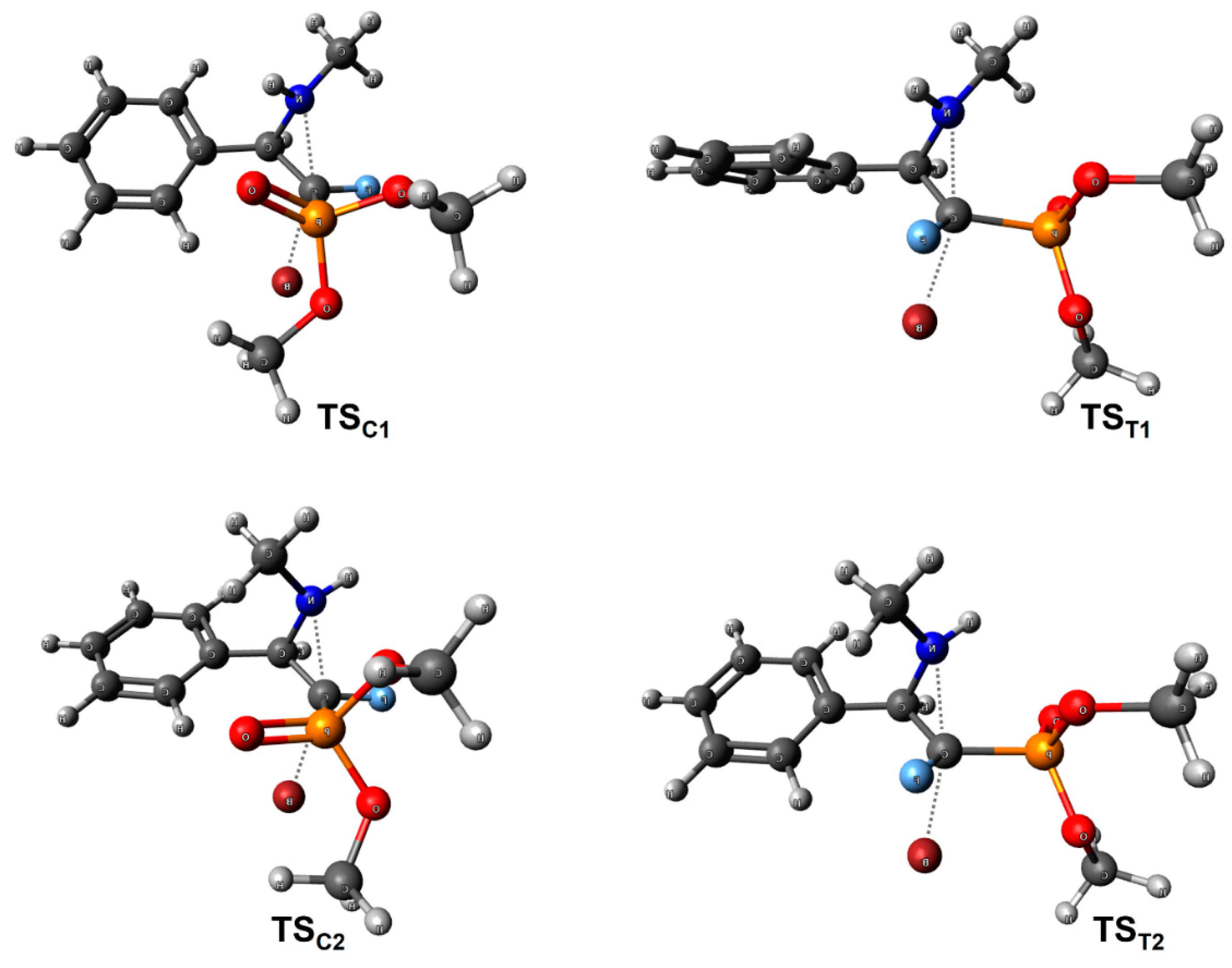
3.2. Theoretical Calculations
Gaussian 16 [
85] was used to fully optimize and calculate the frequencies for all the structures at the ωB97x-D/def2-TZVPD level of theory [
86,
87,
88]. The vibrational frequencies were calculated at the same level of theory, and then their positivity was applied to confirm that each of the calculated structures corresponds to a minimum on the potential energy surface. The polarizable continuum model (PCM) [
89] was used to simulate solvents: DMF (reaction pathways modelling) and chloroform (conformational analysis of NMR solution). Transition structures were located using the Berny algorithm with the NoEigenTest request. Various combinations of conformations for both invertomers (nitrogen atom) were examined to determine minimum energy pathways for all cyclization reactions.
3.3. General Procedure for Synthesis of β-Enaminophosphonates (2–5a,b)/β-Iminophosphonates (6–9a,b)
Compounds
2–
5 and
6–
9 were synthesized according to a previously reported methodology from diethyl 2-oxo-2-phenylethylphosphonate
1 (151 µL, 0.5 mmol) and primary amine (0.5 mmol) [
54]. The reaction mixture was refluxed using a Dean–Stark apparatus. The obtained NMR data based on
1H and
31P{/
1H} NMR for
2–
4a,b and
6–
8a,b are identical with those reported in the literature [
54].
3.4. The Synthesis of β-Enaminophosphonates/β-Iminophosphonates (5a/9a,b)
Compounds 5a/9a,b are prepared as described in the general procedure.
The ratio of enamine 5a and imines 9a,b was determined with the use of 1H and 31P{/1H} NMR from the crude mixture, and was 1:0.53; the ratio of enamine (Z) 5a equaled 1, and imines (E/Z) 9a,b equaled 1:0.1, respectively. For tautomeric mixture 5a/9a,b after column chromatography (silica gel, AcOEt/hexane: 5% → 60%), only the NMR spectra of the main products was given (Z-5a/E-9a 1:0.53). Yellow oil, 157 mg, yield 87%.
(Z)-Diethyl (2-((4-methoxyphenyl)amino)-2-phenylvinyl)phosphonate (Z-5a).
1H NMR (401 MHz): δ = 9.12 (br s, 1H, NH), 7.34–7.30 (m, 2H, Har), 7.29–7.23 (m, 3H, Har), 6.88–6.87 (m, 2H, Har), 6.57–6.56 (m, 2H, Har), 4.23 (d, J = 12.4 Hz, 1H, CHP), 4.13–4.05 (m, 4H, 2x OCH2CH3), 3.64 (s, 3H, Ph(4-OCH3)), 1.32 (td, J = 7.1, 0.5 Hz, 6H, OCH2CH3). 13C NMR (101 MHz): δ = 161.63 (d, J = 6.0 Hz, C=CHP), 155.36 (s, Car(OCH3)), 137.48 (d, J = 19.7 Hz, Cipso), 134.68 (s, Cipso), 130.63, 129.30, 128.30, 123.69 (4x s, CHar), 113.92 (s, CHarCar(OCH3)), 82.17 (d, J = 188.1 CHP), 61.45 (d, J = 6.0 Hz, OCH2CH3), 55.39 (s, Ph(4-OCH3)), 16.20 (d, J = 6.7 Hz, OCH2CH3). 31P{/1H} NMR (122 MHz): δ = 24.73 (s, 1P). MS (EI) m/z = 361.2 [M]+.
(E)-Diethyl (2-((4-methoxyphenyl)imino)-2-phenylethyl)phosphonate (E-9a).
1H NMR (401 MHz): δ = 7.43–7.39 (m, 3H, Har), 7.25–7.19 (m, 2H, Har), 6.88–6.87 (m, 2H, Har), 6.57–6.56 (m, 2H, Har), 4.05–4.00 (m, 2H, OCH2CH3), 3.92–3.84 (m, 2H, OCH2CH3), 3.78 (s, 3H, Ph(4-OCH3)), 3.39 (d, J = 23.3 Hz, 2H, CH2P), 1.10 (td, J = 7.1, 0.5 Hz, 6H, OCH2CH3). 13C NMR (101 MHz): δ = 160.07 (d, J = 7.8 Hz, C=N), 156.39 (s, Car(OCH3)), 143.97 (d, J = 2.3 Hz, Cipso), 138.76 (s, Cipso), 128.32, 128.29, 128.06, 120.83 (4x s, CHar), 114.40 (s, CHarCar(OCH3)), 62.22 (d, J = 6.6 Hz, OCH2CH3), 55.55 (s, Ph(4-OCH3)), 29.51 (d, J = 134.3 Hz, CH2P), 16.46 (d, J = 6.3 Hz, OCH2CH3). 31P{/1H} NMR (122 MHz): δ = 22.12 (s, 1P).
(Z)-Diethyl (2-((4-methoxyphenyl)imino)-2-phenylethyl)phosphonate (Z-9b).
Diagnostic signals: 31P{/1H} NMR (122 MHz): δ = 24.00 (s).
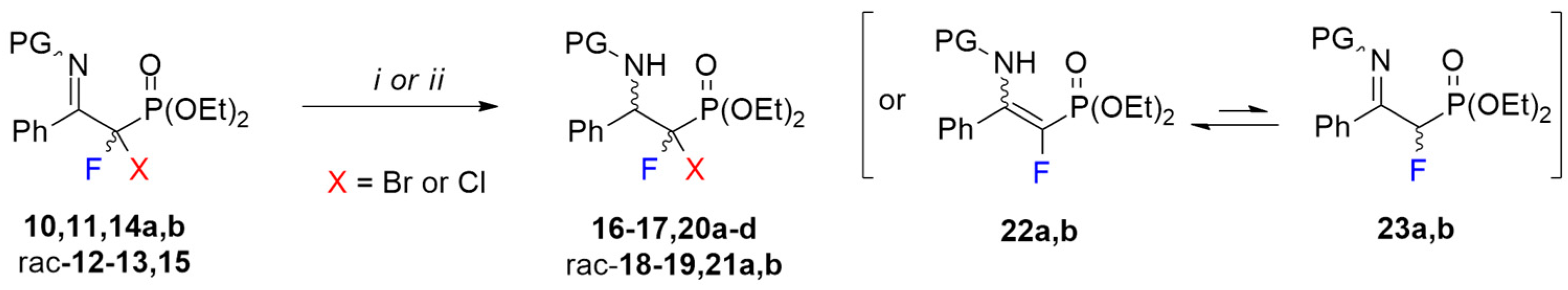
3.5. General Procedure for Synthesis of α,α-Bromofluorinated ß-Iminophosphonates (10–11a,b, rac-12–13)
The solution of Selectfluor (319 mg, 0.9 mmol) in dry, freshly distilled acetonitrile (10 mL) was gently heated to 50 °C and vigorously stirred until the compound was completely dissolved. After, the solution was cooled to ambient temperature and added together with NBS (89 mg, 0.5 mmol) to the mixture of the appropriate β-enaminophosphonate/β-iminophosphonate 2–5/6–9 (0.5 mmol). After 15 min of stirring at room temperature, the solvent was removed under reduced pressure. Next, the residue was dissolved in CHCl3 (2 mL), water (10 mL) was added, and it was extracted (3 × 10 mL CHCl3). The organic layers were combined, dried over Na2SO4, and evaporated to give products as a pale-yellow oil. The crude products were purified by column chromatography (AcOEt/petroleum ether: 5% → 50%).
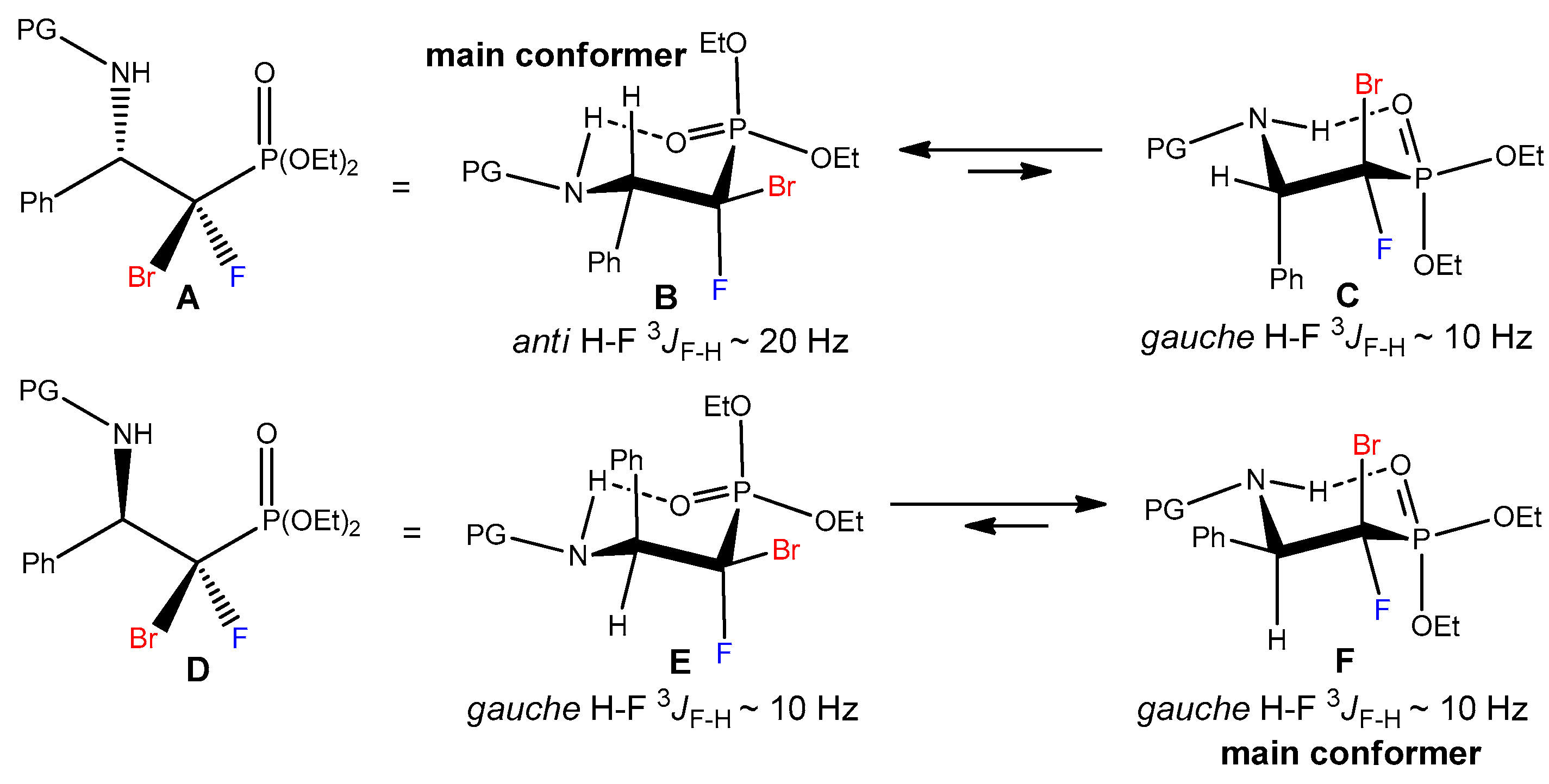
(E)-Diethyl ((1R)-1-bromo-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)imino)ethyl) phosphonate (10a) and (E)-diethyl ((1S)-1-bromo-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)imino)ethyl) phosphonate (10b).
Pale-yellow oil, 210 mg, yield 92%. Isolated as a mixture of diastereomers 10a,b (dr 1:0.54) (153 mg) and single diastereomer 10b (57mg).
10a: 1H NMR (400 MHz): δ = 7.44–7.40 (m, 1H, Har), 7.39–7.36 (m, 2H, Har), 7.33–7.30 (m, 2H, Har), 7.29–7.26 (m, 3H, Har), 7.25–7.20 (m, 2H, Har), 4.47–4.35 (m, 3H, OCH2CH3, CHCH3), 4.34–4.23 (m, 2H, OCH2CH3), 1.38 (d, J = 6.6 Hz, 3H, CHCH3), 1.35 (td, J = 7.0, 0.8 Hz, 3H, OCH2CH3), 1.32 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 164.44 (dd, J = 27.0, 6.0 Hz, C=N), 144.57 (s, Cipso), 131.52 (d, J = 5.0 Hz, Cipso), 128.60, 128.58, 127.87, 126.97, 126.62, 126.47 (6x s, CHar), 100.05 (dd, J = 269.8, 187.5 Hz, CBrF), 65.52 (d, J = 6.2 Hz, OCH2CH3), 65.20 (d, J = 6.7 Hz, OCH2CH3), 61.19 (s, CHCH3), 24.54 (s, CHCH3), 16.64 (d, J = 6.1 Hz, OCH2CH3), 16.61 (d, J = 6.1 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −129.08 (d, J = 82.2 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 7.37 (d, J = 82.1 Hz, 1P). IR (neat): 1648, 1261, 1012, 972, 649 [cm−1]. MS (EI) m/z = 457.3 [M+H]+.
10b: 1H NMR (400 MHz): δ = 7.42–7.37 (m, 3H, Har), 7.31–7.28 (m, 2H, Har), 7.30–7.24 (m, 4H, Har), 7.20–7.16 (m, 1H, Har), 4.42 (q, J = 6.5 Hz, 1H, CHCH3), 4.39–4.26 (m, 2H, OCH2CH3), 4.25–4.05 (m, 2H, OCH2CH3), 1.40 (d, J = 6.5 Hz, 3H, CHCH3), 1.30 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3), 1.22 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 164.64 (dd, J = 28.3, 5.9 Hz, C=N), 144.15 (s, Cipso), 131.70 (d, J = 5.1 Hz, Cipso), 128.50, 128.43, 128.41, 128.33, 126.99, 126.72 (6x s, CHar), 99.78 (dd, J = 269.3, 187.2 Hz, CBrF), 65.51 (d, J = 6.3 Hz, OCH2CH3), 65.05 (d, J = 6.6 Hz, OCH2CH3), 61.40 (s, CHCH3), 23.97 (s, CHCH3), 16.56 (d, J = 6.1 Hz, OCH2CH3), 16.44 (d, J = 6.1 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −128.65 (d, J = 82.3 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 7.93 (d, J = 82.3 Hz, 1P).
(E)-Diethyl ((1S)-1-bromo-1-fluoro-2-phenyl-2-(((R)-1-phenylethyl)imino)ethyl) phosphonate (11a) and (E)-Diethyl ((1R)-1-bromo-1-fluoro-2-phenyl-2-(((R)-1-phenylethyl)imino)ethyl)phosphonate (11b).
Pale-yellow oil, 217 mg, yield 95%. Isolated as a mixture of diastereomers 11a,b (dr 1:0.93), which could not be separated by the chromatography techniques employed in this study.
11a: 1H NMR (400 MHz): δ = 7.43–7.37 (m, 5H, Har), 7.26–7.21 (m, 5H, Har), 4.42–4.35 (m, 3H, OCH2CH3, CHCH3), 4.34–4.25 (m, 2H, OCH2CH3), 1.39 (d, J = 6.5 Hz, 3H, CHCH3), 1.36 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.33 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 164.45 (dd, J = 27.1, 6.1 Hz, C=N), 144.53 (s, Cipso), 131.52 (d, J = 5.1 Hz, Cipso), 129.52, 128.60, 128.45, 126.96, 126.69, 126.43 (6x s, CHar), 100.99 (dd, J = 269.7, 187.4 Hz, CBrF), 65.45 (d, J = 6.1 Hz, OCH2CH3), 65.14 (d, J = 6.7 Hz, OCH2CH3), 61.17 (s, CHCH3), 24.49 (s, CHCH3), 16.56 (d, J = 6.0 Hz, OCH2CH3), 16.45 (d, J = 6.1 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −128.52 (d, J = 82.1 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 7.38 (d, J = 82.1 Hz, 1P). IR (neat): 1644, 1262, 1017, 970, 647 [cm−1]. MS (EI) m/z = 457.3 [M+H]+.
11b: 1H NMR (400 MHz): δ = 7.32–7.27 (m, 5H, Har), 7.23–7.16 (m, 5H, Har), 4.43 (q, J = 6.5 Hz, 1H, CHCH3), 4.27–4.17 (m, 2H, OCH2CH3), 4.16–4.08 (m, 2H, OCH2CH3), 1.41 (d, J = 6.5 Hz, 3H, CHCH3), 1.31 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.23 (td, J = 7.1 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 164.63 (dd, J = 28.4, 5.9 Hz, C=N), 144.12 (s, Cipso), 131.69 (d, J = 5.1 Hz, Cipso), 128.55, 128.41, 128.36, 128.29, 127.01, 126.93 (6x s, CHar), 99.14 (dd, J = 269.1, 187.0 Hz, CBrF), 65.42 (d, J = 6.2 Hz, OCH2CH3), 65.00 (d, J = 6.6 Hz, OCH2CH3), 61.37 (s, CHCH3), 23.92 (s, CHCH3), 16.53 (d, J = 6.1 Hz, OCH2CH3), 16.41 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −128.13 (d, J = 82.3 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 7.42 (d, J = 82.4 Hz, 1P).
rac-(E)-Diethyl ((1R/1S)-1-bromo-1-fluoro-2-((4-methoxybenzyl)imino)-2-phenylethyl)phosphonate (rac-12).
Pale-yellow oil, 222 mg, yield 94%.
1H NMR (400 MHz): δ = 7.45–7.42 (m, 3H, Har), 7.34–7.30 (m, 2H, Har), 7.22–7.15 (m, 2H, Har), 6.85–6.80 (m, 2H, Har), 4.46 (br s, CH2N, 2H), 4.33–4.24 (m, 2H, OCH2CH3), 4.23–4.10 (m, 2H, OCH2CH3), 3.76 (s, 3H, Ph(4-OCH3)), 1.26 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3), 1.25 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 166.67 (dd, J = 27.7, 6.1 Hz, C=N), 158.67 (s, Car(OCH3)), 131.44 (d, J = 5.2 Hz, Cipso), 130.90 (s, Cipso), 129.70, 128.93 (2x s, CHar), 128.60 (d, J = 1.5 Hz, CHar), 128.50 (s, CHar), 113.93 (s, CHarCar(OCH3)), 99.97 (dd, J = 269.7, 187.6 Hz, CBrF), 65.54 (d, J = 6.3 Hz, OCH2CH3), 65.15 (d, J = 6.7 Hz, OCH2CH3), 56.42 (br s, CH2N), 55.39 (s, Ph(4-OCH3)), 16.49 (d, J = 6.1 Hz, OCH2CH3), 16.48 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −129.07 (d, J = 81.2 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 7.98 (d, J = 81.4 Hz, 1P). IR (neat): 1645, 1261, 1247, 1013, 970, 647 [cm−1]. MS (EI) m/z = 392.2 [M-Br]+.
rac-(E)-Diethyl ((1R/1S)-1-bromo-1-fluoro-2-((4-methoxyphenyl)imino)-2-phenylethyl)phosphonate (rac-13).
Brown oil, 200 mg, yield 87%.
1H NMR (400 MHz): δ = 7.31–7.26 (m, 5H, Har), 6.68–6.64 (m, 4H, Har), 4.41–4.30 (m, 4H, 2x OCH2CH3), 3.68 (s, 3H, Ph(4-OCH3)), 1.36 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3), 1.35 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 164.12 (dd, J = 28.4, 5.9 Hz, C=N), 157.44 (s, Car(OCH3)), 140.26 (s, Cipso), 131.94 (d, J = 5.0 Hz Cipso), 129.56, 129.43, 123.19, 118.90 (4x s, CHar), 113.90 (s, CHarCar(OCH3)), 100.17 (dd, J = 270.1, 187.2 Hz, CBrF), 65.58 (d, J = 6.4 Hz, OCH2CH3), 65.29 (d, J = 6.6 Hz, OCH2CH3), 55.35 (s, Ph(4-OCH3)), 16.56 (d, J = 5.9 Hz, OCH2CH3), 16.55 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −128.49 (d, J = 81.7 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 7.30 (d, J = 81.5 Hz, 1P). IR (neat): 1645, 1262, 1246, 1012, 972, 645 [cm−1]. MS (EI) m/z = 459.3 [M+H]+.
3.6. General Procedure for Synthesis of α,α-Chlorofluorinated ß-Iminophosphonates (14a,b, rac-15)
Compounds 14a,b and rac-15 were obtained according to the above-described procedure for bromofluorinated iminophosphonates (10–11a,b, rac-12–13). Selectfluor and N-chlorosuccinimide (NCS) were used as halogenation reagents with appropriate molar equivalents equaled 1.35 and 1.0, respectively.
(E)-Diethyl ((1R)-1-chloro-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)imino)ethyl)phosphonate (14a) and (E)-diethyl ((1S)-1-chloro-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)imino)ethyl)phosphonate (14b)
Pale-yellow oil, 128 mg, yield 62%. Isolated as a mixture of diastereomers 14a,b (dr 0.2:1). The rest of diastereomer 14a (65 mg) was contaminated with difluoro- and dichloroiminophosphonate derivatives.
14a: 1H NMR (400 MHz): δ = 7.45–7.41 (m, 3H, Har), 7.32–7.27 (m, 4H, Har), 7.25–7.21 (m, 3H, Har), 4.49 (q, J = 6.5 Hz, 1H, CHCH3), 4.38–4.32 (m, 2H, OCH2CH3), 4.29–4.25 (m, 2H, OCH2CH3), 1.42 (d, J = 6.5 Hz, 3H, CHCH3), 1.34 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.32 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 163.55 (dd, J = 28.1, 6.5 Hz, C=N), 144.46 (s, Cipso), 131.51 (d, J = 4.7 Hz, Cipso), 128.58, 128.55, 128.43, 128.02, 127.08, 126.58 (6x s, CHar), 105.89 (dd, J = 260.7, 189.9 Hz, CClF), 65.11 (d, J = 6.5 Hz, OCH2CH3), 64.78 (d, J = 6.5 Hz, OCH2CH3), 61.33 (s, CHCH3), 24.51 (s, CHCH3), 16.52 (d, J = 6.1 Hz, OCH2CH3), 16.48 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −125.59 (d, J = 86.2 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 8.02 (d, J = 86.0 Hz, 1P). IR (neat): 1657, 1262, 1011, 969, 666 [cm−1]. MS (EI) m/z = 376.2 [M-Cl]+.
14b: 1H NMR (400 MHz): δ = 7.43–7.37 (m, 3H, Har), 7.31–7.26 (m, 4H, Har), 7.25–7.22 (m, 3H, Har), 4.46 (q, J = 6.5 Hz, 1H, CHCH3), 4.39–4.23 (m, 3H, OCH2CH3, OCHHCH3), 4.22–4.12 (m, 1H, OCHHCH3), 1.44 (d, J = 6.5 Hz, 3H, CHCH3), 1.31 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3), 1.24 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 163.75 (dd, J = 28.6, 6.8 Hz, C=N), 144.16 (s, Cipso), 131.64 (d, J = 4.8 Hz, Cipso), 129.56, 129.53, 128.50, 128.39, 126.99, 126.68 (6x s, CHar), 105.77 (dd, J = 259.9, 193.0 Hz, CClF), 65.35 (d, J = 6.5 Hz, OCH2CH3), 64.96 (d, J = 6.5 Hz, OCH2CH3), 61.45 (br s, CHCH3), 24.16 (s, CHCH3), 16.53 (d, J = 6.0 Hz, OCH2CH3), 16.42 (d, J = 5.9 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −125.39 (d, J = 87.0 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 7.54 (d, J = 87.0 Hz, 1P).
rac- (E)-Diethyl (1R/1S)-1-chloro-1-fluoro-2-((4-methoxybenzyl)imino)-2-phenylethyl)phosphonate (rac-15).
Pale-yellow oil, 195 mg, yield 91%.
1H NMR (400 MHz): δ = 7.43–7.39 (m, 3H, Har), 7.33–7.27 (m, 2H, Har), 7.19–7.14 (m, 2H, Har), 6.84–6.78 (m, 2H, Har), 4.46 (br s, 2H, CH2N), 4.32–4.23 (m, 2H, OCH2CH3), 4.21–4.15 (m, 2H, OCH2CH3), 3.75 (s, 3H, Ph(4-OCH3)), 1.26 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3), 1.24 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 165.78 (dd, J = 27.9, 6.7 Hz, C=N), 158.68 (s, Car(OCH3)), 131.35 (d, J = 4.9 Hz Cipso), 129.18, 129.15, 128.93, 128.51 (4x s, CHar), 128.43 (d, J = 1.3 Hz, CHar), 128.25 (s, CHar), 113.91 (s, CHarCar(OCH3)), 105.94 (dd, J = 260.5, 193.5 Hz, CClF), 65.55 (d, J = 6.9 Hz, OCH2CH3), 65.10 (d, J = 6.6 Hz, OCH2CH3), 56.46 (br s, CH2N), 55.36 (s, Ph(4-OCH3)), 16.48 (d, J = 6.0 Hz, OCH2CH3), 16.45 (d, J = 5.8 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −125.57 (d, J = 85.6 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 8.09 (d, J = 86.0 Hz, 1P). IR (neat): 1648, 1260, 1014, 970, 668 [cm−1]. MS (EI) m/z = 428.8 [M+H]+.
3.7. General Procedure for Synthesis of α,α-Halofluorinated ß-Aminophosphonates (16–17,20a–d, rac-18–19,21a,b)
To the stirred solution of β-iminophosphonate (10–11,14a,b, rac-12–13,15) (0.5 mmol) in MeOH (3 mL), NaBH3CN (188 mg, 3 mmol) and glacial CH3COOH (171 µL, 180 mg, 3 mmol) at room temperature were added. Stirring was continued for 40 min and then the solvent was evaporated. Next, the residue was dissolved in CHCl3 (2 mL), water (10 mL) was added, and the inorganic layer was extracted (3 × 10 mL CHCl3). The organic layers were combined, dried over Na2SO4, and evaporated to give the product as a pale-yellow oil. The crude products were purified by column chromatography (AcOEt/hexane: 5% → 60%).
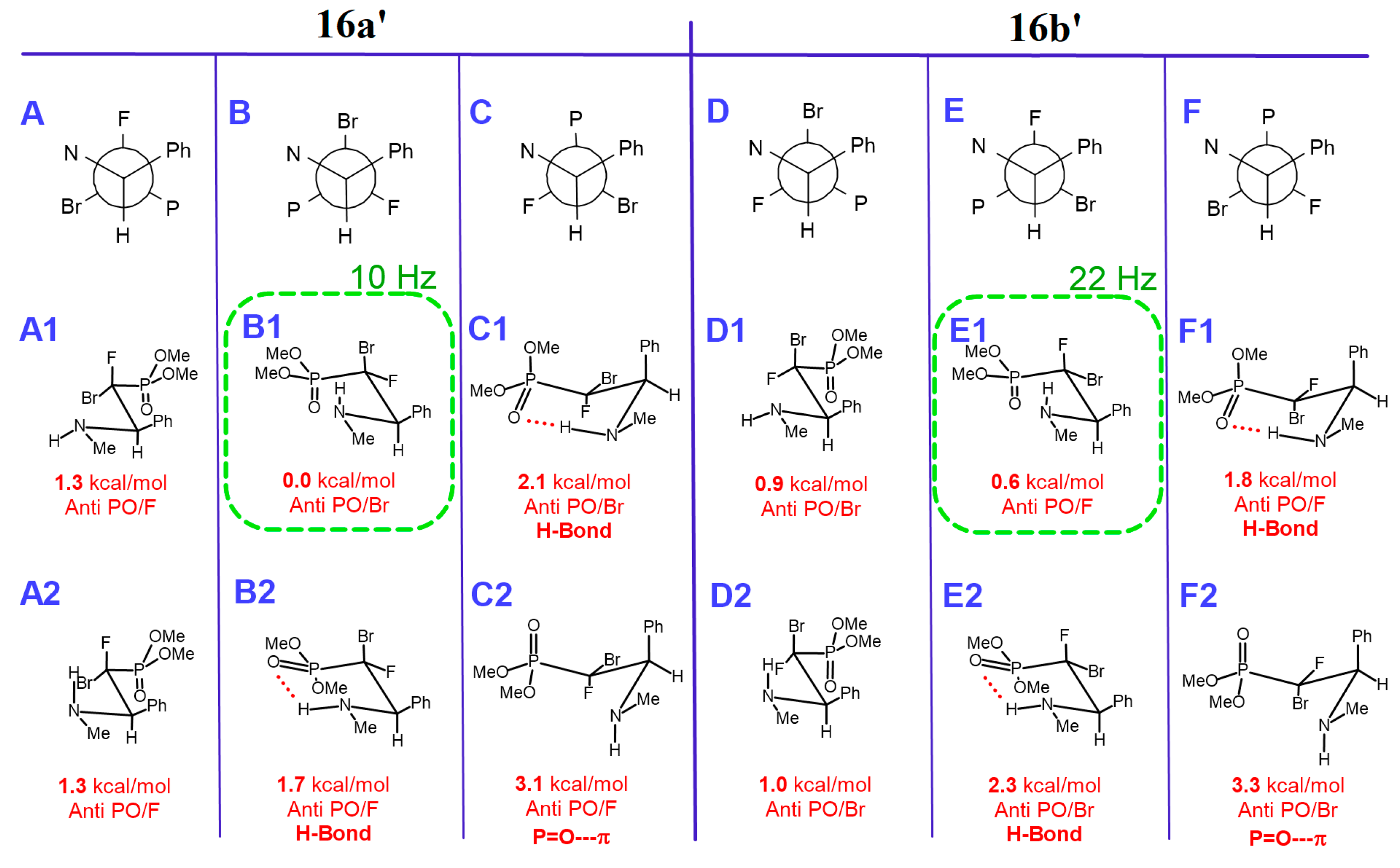
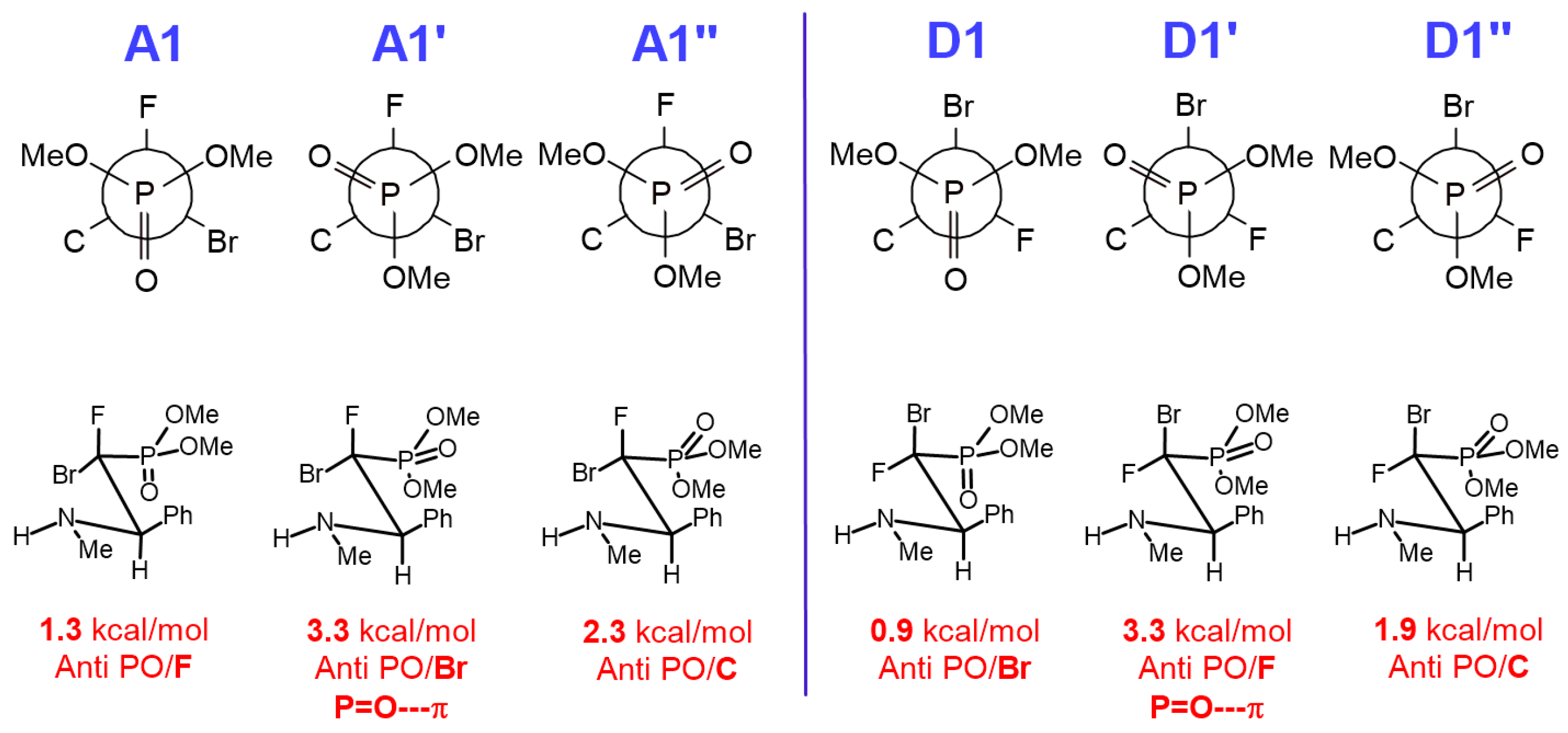
Diethyl ((1S/R, 2S/R)-1-bromo-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (16a-d).
Isolated as a mixture of two major diastereomers 16a,b (dr 1:0.96). Diagnostic signals for traces of diastereomers 16c,d were determined from the crude reaction mixture. Pale-yellow oil, 222 mg, yield 97%.
Diethyl ((1R, 2R)-1-bromo-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (16a).
1H NMR (400 MHz): δ = 7.36–7.33 (m, 5H, Har), 7.28–7.25 (m, 2H, Har), 7.21–7.19 (m, 3H, Har), 4.58 (dd, J = 10.2, 4.7 Hz, 1H, CH(Ph)CF), 4.40–4.29 (m, 2H, OCH2CH3), 4.27–4.23 (m, 2H, OCH2CH3), 3.75 (q, J = 6.4 Hz, 1H, CHCH3), 1.34 (d, J = 6.4 Hz, 3H, CHCH3), 1.28 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.27 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 145.46 (s, Cipso), 135.70 (d, J = 8.6 Hz, Cipso), 128.54, 128.50, 128.41, 128.06, 127.24, 126.91 (6x s, CHar), 107.44 (dd, J = 270.1, 184.3 Hz, CBrF), 65.58 (d, J = 7.3 Hz, OCH2CH3), 65.16 (d, J = 7.3 Hz, OCH2CH3), 64.12 (dd, J = 22.0, 7.5 Hz, CH(Ph)CF), 54.71 (s, CHCH3), 21.89 (s, CHCH3), 16.60 (d, J = 6.1 Hz, OCH2CH3), 16.44 (d, J = 6.2 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −125.06 (dd, J = 82.3, 10.2 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 9.77 (d, J = 82.0 Hz, 1P). IR (neat): 1264, 1024, 977, 648 [cm−1]. MS (EI) m/z = 459.4 [M+H]+.
Diethyl ((1S, 2R)-1-bromo-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (16b).
1H NMR (400 MHz): δ = 7.33–7.29 (m, 5H, Har), 7.26–7.24 (m, 1H, Har), 7.23–7.21 (m, 2H, Har), 7.18–7.16 (m, 2H, Har), 4.37 (dd, J = 22.0, 3.5 Hz, 1H, CH(Ph)CF), 4.22–4.17 (m, 2H, OCH2CH3), 4.17–4.07 (m, 2H, OCH2CH3), 3.74 (q, J = 6.5 Hz, 1H, CHCH3), 1.35 (d, J = 6.4 Hz, 3H, CHCH3), 1.26 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.25 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 145.67 (s, Cipso), 137.14 (dd, J = 6.0, 2.5 Hz, Cipso), 128.44, 128.40, 128.13, 127.64, 127.15, 126.84 (6x s, CHar), 107.62 (dd, J = 274.2, 183.1 Hz, CBrF), 66.47 (dd, J = 18.2, 7.9 Hz, CH(Ph)CF), 64.88 (d, J = 7.2 Hz, OCH2CH3), 64.74 (d, J = 7.2 Hz, OCH2CH3), 55.67 (s, CHCH3), 22.10 (s, CHCH3), 16.52 (d, J = 6.1 Hz, OCH2CH3), 16.42 (d, J = 6.1 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −135.38 (dd, J = 84.6, 22.1 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 9.25 (d, J = 84.7 Hz, 1P).
Diethyl ((1S, 2S)-1-bromo-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (16c).
Diagnostic signals: 19F NMR (283 MHz): δ = −128.26 (dd, J = 78.0, 10.2 Hz). 31P{/1H} NMR (122 MHz): δ = 9.06 (d, J = 77.9 Hz).
Diethyl ((1R, 2S)-1-bromo-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (16d).
Diagnostic signals: 19F NMR (283 MHz): δ = −135.49 (dd, J = 88.4, 20.9 Hz). 31P{/1H} NMR (122 MHz): signal masked by other diastereomers signals.
Diethyl ((1S/1R, 2S/2R)-1-bromo-1-fluoro-2-phenyl-2-(((R)-1-phenylethyl)amino)ethyl)phosphonate (17a-d).
Isolated as a mixture of two major diastereomers 17a,b (dr 1:0.8). Diagnostic signals for traces of diastereomers 17c,d were determined from the crude reaction mixture. Pale-yellow oil, 217 mg, yield 95%.
Diethyl ((1S, 2S)-1-bromo-1-fluoro-2-phenyl-2-(((R)-1-phenylethyl)amino)ethyl)phosphonate (17a).
1H NMR (400 MHz): δ = 7.35–7.33 (m, 5H, Har), 7.28–7.26 (m, 2H, Har), 7.22–7.19 (m, 3H, Har), 4.58 (dd, J = 10.2, 4.7 Hz, 1H, CH(Ph)CF), 4.36–4.32 (m, 2H, OCH2CH3), 4.26–4.23 (m, 2H, OCH2CH3), 3.75 (q, J = 6.4 Hz, 1H, CHCH3), 1.35 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.32 (d, J = 6.4 Hz, 3H, CHCH3), 1.31 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 145.41 (s, Cipso), 135.63 (d, J = 8.6 Hz, Cipso), 128.52, 128.49, 128.42, 128.04, 127.22, 126.81 (6x s, CHar), 107.39 (dd, J = 270.1, 184.4 Hz, CBrF), 65.56 (d, J = 7.0 Hz, OCH2CH3), 65.15 (d, J = 7.3 Hz, OCH2CH3), 64.05 (dd, J = 21.9, 7.5 Hz, CH(Ph)CF), 54.65 (s, CHCH3), 21.85 (s, CHCH3), 16.59 (d, J = 6.0 Hz, OCH2CH3), 16.43 (d, J = 6.1 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −125.04 (dd, J = 82.1, 10.1 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 9.25 (d, J = 82.0 Hz, 1P). IR (neat): 1262, 1021, 978, 646 [cm−1]. MS (EI) m/z = 459.4 [M-H]+.
Diethyl ((1R, 2S)-1-bromo-1-fluoro-2-phenyl-2-(((R)-1-phenylethyl)amino)ethyl)phosphonate (17b).
1H NMR (400 MHz): δ = 7.32–7.29 (m, 5H, Har), 7.26–7.24 (m, 2H, Har), 7.19–7.16 (m, 3H, Har), 4.42–4.37 (m, 2H, OCH2CH3), 4.34 (dd, J = 22.1, 3.2 Hz, 1H, CH(Ph)CF), 4.23–4.15 (m, 2H, OCH2CH3), 3.74 (q, J = 6.5 Hz, 1H, CHCH3), 1.33 (d, J = 6.4 Hz, 3H, CHCH3), 1.27 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.26 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 145.62 (s, Cipso), 137.08 (dd, J = 6.2, 2.7 Hz, Cipso), 128.45, 128.40, 128.31, 128.11, 127.13, 126.88 (6x s, CHar), 107.57 (dd, J = 274.0, 182.8 Hz, CBrF), 66.42 (dd, J = 18.2, 8.0 Hz, CH(Ph)CF), 64.85 (d, J = 7.2 Hz, OCH2CH3), 64.72 (d, J = 7.1 Hz, OCH2CH3), 55.63 (s, CHCH3), 22.06 (s, CHCH3), 16.51 (d, J = 6.1 Hz, OCH2CH3), 16.42 (d, J = 6.1 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −135.36 (dd, J = 84.6, 22.0 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 8.73 (d, J = 84.6 Hz, 1P).
Diethyl((1R, 2R)-1-bromo-1-fluoro-2-phenyl-2-(((R)-1-phenylethyl)amino)ethyl)phosphonate (17c).
Diagnostic signals 19F NMR (283 MHz): δ = −128.25 (dd, J = 77.9, 10.2 Hz). 31P{/1H} NMR (122 MHz): δ = 8.56 (d, J = 78.0 Hz).
Diethyl((1R, 2S)-1-bromo-1-fluoro-2-phenyl-2-(((R)-1-phenylethyl)amino)ethyl)phosphonate (17d).
19F NMR (283 MHz): signal masked by other diastereomers signals. Diagnostic signals 31P{/1H} NMR (122 MHz): δ = 8.40 (d, J = 86.9 Hz).
rac-Diethyl ((1R, 2R)-1-bromo-1-fluoro-2-((4-methoxybenzyl)amino)-2-phenylethyl)phosphonate (rac-18a) and rac-diethyl ((1R, 2S)-1-bromo-1-fluoro-2-((4-methoxybenzyl)amino)-2-phenylethyl)phosphonate (rac-18b).
Isolated as a mixture of two diastereomers rac-18a,b (dr 1:0.75), which could not be separated by the chromatography techniques employed in this study. Pale-yellow oil, 230 mg, yield 97%.
rac-18a: 1H NMR (400 MHz): δ = 7.46–7.44 (m, 3H, Har), 7.38–7.36 (m, 2H, Har), 7.14–7.10 (m, 2H, Har), 6.83–6.81 (m, 2H, Har), 4.37–4.26 (m, 3H, OCH2CH3, CH(Ph)CFP), 4.20–4.15 (m, 2H, OCH2CH3), 3.76 (s, 3H, Ph(4-OCH3)), 3.72 (br d, J = 12.9 Hz, 1H, CHHN), 3.48 (br d, J = 13.1 Hz, 1H, CHHN), 1.30 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3), 1.22 (td, J = 7.0, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 158.84 (s, Car(OCH3)), 135.13 (d, J = 8.5 Hz, Cipso), 131.52 (s, Cipso), 129.85 (d, J = 1.5 Hz, CHar), 129.71, 128.65, 128.12 (3x s, CHar), 113.78 (s, CHarCar(OCH3)), 106.46 (dd, J = 270.2, 183.8 Hz, CBrF), 65.79 (d, J = 7.0 Hz, OCH2CH3), 65.24 (dd, J = 21.5, 7.9 Hz, CH(Ph)CF), 65.03 (d, J = 7.1 Hz, OCH2CH3), 55.32 (s, Ph(4-OCH3)), 50.02 (s, CH2N), 16.48 (d, J = 5.9 Hz, OCH2CH3), 16.33 (d, J = 5.9 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −126.81 (dd, J = 79.5, 9.4 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 9.52 (d, J = 79.5 Hz, 1P). IR (neat): 1261, 1245, 1020, 975, 647 [cm−1]. MS (EI) m/z = 475.3 [M-H]+.
rac-18b: 1H NMR (400 MHz): δ = 7.44–7.41 (m, 3H, Har), 7.35–7.32 (m, 2H, Har), 7.17–7.14 (m, 2H, Har), 6.80–6.78 (m, 2H, Har), 4.28–4.22 (m, 3H, OCH2CH3, CH(Ph)CFP), 4.13–4.04 (m, 2H, OCH2CH3), 3.76 (s, 3H, Ph(4-OCH3)), 3.65 (br d, J = 13.0 Hz, 1H, CHHN), 3.47 (br d, J = 13.0 Hz, 1H, CHHN), 1.22 (td, J = 7.1, 1.0 Hz, 3H, OCH2CH3), 1.21 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 158.80 (s, Car(OCH3)), 136.37 (dd, J = 5.9, 2.5 Hz, Cipso), 131.54 (s, Cipso), 129.95 (d, J = 1.5 Hz, CHar), 129.59, 128.55, 128.14 (3x s, CHar), 113.77 (s, CHarCar(OCH3)), 106.64 (dd, J = 272.9, 184.4 Hz, CBrF), 67.72 (dd, J = 18.4, 8.2 Hz, CH(Ph)CF), 64.99 (d, J = 7.0 Hz, OCH2CH3), 64.57 (d, J = 7.3 Hz, OCH2CH3), 55.31 (s, Ph(4-OCH3)), 50.76 (s, CH2N), 16.47 (d, J = 5.9 Hz, OCH2CH3), 16.36 (d, J = 5.9 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −135.34 (dd, J = 85.7, 20.6 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 8.93 (d, J = 85.7 Hz, 1P).
rac-Diethyl ((1R, 2R)-1-bromo-1-fluoro-2-((4-methoxyphenyl)amino)-2-phenylethyl)phosphonate (rac-19a) and rac-diethyl ((1R, 2S)-1-bromo-1-fluoro-2-((4-methoxyphenyl)amino)-2-phenylethyl)phosphonate (rac-19b).
Isolated as a mixture of two diastereomers rac-19a,b (dr 1:0.86), which could not be separated by the chromatography techniques employed in this study. Pale yellow oil, 209 mg, yield 91%.
rac-19a: 1H NMR (400 MHz): δ = 7.34–7.26 (m, 5H, Har), 6.66–6.64 (m, 2H, Har), 6.59–6.57 (m, 2H, Har), 4.37–4.26 (m, 3H, OCH2CH3, CH(Ph)CFP), 4.17–4.10 (m, 2H, OCH2CH3), 3.65 (s, 3H, Ph(4-OCH3)), 1.35 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.25 (td, J = 7.1, 0.7 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 152.95 (s, Car(OCH3)), 139.48 (s, Cipso), 135.71 (d, J = 8.8 Hz, Cipso), 129.17 (d, J = 2.1 Hz, CHar), 128.66, 128.51, 128.19 (3x s, CHar), 114.84 (s, CHarCar(OCH3)), 105.61 (dd, J = 271.6, 183.6 Hz, CBrF), 65.87 (d, J = 6.9 Hz, OCH2CH3), 65.65 (d, J = 7.4 Hz, OCH2CH3), 63.30 (dd, J = 23.4, 6.9 Hz, CH(Ph)CF), 55.68 (s, Ph(4-OCH3)), 16.57 (d, J = 5.7 Hz, OCH2CH3), 16.43 (d, J = 5.6 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −126.36 (dd, J = 78.1, 9.7 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 9.22 (d, J = 77.9 Hz, 1P). IR (neat): 1260, 1249, 1023, 974, 646 [cm−1]. MS (EI) m/z = 459.2 [M-H]+.
rac-19b: 1H NMR (400 MHz): δ = 7.51–7.43 (m, 5H, Har), 6.70–6.67 (m, 2H, Har), 6.57–6.53 (m, 2H, Har), 4.24–4.17 (m, 3H, OCH2CH3, CH(Ph)CFP), 4.10–4.00 (m, 2H, OCH2CH3), 3.66 (s, 3H, Ph(4-OCH3)), 1.27 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.16 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 152.82 (s, Car(OCH3)), 140.04 (s, Cipso), 136.51 (d, J = 4.8 Hz, Cipso), 129.36 (d, J = 1.7 Hz, CHar), 128.85, 128.61, 128.49 (3x s, CHar), 115.68 (s, CHarCar(OCH3)), 105.37 (dd, J = 274.0, 181.3 Hz, CBrF), 66.49 (dd, J = 19.7, 7.6 Hz, CH(Ph)CF), 65.06 (d, J = 6.6 Hz, OCH2CH3), 64.79 (d, J = 6.7 Hz, OCH2CH3), 55.69 (s, Ph(4-OCH3)), 16.36 (d, J = 5.7 Hz, OCH2CH3), 16.24 (d, J = 5.9 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −135.34 (dd, J = 82.1, 18.6 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 8.17 (d, J = 82.1 Hz, 1P).
Diethyl ((1S/1R, 2S/2R)-1-chloro-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (20a-d).
Isolated as a mixture of four diastereomers 20a–d (dr 1:0.83:0.07:0.11). Pale-yellow oil, 196 mg, yield 95%.
Diethyl ((1R, 2R)-1-chloro-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (20a).
1H NMR (400 MHz): δ = 7.35–7.32 (m, 5H, Har), 7.27–7.25 (m, 2H, Har), 7.21–7.19 (m, 3H, Har), 4.71 (dd, J = 8.9, 4.6 Hz, 1H, CH(Ph)CF), 4.38–4.31 (m, 2H, OCH2CH3), 4.29–4.26 (m, 2H, OCH2CH3), 3.74 (q, J = 6.4 Hz, CHCH3), 1.36 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.34 (d, J = 6.4 Hz, 3H, CHCH3), 1.33 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 145.40 (s, Cipso), 135.16 (d, J = 8.2 Hz, Cipso), 128.49, 128.45, 128.40, 128.09, 127.21, 126.76 (6x s, CHar), 110.45 (dd, J = 260.4, 191.5 Hz, CClF), 65.32 (d, J = 6.9 Hz, OCH2CH3), 64.97 (d, J = 7.2 Hz, OCH2CH3), 63.80 (dd, J = 23.3, 8.1 Hz, CH(Ph)CF), 54.76 (s, CHCH3), 21.68 (s, CHCH3), 16.54 (d, J = 6.0 Hz, OCH2CH3), 16.42 (d, J = 6.3 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −123.06 (dd, J = 87.4, 8.8 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 9.61 (d, J = 87.4 Hz, 1P). IR (neat): 1264, 1025, 984, 673 [cm−1]. MS (EI) m/z = 413.2 [M]+.
Diethyl ((1S, 2R)-1-chloro-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (20b).
1H NMR (400 MHz): δ = 7.32–7.29 (m, 5H, Har), 7.25–7.24 (m, 2H, Har), 7.23–7.21 (m, 2H, Har), 7.19–7.18 (m, 1H, Har), 4.41 (dd, J = 21.3, 3.3 Hz, 1H, CH(Ph)CF), 4.24–4.19 (m, 2H, OCH2CH3), 4.18–4.04 (m, 2H, OCH2CH3), 3.73 (q, J = 6.6 Hz, 1H, CHCH3), 1.33 (d, J = 6.3 Hz, 3H, CHCH3), 1.29 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.26 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 145.55 (s, Cipso), 136.48 (dd, J = 6.1, 2.7 Hz, Cipso), 128.45, 128.35, 128.30, 128.15, 127.12, 126.84 (6x s, CHar), 111.08 (dd, J = 265.2, 190.1 Hz, CClF), 65.83 (dd, J = 18.6, 8.7 Hz, CH(Ph)CF), 64.80 (d, J = 7.2 Hz, OCH2CH3), 64.68 (d, J = 7.4 Hz, OCH2CH3), 55.52 (s, CHCH3), 21.96 (s, CHCH3), 16.48 (d, J = 6.1 Hz, OCH2CH3), 16.43 (d, J = 6.2 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −134.74 (dd, J = 89.0, 21.5 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 9.02 (d, J = 89.2 Hz, 1P).
Diethyl ((1S, 2S)-1-chloro-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (20c).
Diagnostic signals 19F NMR (283 MHz): δ = −125.91 (dd, J = 83.2, 9.2 Hz). 31P{/1H} NMR (122 MHz): δ = 8.89 (d, J = 83.3 Hz).
Diethyl ((1R, 2S)-1-chloro-1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (20d).
Diagnostic signals 19F NMR (283 MHz): δ = −134.88 (dd, J = 91.3, 21.0 Hz). 31P{/1H} NMR (122 MHz): δ = 8.66 (d, J = 91.5 Hz).
rac-Diethyl ((1R, 2R)-1-chloro-1-fluoro-2-((4-methoxybenzyl)amino)-2-phenylethyl)phosphonate (rac-21a) and rac-diethyl ((1R, 2S)-1-chloro-1-fluoro-2-((4-methoxybenzyl)amino)-2-phenylethyl)phosphonate (rac-21b).
Isolated as a mixture of two diastereomers rac-21a,b (dr 1:0.92), which could not be separated by the chromatography techniques employed in this study. Pale-yellow oil, 206 mg, yield 96%.
rac-21a: 1H NMR (400 MHz): δ = 7.45–7.42 (m, 3H, Har), 7.39–7.36 (m, 2H, Har), 7.14–7.11 (m, 2H, Har), 6.83–6.81 (m, 2H, Har), 4.45 (dd, J = 8.4, 4.4 Hz, 1H, CH(Ph)CFP), 4.36–4.25 (m, 2H, OCH2CH3), 4.16–4.08 (m, 2H, OCH2CH3), 3.75 (s, 3H, Ph(4-OCH3)), 3.70 (br d, J = 12.9 Hz, 1H, CHHN), 3.47 (d, J = 12.9 Hz, 1H, CHHN), 1.28 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.23 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 158.86 (s, Car(OCH3)), 134.75 (d, J = 8.3 Hz, Cipso), 131.48 (s, Cipso), 129.83 (d, J = 1.3 Hz, CHar), 129.71, 128.64, 128.18 (3x s, CHar), 113.82 (s, CHarCar(OCH3)), 109.70 (dd, J = 260.8, 191.2 Hz, CClF), 65.57 (d, J = 6.9 Hz, OCH2CH3), 65.07 (dd, J = 22.1, 8.6 Hz, CH(Ph)CF), 64.92 (d, J = 7.1 Hz, OCH2CH3), 55.31 (s, Ph(4-OCH3)), 50.18 (s, CH2N), 16.45 (d, J = 5.9 Hz, OCH2CH3), 16.32 (d, J = 5.9 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −124.54 (dd, J = 84.8, 8.3 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 9.37 (d, J = 84.7 Hz, 1P). IR (neat): 1263, 1253, 1024, 981, 672 [cm−1]. MS (EI) m/z = 430.8 [M-H]+.
rac-21b: 1H NMR (400 MHz): δ = 7.43–7.40 (m, 3H, Har), 7.36–7.33 (m, 2H, Har), 7.18–7.14 (m, 2H, Har), 6.80–6.78 (m, 2H, Har), 4.23–4.17 (m, 3H, OCH2CH3, CH(Ph)CFP), 4.10–3.98 (m, 2H, OCH2CH3), 3.76 (s, 3H, Ph(4-OCH3)), 3.65 (br d, J = 12.9 Hz, 1H, CHHN), 3.46 (d, J = 12.9 Hz, 1H, CHHN), 1.25 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3), 1.22 (td, J = 7.0, 0.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 158.83 (s, Car(OCH3)), 135.85 (dd, J = 6.0, 2.4 Hz, Cipso), 131.52 (s, Cipso), 129.90 (d, J = 1.3 Hz, CHar), 129.60, 128.55, 128.21 (3x s, CHar), 113.81 (s, CHarCar(OCH3)), 110.35 (dd, J = 264.0, 191.1 Hz, CClF), 67.24 (dd, J = 18.8, 8.9 Hz, CH(Ph)CF), 64.99 (d, J = 7.1 Hz, OCH2CH3), 64.61 (d, J = 7.2 Hz, OCH2CH3), 55.30 (s, Ph(4-OCH3)), 50.70 (s, CH2N), 16.42 (d, J = 5.9 Hz, OCH2CH3), 16.35 (d, J = 5.9 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −134.91 (dd, J = 90.6, 20.5 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 8.76 (d, J = 90.8 Hz, 1P).
3.8. Synthesis of α-Fluorinated ß-Enaminophosphonate/ß-Iminophosphonate (22a,b/23a,b)
To a solution of the β-iminophosphonate (10a,b) (137 mg, 0.3 mmol) in anhydrous THF (2 mL), LiAlH4 (17 mg, 0.45 mmol) at 0 °C was added. The reaction was warmed to room temperature, and then stirred for 40 min. After this time, solvent was evaporated and chloroform (5 mL) was added to the residue. The crude mixture was filtered through a syringe filter and purified using column chromatography (AcOEt/hexane: 10% → 50%) to give yellow oil with 83% yield (94 mg), as a mixture of enamine (22a,b) and imine (23a,b): (E-22a/Z-22b ratio 1:0.3; 22a,b/23a,b ratio 1:0.05)
(E)-Diethyl (1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)vinyl)phosphonate (E-22a).
1H NMR (401 MHz): δ = 7.38–7.35 (m, 2H, Har), 7.28–7.25 (m, 2H, Har), 7.24–7.20 (m, 2H, Har), 7.19–7.14 (m, 2H, Har), 7.05–7.02 (m, 2H, Har), 6.48 (dd, J = 10.5 Hz, 4.6 Hz, NH), 4.09–3.76 (m, 5H, 2x OCH2CH3, CHCH3), 1.41 (d, J = 6.9 Hz, 3H, CHCH3), 1.35 (td, J = 7.1, 0.7 Hz, 3H, OCH2CH3), 1.26 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 151.41 (dd, J = 30.1 Hz, 17.3 Hz, C(Ph) = CF), 144.55 (s, Cipso), 129.52, 129.30, 128.39, 128.33, 126.49, 125.78, (6x s, CHar), 62.77 (d, J = 4.8 Hz, OCH2CH3), 62.56 (d, J = 4.6 Hz, OCH2CH3), 54.48 (s, CHCH3), 23.41 (s, CHCH3), 16.37 (d, J = 6.7 Hz, OCH2CH3), 16.20 (d, J = 6.8 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −175.62 (dd, J = 91.7, 4.6 Hz, 1F). 31P{/1H} NMR (162 MHz): δ = 12.10 (d, J = 91.8 Hz, 1P). MS (EI) m/z = 377.1 [M]+.
(Z)-Diethyl(1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)vinyl)phosphonate (Z-22b).
Diagnostic signals 19F NMR (283 MHz): δ = −163.94 (dd, J = 92.6, 6.1 Hz). 31P{/1H} NMR (162 MHz): δ = 9.42 (d, J = 92.5 Hz).
(E/Z)-Diethyl(1-fluoro-2-phenyl-2-(((S)-1-phenylethyl)imino)ethyl)phosphonate (23a,b).
Diagnostic signals 19F NMR (283 MHz): −205.17 (dd, J = 78.4, 46.2 Hz), −205.73 (dd, J = 79.1, 46.2 Hz) 31P{/1H} NMR (162 MHz): signals masked by other tautomers signals.
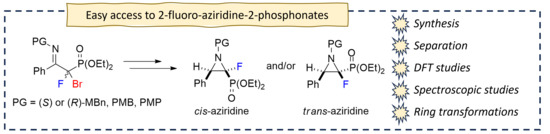
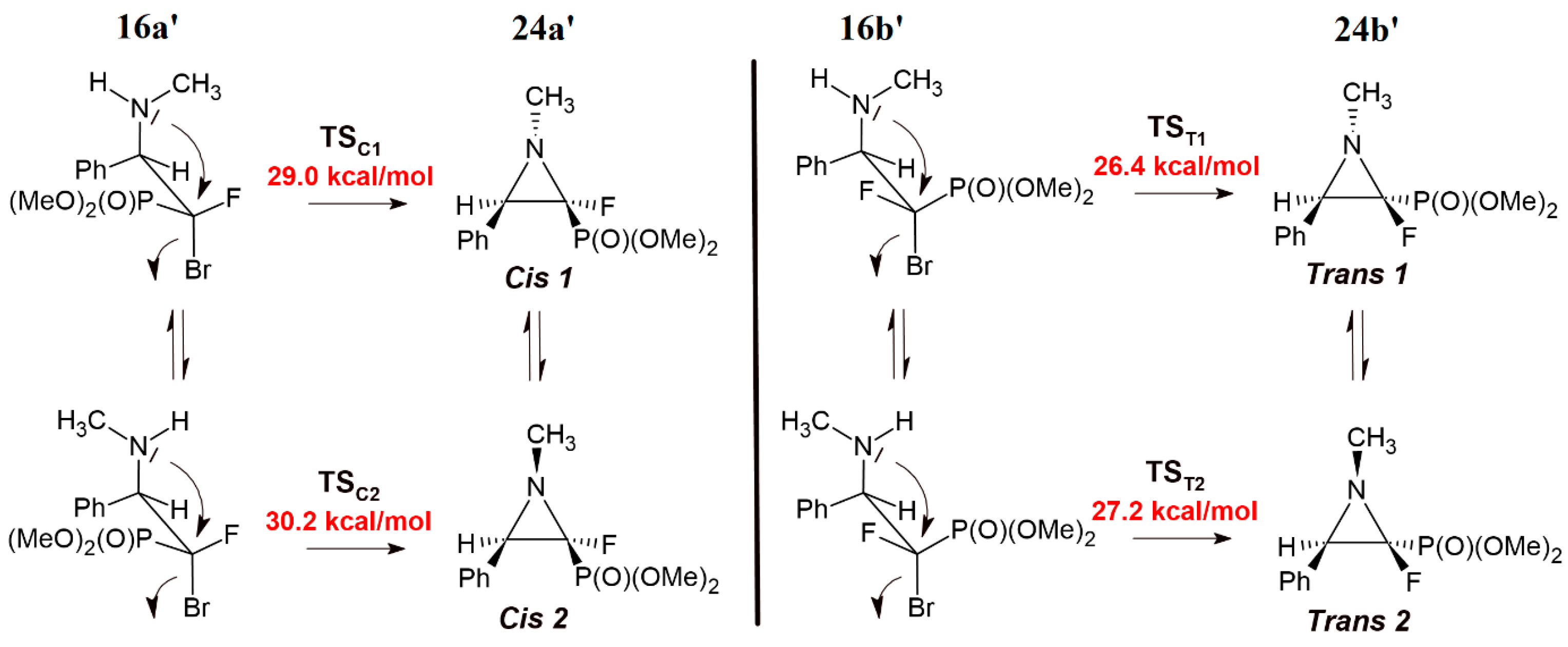
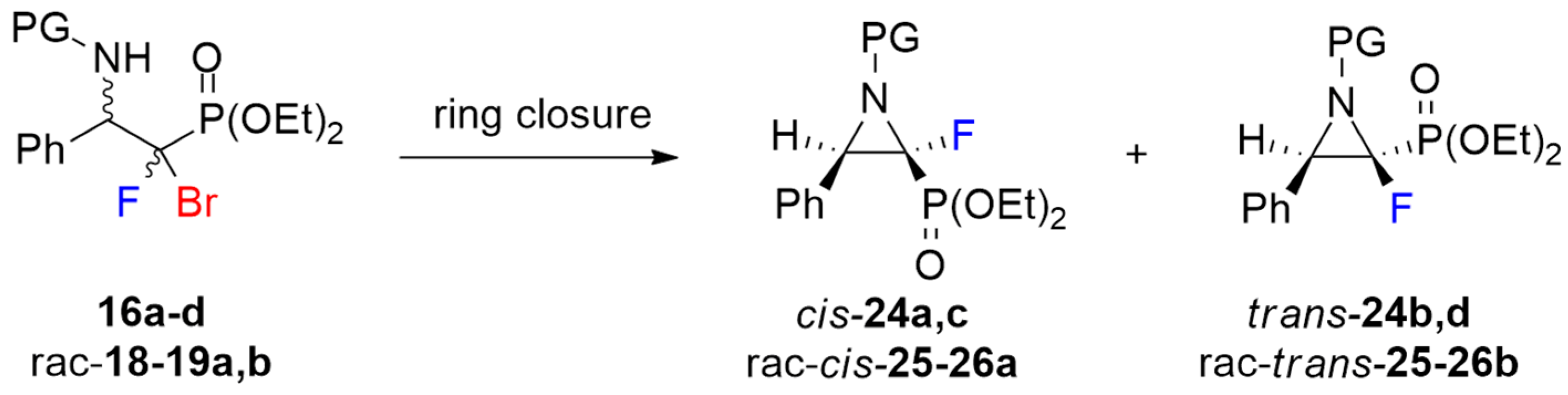
3.9. General Procedure for Synthesis of 2-Fluorinated Aziridine-2-phosphonates (24a–d, rac-25–26a,b)
To a solution of the β-aminophosphonate (16a–d, rac-b) (0.5 mmol) in anhydrous DMF (3 mL), triethylamine (84 µL, 61 mg, 0.6 mmol) was added. The reaction mixture was heated at 70 °C for 4 h at inert atmosphere. After completion of the reaction (monitored by 19F NMR), the solvent was removed under reduced pressure. The mixture was purified by column chromatography (AcOEt/petroleum ether 5% → 50%) with previously deactivated silica gel (short pad 1 cm, 1% triethylamine in hexane, 20 mL).
Diethyl ((2S/2R, 3R/3S)-2-fluoro-3-phenyl-1-((S)-1-phenylethyl)aziridin-2-yl)phosphonate (24a-d).
Crude reaction mixture: 24a–d (dr 0.78:1:0.13:0.08). Isolated as a mixture of four diastereomers 24a–d (dr 0.70:1:0.17:0.04) (97mg) and single diastereomer 24a (32mg). Diagnostic signals for traces of diastereomers 24c–d were determined from the crude reaction mixture. Pale-yellow oil, 129 mg, yield 68%.
Diethyl ((2S,3R)-2-fluoro-3-phenyl-1-((S)-1-phenylethyl)aziridin-2-yl)phosphonate (cis-24a).
1H NMR (401 MHz): δ = 7.49–7.43 (m, 1H, Har), 7.30–7.21 (m, 8H, Har), 7.15–7.10 (m, 1H, Har), 4.08–3.96 (m, 3H, OCH2CH3, OCHHCH3), 3.92–3.84 (m, 1H, OCHHCH3), 3.81 (q, J = 6.4 Hz, 1H, CHCH3), 3.18 (d, J = 8.7 Hz, 1H, CH(Ph)CPF), 1.61 (d, J = 6.5 Hz, 3H, CHCH3), 1.19 (t, J = 7.3 Hz, 3H, OCH2CH3), 1.17 (t, J = 7.2 Hz, 3H, OCH2CH3). 1H{/19F} NMR (401 MHz): δ = 7.50–7.44 (m, 1H, Har), 7.31–7.21 (m, 8H, Har), 7.15–7.10 (m, 1H, Har), 4.08–3.97 (m, 3H, OCH2CH3, OCHHCH3), 3.92–3.83 (m, 1H, OCHHCH3), 3.82 (q, J = 6.6 Hz, 1H, CHCH3), 3.18 (br s, 1H, CH(Ph)CPF), 1.61 (d, J = 6.5 Hz, 3H, CHCH3), 1.20 (t, J = 7.3 Hz, 3H, OCH2CH3), 1.17 (t, J = 7.3 Hz, 3H, OCH2CH3). 1H{/31P} NMR (401 MHz): δ = 7.49–7.42 (m, 1H, Har), 7.30–7.21 (m, 8H, Har), 7.14–7.10 (m, 1H, Har), 4.08–3.95 (m, 3H, OCH2CH3, OCHHCH3), 3.91–3.84 (m, 1H, OCHHCH3), 3.81 (q, J = 6.4 Hz, 1H, CHCH3), 3.18 (d, J = 8.7 Hz, 1H, CH(Ph)CPF), 1.61 (d, J = 6.5 Hz, 3H, CHCH3), 1.20 (t, J = 7.3 Hz, 3H, OCH2CH3), 1.17 (t, J = 7.2 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 143.12 (s, Cipso), 133.87 (s, Cipso), 128.53, 128.38, 128.09, 127.74, 127.60, 127.43 (6x s, CHar), 87.05 (dd, J = 274.2, 272.1 Hz, CFP), 63.35 (d, J = 6.2 Hz, OCH2CH3), 63.35 (d, J = 6.2 Hz OCH2CH3), 60.58 (d, J = 13.3 Hz, CHCH3), 48.96 (dd, J = 19.1, 1.6 Hz, CH(Ph)CFP), 23.30 (s, CHCH3), 16.31 (d, J = 6.3 Hz, OCH2CH3), 16.22 (d, J = 6.2 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −182.45 (dd, J = 118.5, 8.7 Hz, 1F) 31P{/1H} NMR (122 MHz): δ = 9.64 (d, J = 118.8 Hz, 1P).
Diethyl ((2R,3R)-2-fluoro-3-phenyl-1-((S)-1-phenylethyl)aziridin-2-yl)phosphonate (trans-24b).
1H NMR (401 MHz): δ = 7.38–7.34 (m, 2H, Har), 7.29–7.24 (m, 4H, Har), 7.23–7.19 (m, 3H, Har), 7.17–7.13 (m, 1H, Har), 4.35 (quint, J = 7.2 Hz, 2H, OCH2CH3), 4.31–4.24 (m, 1H, OCHHCH3), 4.23–4.13 (m, 2H, OCHHCH3, CHCH3), 3.26 (t, J = 4.2 Hz, 1H, CH(Ph)CFP), 1.64 (d, J = 6.5 Hz, 3H, CHCH3), 1.44 (td, J = 7.1, 0.7 Hz, 3H, OCH2CH3), 1.33 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3). 1H{/19F} NMR (401 MHz): δ = 7.37–7.33 (m, 2H, Har), 7.28–7.24 (m, 4H, Har), 7.23–7.19 (m, 3H, Har), 7.18–7.13 (m, 1H, Har), 4.35 (quint, J = 7.2 Hz, 2H, OCH2CH3), 4.32–4.24 (m, 1H, OCHHCH3), 4.23–4.12 (m, 2H, OCHHCH3, CHCH3), 3.26 (br d, J = 4.3 Hz, 1H, CH(Ph)CFP), 1.64 (d, J = 6.5 Hz, 3H, CHCH3), 1.44 (td, J = 7.0, 0.7 Hz, 3H, OCH2CH3), 1.33 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3). 1H{/31P} NMR (401 MHz): δ = 7.37–7.32 (m, 2H, Har), 7.29–7.23 (m, 4H, Har), 7.22–7.18 (m, 3H, Har), 7.17–7.14 (m, 1H, Har), 4.35 (q, J = 7.1 Hz, 2H, OCH2CH3), 4.32–4.24 (m, 1H, OCHHCH3), 4.22–4.14 (m, 2H, OCHHCH3, CHCH3), 3.26 (d, J = 4.5 Hz, 1H, CH(Ph)CFP), 1.64 (d, J = 6.5 Hz, 3H, CHCH3), 1.44 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.33 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 143.14 (s, Cipso), 133.62 (dd, J = 5.2, 1.5 Hz, Cipso), 128.39, 128.10, 127.73 (3x s, CHar), 127.61 (t, J = 0.9 Hz, CHar), 127.30, 127.02 (2x s, CHar), 84.27 (dd, J = 258.7, 233.2 Hz, CFP), 64.15 (dd, J = 7.8, 1.1 Hz, OCH2CH3), 63.54 (dd, J = 6.0, 0.5 Hz, OCH2CH3), 61.18 (dd, J = 5.0, 3.1 Hz, CHCH3), 47.46 (dd, J = 12.7, 5.8 Hz, CH(Ph)CFP), 23.64 (s, CHCH3), 16.39 (d, J = 6.4 Hz, OCH2CH3), 16.38 (d, J = 6.5 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −168.76 (dt, J = 113.8, 4.8 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 10.83 (d, J = 114.0 Hz, 1P). IR (neat): 1259, 1162, 1019, 957, 759 [cm−1]. HRMS (ESI): m/z calcd for C20H26FNO3P, [M + H]+: 378.1634 found: 378.1628.
Diethyl ((2R,3S)-2-fluoro-3-phenyl-1-((S)-1-phenylethyl)aziridin-2-yl)phosphonate (cis-24c).
Diagnostic signals 19F NMR (283 MHz): δ = −178.36 (dd, J = 114.2, 8.5 Hz). 31P{/1H} NMR (122 MHz): δ = 8.94 (d, J = 114.1 Hz).
Diethyl ((2S,3S)-2-fluoro-3-phenyl-1-((S)-1-phenylethyl)aziridin-2-yl)phosphonate (trans-24d).
Diagnostic signals 19F NMR (283 MHz): δ = −169.11 (dt, J = 110.8, 4.7 Hz). 31P{/1H} NMR (122 MHz): δ = 10.22 (d, J = 110.6 Hz).
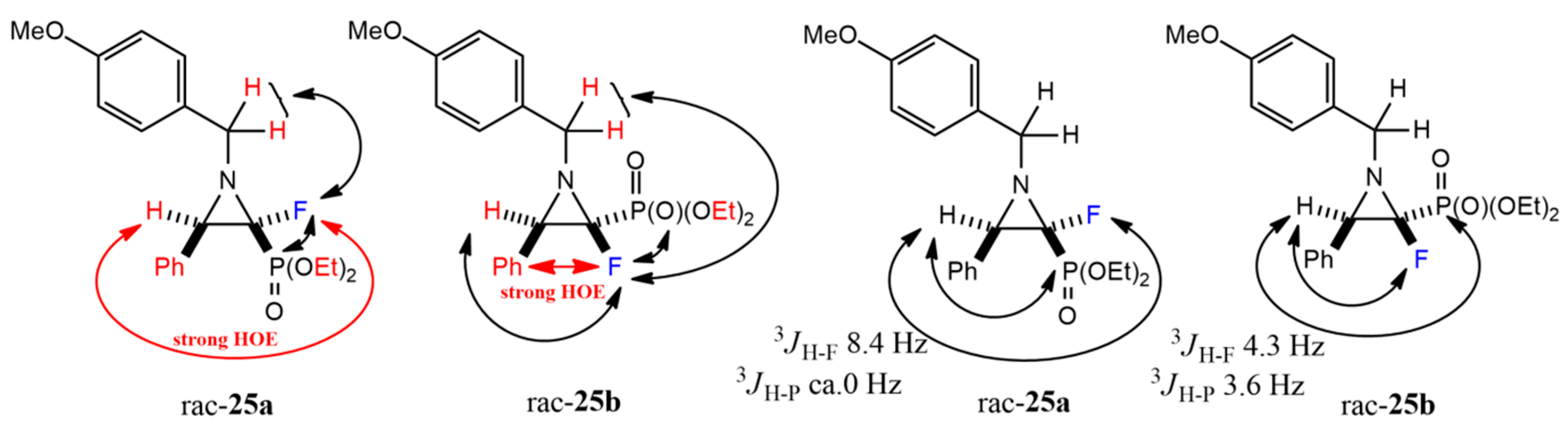
rac-Diethyl ((2S,3R)-2-fluoro-1-(4-methoxybenzyl)-3-phenylaziridin-2-yl)phosphonate (rac-cis-25a) and rac-diethyl ((2R,3R)-2-fluoro-1-(4-methoxybenzyl)-3-phenylaziridin-2-yl)phosphonate (rac-trans-25b).
Crude reaction mixture: 25a,b (dr 0.68:1). Isolated as a mixture of diastereomers 25a,b (dr 0.52:1) (94mg) and single diastereomer 25b (28mg). Pale yellow oil, 122 mg, yield 62%.
rac-cis-25a: 1H NMR (401 MHz): δ = 7.45–7.40 (m, 3H, Har), 7.36–7.29 (m, 2H, Har), 7.23–7.19 (m, 2H, Har), 6.92–6.88 (m, 2H, Har), 4.15 (br d, J = 13.6, 1H, CHHN), 3.99–3.85 (m, 4H, 2x OCH2CH3), 4.01 (br d, J = 13.6, 1H, CHHN), 3.82 (s, 3H, Ph(4-OCH3)), 3.26 (d, J = 8.5 Hz, 1H, CH(Ph)CFP), 1.11 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.09 (td, J = 7.1, 0.7 Hz, 3H, OCH2CH3). 1H{/19F} NMR (401 MHz): δ = 7.45–7.41 (m, 2H, Har), 7.36–7.30 (m, 2H, Har), 7.24–7.19 (m, 2H, Har), 6.91–6.86 (m, 2H, Har), 4.15 (br d, J = 13.5, 1H, CHHN), 3.97–3.85 (m, 4H, 2x OCH2CH3), 4.02 (br d, J = 13.6, 1H, CHHN), 3.81 (s, 3H, Ph(4-OCH3)), 3.25 (br s, 1H, CH(Ph)CFP), 1.11 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 1.10 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3). 1H{/31P} NMR (401 MHz): δ = 7.45–7.41 (m, 2H, Har), 7.36–7.30 (m, 2H, Har), 7.24–7.19 (m, 2H, Har), 6.91–6.86 (m, 2H, Har), 4.15 (d, J = 12.8 Hz, 1H, CHHN), 3.98–3.84 (m, 4H, 2x OCH2CH3), 4.02 (d, J = 13.0 Hz, 1H, CHHN), 3.83 (s, 3H, Ph(4-OCH3)), 3.27 (d, J = 8.4 Hz, 1H, CH(Ph)CFP), 1.11 (t, J = 7.0 Hz, 3H, OCH2CH3), 1.09 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 159.00 (s, Car(OCH3)), 133.44–133.38 (m, Cipso), 131.86 (s, Cipso), 129.21, 128.17, 127.86, 127.56 (4x s, CHar), 113.80 (s, CHarCar(OCH3)), 86.67 (dd, J = 274.0, 272.5 Hz, CFP), 63.15 (d, J = 6.1 Hz, OCH2CH3), 63.03 (d, J = 6.2 Hz, OCH2CH3), 55.23 (s, Ph(4-OCH3)), 53.30 (d, J = 15.1 Hz, CH2N), 49.08 (dd, J = 19.2, 1.3 Hz, CH(Ph)CFP), 16.12 (d, J = 6.0 Hz, OCH2CH3), 16.11 (d, J = 6.1 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −180.27 (dd, J = 116.7, 8.4 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 9.97 (d, J = 116.9 Hz, 1P).
rac-trans-25b: 1H NMR (401 MHz): δ = 7.39 (d, J = 7.7 Hz, 4H, Har), 7.36–7.30 (m, 3H, Har), 6.87 (d, J = 8.6 Hz, 2H, Har), 4.46 (dd, J = 13.6, 2.8 Hz, 1H, CHHN), 4.34–4.27 (m, 1H, OCHHCH3), 4.25–4.18 (m, 3H, OCHHCH3, OCH2CH3), 4.08 (dd, J = 13.6, 5.1 Hz, 1H, CHHN), 3.80 (s, 3H, Ph(4-OCH3)), 3.45 (t, J = 4.1 Hz, 1H, CH(Ph)CFP), 1.37 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3), 1.34 (td, J = 7.1, 0.7 Hz, 3H, OCH2CH3). 1H{/19F} NMR (401 MHz): δ = 7.40–7.35 (m, 4H, Har), 7.37–7.32 (m, 3H, Har), 6.88 (d, J = 8.8 Hz, 2H, Har), 4.47 (d, J = 13.6, 1H, CHHN), 4.32–4.26 (m, 1H, OCHHCH3), 4.25–4.17 (m, 3H, OCHHCH3, OCH2CH3), 4.08 (d, J = 13.5, 1H, CHHN), 3.81 (s, 3H, Ph(4-OCH3)), 3.45 (d, J = 4.1 Hz, 1H, CH(Ph)CFP), 1.38 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3), 1.35 (td, J = 7.1, 0.7 Hz, 3H, OCH2CH3). 1H{/31P} NMR (401 MHz): δ = 7.40–7.36 (m, 4H, Har), 7.37–7.32 (m, 3H, Har), 6.88 (d, J = 8.7 Hz, 2H, Har), 4.47 (dd, J = 13.6, 2.8 Hz, 1H, CHHN), 4.33–4.26 (m, 1H, OCHHCH3), 4.25–4.18 (m, 3H, OCHHCH3, OCH2CH3), 4.08 (dd, J = 13.6, 5.1 Hz, 1H, CHHN), 3.81 (s, 3H, Ph(4-OCH3)), 3.45 (d, J = 4.3 Hz, 1H, CH(Ph)CFP), 1.38 (t, J = 7.1, 3H, OCH2CH3), 1.35 (t, J = 7.1, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 158.79 (s, Car(OCH3)), 133.58 (br d, J = 5.2 Hz, Cipso), 131.93 (s, Cipso), 129.99, 128.15, 127.84, 127.67 (4x s, CHar), 113.71 (s, CHarCar(OCH3)), 83.95 (dd, J = 259.6, 231.6 Hz, CFP), 63.71 (d, J = 7.0 Hz, OCH2CH3), 63.50 (d, J = 6.0 Hz, OCH2CH3), 55.53 (dd, J = 5.4, 3.5 Hz, CH2N), 55.19 (s, Ph(4-OCH3)), 48.43 (dd, J = 12.9, 6.0 Hz, CH(Ph)CFP), 16.27 (d, J = 5.9 Hz, OCH2CH3), 16.25 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −170.98 (dq, J = 111.7, 4.3 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 10.95 (d, J = 111.6 Hz, 1P). IR (neat): 1247, 1164, 1097, 1018, 978, 765 [cm−1]. HRMS (ESI): m/z calcd for C20H26FNO4P, [M + H]+: 394.1583 found: 394.1587.
rac-Diethyl ((2S,3R)-2-fluoro-1-(4-methoxyphenyl)-3-phenylaziridin-2-yl)phosphonate (rac-cis-26a) and rac diethyl ((2R,3R)-2-fluoro-1-(4-methoxyphenyl)-3-phenylaziridin-2-yl)phosphonate (rac-trans-26b).
Crude reaction mixture: 26a,b (dr 0.09:1). Isolated as single diastereomer 26b. Diagnostic signals for traces of diastereomers 26a were determined from the crude reaction mixture. Pale-yellow oil, 85 mg, yield 45%.
rac-cis-26a: Diagnostic signals 19F NMR (283 MHz): δ = −171.22 (dd, J = 117.5, 7.9 Hz). 31P{/1H} NMR (122 MHz): δ = 8.63 (d, J = 117.4 Hz).
rac-trans-26b: 1H NMR (401 MHz): δ = 7.51–7.48 (m, 2H, Har), 7.40–7.34 (m, 3H, Har), 7.13–7.06 (m, 2H, Har), 6.86–6.82 (m, 2H, Har), 4.19–3.98 (m, 4H, 2x OCH2CH3), 3.84 (t, J = 4.1 Hz, 1H, CH(Ph)CFP), 3.77 (s, 3H, Ph(4-OCH3)), 1.31 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3), 1.17 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3). 1H{/19F} NMR (401 MHz): δ = 7.52–7.47 (m, 2H, Har), 7.42–7.35 (m, 3H, Har), 7.11–7.06 (m, 2H, Har), 6.85–6.80 (m, 2H, Har), 4.19–3.96 (m, 4H, 2x OCH2CH3), 3.84 („d”, J = 4.2 Hz, 1H, CH(Ph)CFP), 3.76 (s, 3H, Ph(4-OCH3)), 1.31 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3), 1.17 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3). 1H{/31P} NMR (401 MHz): δ = 7.50–7.46 (m, 2H, Har), 7.41–7.34 (m, 3H, Har), 7.11–7.05 (m, 2H, Har), 6.85–6.82 (m, 2H, Har), 4.20–3.99 (m, 4H, 2x OCH2CH3), 3.83 (d, J = 4.5 Hz, 1H, CH(Ph)CFP), 3.77 (s, 3H, Ph(4-OCH3)), 1.30 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.16 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 156.13 (s, Car(OCH3)), 132.83 (dd, J = 5.3, 1.2 Hz, Cipso), 134.22 (s, Cipso), 129.30 (d, J = 3.2 Hz, CHar), 128.57, 128.38, 127.79 (3x s, CHar), 114.24 (s, CHarCar(OCH3)), 83.91 (dd, J = 260.8, 238.3 Hz, CFP), 63.63 (d, J = 6.9 Hz, OCH2CH3), 63.21 (d, J = 6.0 Hz, OCH2CH3), 55.49 (s, Ph(4-OCH3)), 46.12 (dd, J = 13.1 Hz, 5.4 Hz, CH(Ph)CFP), 16.29 (d, J = 6.1 Hz, OCH2CH3), 16.17 (d, J = 5.6 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −169.58 (dd, J = 121.4, 4.3 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 8.82 (d, J = 121.5 Hz, 1P). HRMS (ESI): m/z calcd for C19H24FNO4P, [M + H]+: 380.1427 found: 380.1421.
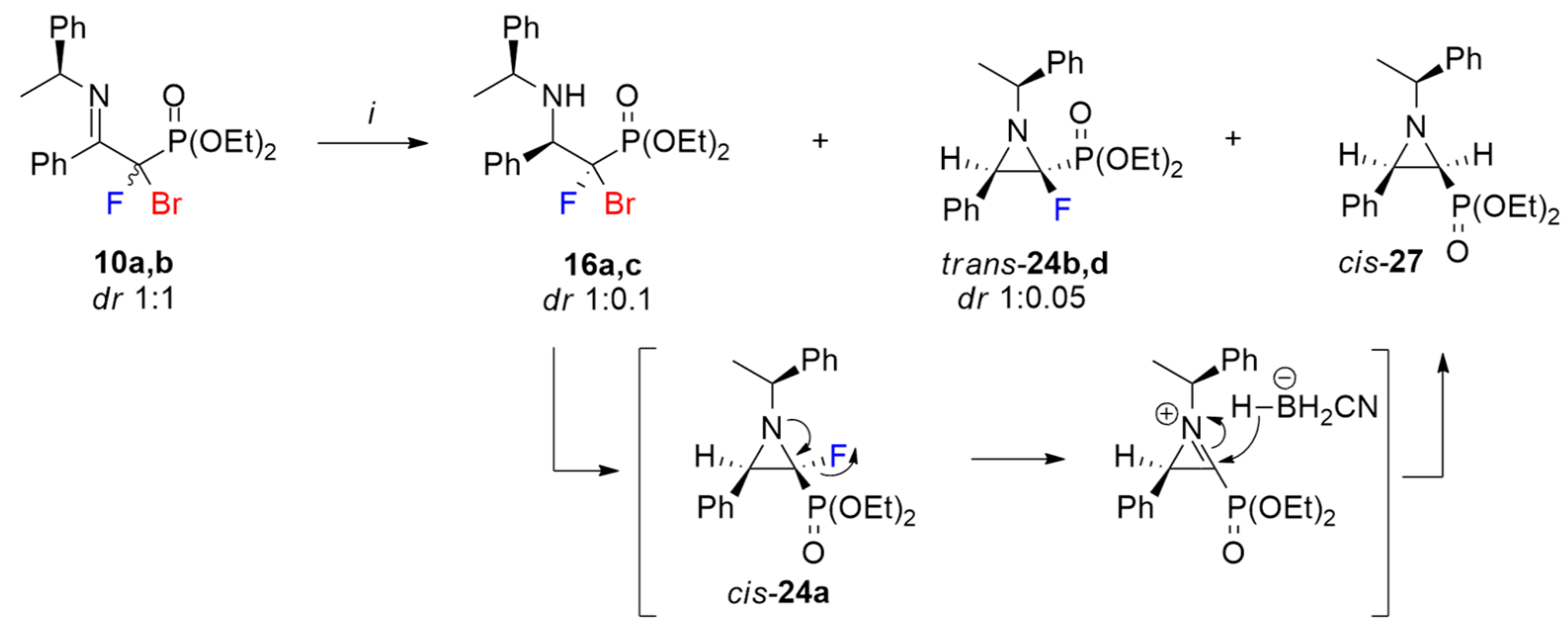
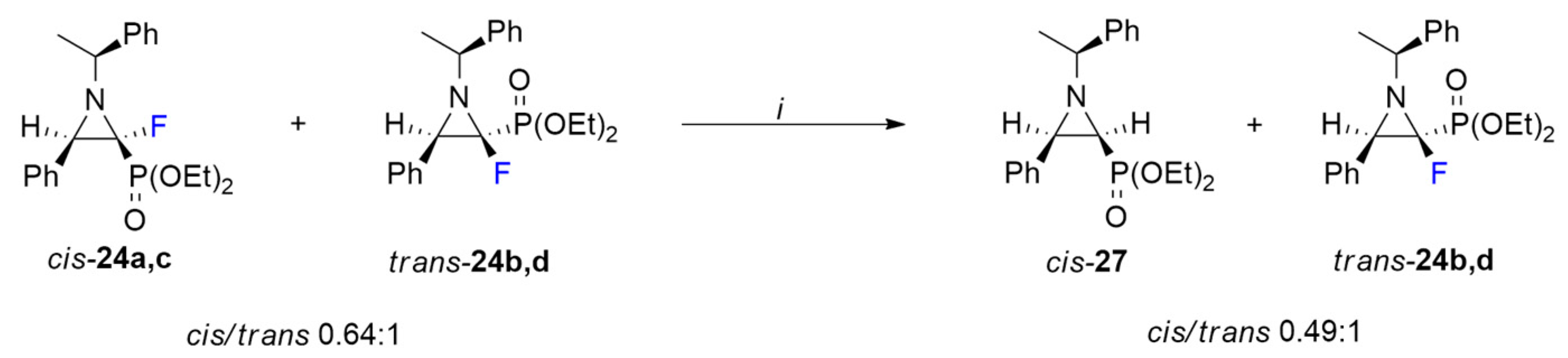
3.10. Separation Method for Chiral Aziridines and Synthesis of Non-Fluorinated Aziridine-2-phosphonate: Diethyl ((2S,3R)-3-Phenyl-1-((S)-1-phenylethyl)aziridin-2-yl)phosphonate (cis-27)
Method A. Imine 10a-d (dr 1:1, 228 mg, 0.5 mmol) was dissolved in anhydrous methanol (5 mL) and NaBH3CN (250 mg, 4 mmol), and glacial CH3COOH (88 µL, 92 mg, 1.5 mmol) was added. The reaction mixture was refluxed for 7h and next the solvent was evaporated. Then, the residue was dissolved in CH2Cl2 (3 mL) and extracted with a saturated solution of NaHCO3 and brine. The organic layers were combined, dried over Na2SO4, and evaporated to give a mixture of products 16a,c, trans-24b,d, and cis-27. The product cis-27 was purified by column chromatography (AcOEt/hexane: 5% → 60%) as a pale-yellow oil (58 mg, yield 32%). Amine 16a,c (dr 1:0.1) and aziridine trans-24b,d (dr 1:0.05) were isolated as a mixture (0.3:1, 84 mg).
Method B. To the mixture of cis- and trans-aziridines (24a-d; 0.64(dr 1:0.07)/1(dr 1:0.09), 113 mg, 0.3 mmol) dissolved in methanol (3 mL), Pd/C (10 mol%, 2 mg) and NaBH4 (23 mg, 0.6 mmol) were added. The reaction was stirred at 70 °C for 3h. Then, the solvent was evaporated, the residue was dissolved in CHCl3 (3 mL), water (10 mL) was added, and it was extracted (3 × 10 mL CHCl3). The organic layers were dried over anhydrous Na2SO4 and evaporated to give crude products trans-24b,d/cis-27 (1:0.49) separated using column chromatography with previously deactivated silica gel (short pad 1 cm, 1% triethylamine in hexane, 20 mL). Cis-27 was isolated as a single diastereomer (42 mg, yield 39%) and trans-24b,d was isolated as a mixture of diastereomers (dr 1:0.07, 49 mg).
cis-27: 1H NMR (401 MHz): δ = 7.44–7.40 (m, 2H, Har), 7.31–7.26 (m, 4H, Har), 7.23–7.18 (m, 2H, Har), 7.17–7.10 (m, 2H, Har), 3.99– 3.90 (m, 2H, OCH2CH3), 3.85–3.78 (m, 1H, OCHHCH3), 3.64–3.54 (m, 1H, OCHHCH3), 2.95 (t, J = 6.7 Hz, 1H, CH(Ph)CHP), 2.74 (q, J = 6.5 Hz, 1H, CHCH3), 1.99 (dd, J = 17.8, 6.8 Hz, 1H, CHP), 1.56 (d, J = 6.5 Hz, 3H, CHCH3), 1.17 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3), 1.07 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3). 1H{/31P} NMR (401 MHz): δ = 7.46–7.40 (m, 2H, Har), 7.32–7.25 (m, 4H, Har), 7.24–7.18 (m, 2H, Har), 7.17–7.11 (m, 2H, Har), 4.00–3.92 (m, 2H, OCH2CH3), 3.86–3.78 (m, 1H, OCHHCH3), 3.65–3.56 (m, 1H, OCHHCH3), 2.96 (d, J = 6.7 Hz, 1H, CH(Ph)CHP), 2.75 (q, J = 6.5 Hz, 1H, CHCH3), 2.00 (d, J = 6.8 Hz, 1H, CHP), 1.57 (d, J = 6.6 Hz, 3H, CHCH3), 1.18 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.08 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 143.56 (s, Cipso), 135.94 (d, J = 2.0 Hz, Cipso), 128.54, 128.06, 127.69, 127.51, 127.18, 127.09, (6 x s, CHar), 71.85 (d, J = 6.1 Hz, CHCH3), 62.10 (d, J = 6.4 Hz, OCH2CH3), 61.81 (d, J = 6.4 Hz, OCH2CH3), 46.27 (d, J = 5.7 Hz, CH(Ph)CHP), 39.86 (s, CHP), 23.12 (s, CHCH3), 16.39 (d, J = 6.5 Hz, OCH2CH3), 16.36 (d, J = 6.0 Hz, OCH2CH3). 31P{/1H} NMR (162 MHz): δ = 21.44 (s, 1P). HRMS (ESI): m/z calcd for C20H27NO3P, [M + H]+: 360.1729 found: 360.1723.
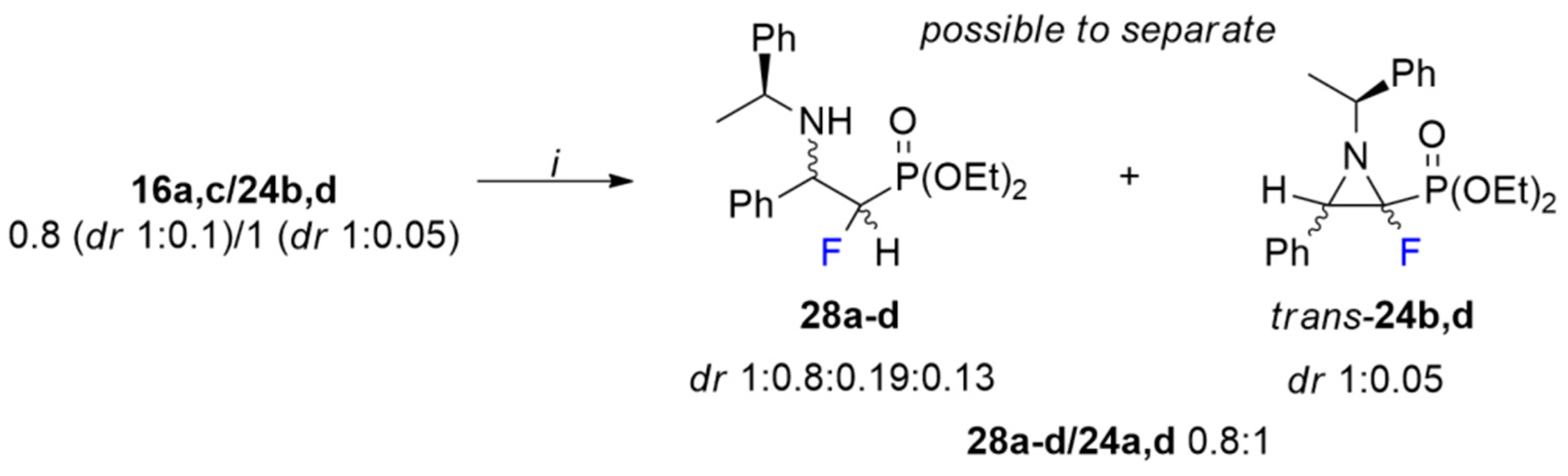
3.11. Isolation of Trans-Aziridine 24. Synthesis of α-Fluorinated ß-Aminophosphonate: Diethyl ((1S/R, 2S/R)-1-Fluoro-2-phenyl-2-(((S)-1-phenylethyl)amino)ethyl)phosphonate (28a–d)
To the mixture of amine (16a,c) and aziridine (24b,d) (0.8 (dr 1:0.1): 1(dr 1:0.05), 186 mg) dissolved in methanol (2 mL), NaBH4 (15 mg, 0.6 mmol) and Pd/C (10 mol%, 2 mg) were added. The reaction was stirred at room temperature for 20 min. Then, the crude mixture was filtrated through Celite with MeOH as a mobile phase and concentrated under vacuum. The residue was dissolved in CH2Cl2 (2 mL) and extracted with brine. The organic layers were dried over anhydrous Na2SO4 and evaporated to give a mixture of monofluorinated amine 28a–d and aziridine 24b,d (0.8:1, respectively). The crude products 24b,d/28a–d (28a–d: dr 1:0.8:0.19:0.13) were separated using column chromatography with previously deactivated silica gel (1% triethylamine in hexane, 20 mL). Amine was isolated as a mixture of diastereomers 28a–d (dr 1:0.75:0.09:0.05, 70 mg, pale-yellow oil) and aziridine was separated as a mixture of diastereomers 24b,d (dr 1:0.05, 75 mg).
28a: 1H NMR (401 MHz): δ = 7.43–7.34 (m, 5H, Har), 7.33–7.26 (m, 5H, Har), 4.85 (ddd, J = 45.2, 5.1, 3.9 Hz, 1H, CHFP), 4.38 (ddd, J = 23.5, 6.2, 5.1 Hz, 1H, CHCFP), 4.02–3.76 (m, 4H, 2x OCH2CH3), 3.67 (q, J = 6.4 Hz, 1H, CHCH3), 1.36 (d, J = 6.5 Hz, 3H, CHCH3), 1.27 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3), 1.26 (td, J = 7.1, 0.5 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 145.61 (s, Cipso) 138.58 (d, J = 6.6, 1.3 Hz, Cipso), 128.37, 128.34, 128.25, 127.85, 126.91, 126.58 (6x s, CHar), 92.58 (dd, J = 188.3, 167.2 Hz, CFP), 63.01 (d, J = 6.9 Hz, OCH2CH3), 62.10 (d, J = 6.9 Hz, OCH2CH3), 59.76 (dd, J = 17.9, 3.5 Hz, CHCFP), 54.55 (s, CHCH3), 22.08 (s, CHCH3), 16.33 (d, J = 5.9 Hz, OCH2CH3), 16.15 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −213.48 (ddd, J = 72.0, 45.6, 21.2 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 16.01 (d, J = 72.2 Hz, 1P). IR (neat): 1253, 1019, 969 [cm−1]. MS (EI) m/z = 379.4 [M]+.
28b: 1H NMR (401 MHz): δ = 7.50–7.43 (m, 5H, Har), 7.35–7.32 (m, 5H, Har), 4.99 (ddd, J = 45.8, 5.8, 3.7 Hz, 1H, CHFP), 4.28–4.06 (m, 5H, 2x OCH2CH3, CHCFP), 3.71 (q, J = 6.4 Hz, 1H, CHCH3), 1.34 (d, J = 6.5 Hz, 3H, CHCH3), 1.18 (dd, J = 7.1, 0.5 Hz, 3H, OCH2CH3), 1.15 (dd, J = 7.1, 0.6 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 145.45 (s, Cipso) 138.43 (dd, J = 8.6, 3.6 Hz, Cipso), 128.32, 128.29, 128.20, 127.76, 126.82, 126.57 (6x s, CHar), 90.89 (dd, J = 187.8, 165.4 Hz, CFP), 63.38 (d, J = 6.4 Hz, OCH2CH3), 62.55 (d, J = 6.7 Hz, OCH2CH3), 59.66 (dd, J = 19.9, 4.6 Hz, CHCFP), 54.45 (s, CHCH3), 21.50 (s, CHCH3), 16.24 (d, J = 5.9 Hz, OCH2CH3), 16.21 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (283 MHz): δ = −215.99 (ddd, J = 76.4, 45.3, 23.5 Hz, 1F). 31P{/1H} NMR (122 MHz): δ = 16.16 (d, J = 76.4 Hz, 1P). MS (EI) m/z = 379.4 [M]+.
28c: Diagnostic signals 19F NMR (283 MHz): δ = −212.07 (ddd, J = 70.3, 45.3, 16.4 Hz). 31P{/1H} NMR (122 MHz): δ = 15.99 (d, J = 70.1 Hz).
28d: Diagnostic signals 19F NMR (283 MHz): δ = −216.77 (ddd, J = 79.8, 45.2, 24.3 Hz). 31P{/1H} NMR (122 MHz): δ = 15.88 (d, J = 79.6 Hz).
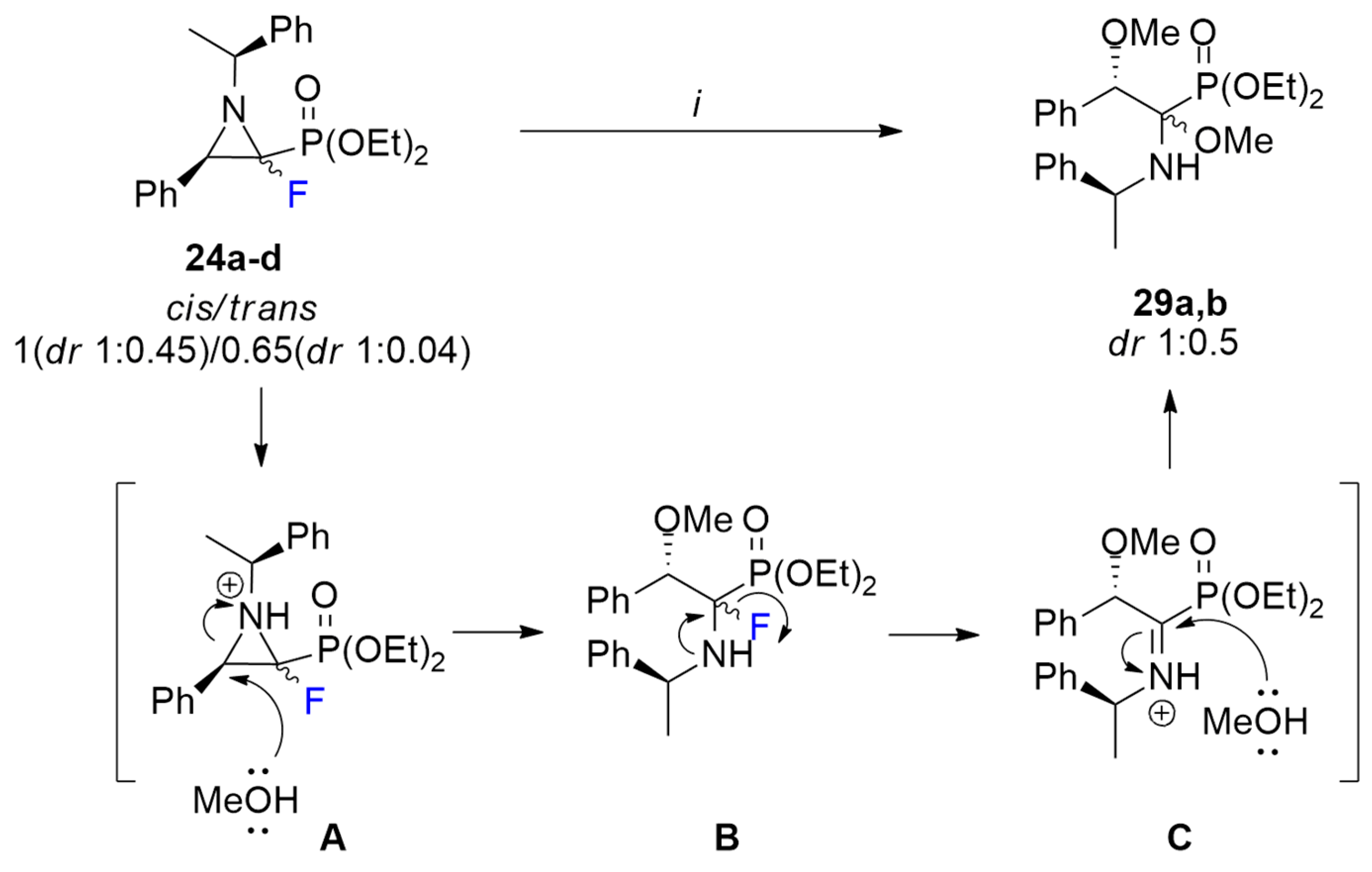
3.12. Ring Opening of Fluorinated Aziridine-2-phosphonate: Synthesis of Diethyl ((1R, 2S)-1,2-Dimethoxy-2-phenyl-1-(((S)-1-phenylethyl)amino)ethyl)phosphonate (29a) and Diethyl ((1S, 2S)-1,2-Dimethoxy-2-phenyl-1-(((S)-1-phenylethyl)amino)ethyl)phosphonate (29b)
To the mixture of aziridines 24a–d (cis/trans 1(dr 1:0.45):0.65(dr 1:0.04), 211 mg, 0.5 mmol) dissolved in MeOH (2 mL), H2SO4 (98%; 27 µL, 49 mg, 0.5 mmol) was added dropwise. The reaction was stirred at 70 °C for 2h. Next, the solution was concentrated, and the crude mixture was neutralized with aqueous NaHCO3, extracted with CH2Cl2 (3 × 6 mL), and washed with brine (6 mL). The organic layers were combined and dried over anhydrous Na2SO4. After evaporation of the solvent, the crude product was purified by column chromatography to give a mixture of diastereomers 29a,b (dr 1:0.5) as a pale-yellow oil, 149 mg, yield 63%.
29a: 1H NMR (401 MHz): δ = 7.42–7.39 (m, 5H, Har), 7.35–7.31 (m, 5H, Har), 4.33 (d, J = 11.9 Hz, 1H, CHOCH3), 4.02–3.89 (m, 4H, 2x OCH2CH3), 3.50 (q, J = 6.7 Hz, 1H, CHCH3), 3.48 (d, J = 0.7 Hz, 3H, C(P)OCH3), 3.28 (s, 3H, CHOCH3), 1.25 (d, J = 6.4 Hz, 3H, CHCH3), 1.18 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.11 (t, J = 7.0 Hz, 3H, OCH2CH3). 1H{/31P} NMR (401 MHz): δ = 7.43–7.39 (m, 5H, Har), 7.37–7.32 (m, 5H, Har), 4.32 (s, 1H, CHOCH3), 4.00–3.89 (m, 4H, 2x OCH2CH3), 3.51 (q, J = 6.7 Hz, 1H, CHCH3), 3.49 (s, 3H, C(P)OCH3), 3.29 (s, 3H, CHOCH3), 1.25 (d, J = 6.4 Hz, 3H, CHCH3), 1.19 (t, J = 7.0 Hz, 3H, OCH2CH3), 1.11 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 144.55 (s, Cipso), 138.47 (d, J = 5.5 Hz, Cipso), 104.47 (d, J = 196.3 Hz, CP), 63.38 (d, J = 14.7 Hz, CHOCH3), 63.02 (d, J = 6.7 Hz, OCH2CH3), 62.70 (d, J = 6.9 Hz, OCH2CH3), 53.73 (s, CHCH3), 52.79 (d, J = 4.0 Hz, CHOCH3), 50.49 (d, J = 8.9 Hz, C(P)OCH3), 20.91 (s, CHCH3), 16.48 (d, J = 5.9 Hz, OCH2CH3), 16.31 (d, J = 6.1 Hz, OCH2CH3). 31P{/1H} NMR (122 MHz): δ = 18.20 (s, 1P). HRMS (ESI): m/z calcd for C22H33NO5P, [M + H]+: 422.2096 found: 422.2092.
29b: 1H NMR (401 MHz): δ = 7.28–7.24 (m, 5H, Har), 7.22–7.17 (m, 5H, Har), 4.13–4.02 (m, 5H, 2x OCH2CH3, CHOCH3), 3.42 (q, J = 6.5 Hz, 1H, CHCH3), 3.34 (br s, 3H, C(P)OCH3), 3.11 (br s, 3H, CHOCH3), 1.24 (d, J = 6.6 Hz, 3H, CHCH3), 1.17 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.04 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz): δ = 145.03 (s, Cipso), 138.21 (d, J = 3.1 Hz, Cipso), 103.89 (d, J = 196.3 Hz, CP), 63.09 (d, J = 6.9 Hz, OCH2CH3), 62.58 (d, J = 6.8 Hz, OCH2CH3), 62.24 (d, J = 7.2 Hz, CHOCH3), 54.16 (s, CHCH3), 52.27 (d, J = 3.0 Hz, CHOCH3), 49.65 (d, J = 10.4 Hz, C(P)OCH3), 21.85 (s, CHCH3), 16.25 (d, J = 6.1 Hz, OCH2CH3), 16.15 (d, J = 6.2 Hz, OCH2CH3). 31P{/1H} NMR (122 MHz): δ = 17.32 (s, 1P). IR (neat): 1494, 1452, 1276, 1259, 1233, 1024, 969 [cm−1].




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
