
Molecular Dynamics Simulations on the Adsorbed Monolayers of N-Dodecyl Betaine at the Air–Water Interface

 , and
, and
Abstract
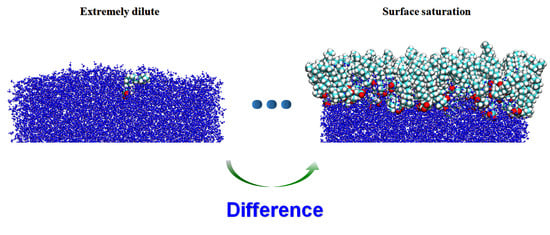
:
1. Introduction
2. Results and Discussion
2.1. Distribution of Water Molecules around Polar Heads
2.2. Hydrogen Bonds between Water Molecules and Polar Heads
2.3. Tilted Angle at the Interface
2.4. Gauche Defects of NDB Molecule
2.5. The Thickness of Different NDB Monolayers
2.6. Weak Interaction Analysis
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Clendennen, S.K.; Boaz, N.W. Chapter 14—Betaine amphoteric surfactants—Synthesis, properties, and applications. In Biobased Surfactants, 2nd ed.; Hayes, D.G., Solaiman, D.K.Y., Ashby, R.D., Eds.; AOCS Press: London, UK, 2019; pp. 447–469. [Google Scholar]
- Di Gioacchino, M.; Bruni, F.; Ricci, M.A. Aqueous solution of betaine: Hydration and aggregation. J. Mol. Liq. 2020, 318, 114253. [Google Scholar] [CrossRef]
- Kelleppan, V.T.; King, J.P.; Butler, C.S.G.; Williams, A.P.; Tuck, K.L.; Tabor, R.F. Heads or tails? The synthesis, self-assembly, properties and uses of betaine and betaine-like surfactants. Adv. Colloid Interface Sci. 2021, 297, 102528. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xiao, J.; Garamus, V.M.; Almásy, L.; Willumeit, R.; Mu, B.; Zou, A. Interaction of the biosurfactant, surfactin with betaines in aqueous solution. Langmuir 2013, 29, 10648–10657. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.D.; Garrett, P.R.; Rennie, G.K.; Thomas, R.K.; Penfold, J. Structure of an adsorbed layer of n-dodecyl-n,n-dimethylamino acetate at the air/solution interface as determined by neutron reflection. J. Phys. Chem. B 1997, 101, 7121–7126. [Google Scholar] [CrossRef]
- Erfani, A.; Khosharay, S.; Flynn, N.H.; Ramsey, J.D.; Aichele, C.P. Effect of zwitterionic betaine surfactant on interfacial behavior of bovine serum albumin (bsa). J. Mol. Liq. 2020, 318, 114067. [Google Scholar] [CrossRef]
- McCoy, T.M.; King, J.P.; Moore, J.E.; Kelleppan, V.T.; Sokolova, A.V.; de Campo, L.; Manohar, M.; Darwish, T.A.; Tabor, R.F. The effects of small molecule organic additives on the self-assembly and rheology of betaine wormlike micellar fluids. J. Colloid Interface Sci. 2019, 534, 518–532. [Google Scholar] [CrossRef]
- Gao, S.; Song, Z.; Lan, F.; Li, P.; Zhang, A.; Hu, J.; Jiang, Q. Studies on physicochemical properties and aggregation behavior of two pairs of betaine surfactants. Ind. Eng. Chem. Res 2019, 58, 22260–22272. [Google Scholar] [CrossRef]
- Hussain, S.M.S.; Fogang, L.T.; Kamal, M.S. Synthesis and performance evaluation of betaine type zwitterionic surfactants containing different degrees of ethoxylation. J. Mol. Struct. 2018, 1173, 983–989. [Google Scholar] [CrossRef]
- Chandrakar, A.; Bhargava, B.L. Aqueous solutions of hydroxyl-functionalized ionic liquids: Molecular dynamics studies. J. Mol. Graph. Model. 2020, 101, 107721. [Google Scholar] [CrossRef]
- Dash, S.; Chowdhury, U.D.; Bhargava, B.L. The effect of external salts on the aggregation of the multiheaded surfactants: All-atom molecular dynamics studies. J. Mol. Graph. Model. 2022, 111, 108110. [Google Scholar] [CrossRef]
- Nadimi, H.; Housaindokht, M.R.; Moosavi, F. The effect of anion on aggregation of amino acid ionic liquid: Atomistic simulation. J. Mol. Graph. Model. 2020, 101, 107733. [Google Scholar] [CrossRef]
- Peredo-Mancilla, D.; Dominguez, H. Adsorption of phenol molecules by sodium dodecyl sulfate (sds) surfactants deposited on solid surfaces: A computer simulation study. J. Mol. Graph. Model. 2016, 65, 108–112. [Google Scholar] [CrossRef]
- Gao, S.; Bao, X.; Yu, L.; Wang, H.; Li, J.; Chen, X. Molecular dynamics study of “quasi-gemini” surfactant at n-decane/water interface: The synergistic effect of hydrophilic headgroups and hydrophobic tails of surfactants on the interface properties. Colloids Surf. A 2022, 634, 127899. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, X.; Dong, L.; Liu, G.; Xia, Q.; Yuan, S. Molecular dynamics study on the effect of surfactant mixture on their packing states in mixed micelles. Colloids Surf. A 2021, 631, 127714. [Google Scholar] [CrossRef]
- Liu, Z.-Y.; Xu, Z.; Zhou, H.; Wang, Y.; Liao, Q.; Zhang, L.; Zhao, S. Interfacial behaviors of betaine and binary betaine/carboxylic acid mixtures in molecular dynamics simulation. J. Mol. Liq. 2017, 240, 412–419. [Google Scholar] [CrossRef]
- Cai, H.-Y.; Zhang, Y.; Liu, Z.-Y.; Li, J.-G.; Gong, Q.-T.; Liao, Q.; Zhang, L.; Zhao, S. Molecular dynamics simulation of binary betaine and anionic surfactant mixtures at decane—Water interface. J. Mol. Liq. 2018, 266, 82–89. [Google Scholar] [CrossRef]
- Su, L.; Sun, J.; Ding, F.; Gao, X.; Zheng, L. Effect of molecular structure on synergism in mixed zwitterionic/anionic surfactant system: An experimental and simulation study. J. Mol. Liq. 2021, 322, 114933. [Google Scholar] [CrossRef]
- Ergin, G.; Lbadaoui-Darvas, M.; Takahama, S. Molecular structure inhibiting synergism in charged surfactant mixtures: An atomistic molecular dynamics simulation study. Langmuir 2017, 33, 14093–14104. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Liu, G.; Wang, H.; Xia, Q.; Yuan, S. Exploring relationship of the state of n-dodecyl betaine in the solution monomer, at the interface and in the micelle via configurational entropy. Colloids Surf. A 2020, 600, 124975. [Google Scholar] [CrossRef]
- Wei, Y.; Gao, F.; Wang, H.; Liu, G.; Xia, Q.; Yuan, S. A molecular dynamics study combining with entropy calculation on the packing state of hydrophobic chains in micelle interior. J. Mol. Liq. 2019, 283, 860–866. [Google Scholar] [CrossRef]
- van der Spoel, D.; van Maaren, P.J.; Larsson, P.; Tîmneanu, N. Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. J. Phys. Chem. B 2006, 110, 4393–4398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Maginn, E.J. Direct correlation between ionic liquid transport properties and ion pair lifetimes: A molecular dynamics study. J. Phys. Chem. Lett. 2015, 6, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.L.; McCabe, C.; Cummings, P.T. Molecular simulations of liquid-liquid interfacial properties: Water--n-alkane and water-methanol-n-alkane systems. Phys. Rev. E 2003, 67, 011603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Wang, H.; Xia, Q.; Yuan, S. A molecular dynamics study on the dependence of phase behaviors and structural properties of two-dimensional interfacial monolayer on surface area. Appl. Surf. Sci. 2018, 459, 741–748. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Yue, W. Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation. Phys. Rev. B 1986, 33, 8800–8802. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lindahl, E.; Abraham, M.; Hess, B.; van der Spoel, D. Gromacs 2019 Source Code. Available online: https://doi.org/10.5281/zenodo.2424362 (accessed on 21 July 2023).
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The resp model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | 0.44 nm2 | 0.53 nm2 | 0.79 nm2 | 1 nm2 | 2.56 nm2 | 4 nm2 | 16 nm2 | 64 nm2 |
| Hydrogen bond number | 4.14 ± 0.08 | 4.21 ± 0.09 | 4.31 ± 0.11 | 4.36 ± 0.13 | 4.47 ± 0.19 | 4.61 ± 0.24 | 4.85 ± 0.45 | 4.98 ± 0.87 |
| Coordination number | 5.31 ± 0.11 | 5.47 ± 0.13 | 5.62 ± 0.16 | 5.70 ± 0.19 | 5.89 ± 0.29 | 6.12 ± 0.36 | 6.60 ± 0.64 | 6.89 ± 1.21 |
| Lifetime | 14.84 ps | 14.17 ps | 12.94 ps | 12.31 ps | 10.78 ps | 9.75 ps | 8.63 ps | 8.10 ps |
| Interfacial | Thickness | (nm) | ||||||
|---|---|---|---|---|---|---|---|---|
| System | 0.44 nm2 | 0.53 nm2 | 0.79 nm2 | 1 nm2 | 2.25 nm2 | 4 nm2 | 16 nm2 | 64 nm2 |
| Thickness | 1.50 | 1.48 | 1.40 | 1.33 | 0.97 | 0.74 | 0.62 | 0.60 |
| 0.44 nm2 | 0.53 nm2 | 0.79 nm2 | 1 nm2 | 2.56 nm2 | 4 nm2 | 16 nm2 | 64 nm2 | |
| NDB | 288 | 242 | 162 | 128 | 50 | 32 | 8 | 2 |
| H2O | 16,586 | 16,667 | 16,824 | 16,904 | 17,042 | 17,074 | 17,115 | 17,128 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Cao, L.; Jiang, Y.; Huang, Z.; Liu, G.; Wei, Y.; Xia, Q. Molecular Dynamics Simulations on the Adsorbed Monolayers of N-Dodecyl Betaine at the Air–Water Interface. Molecules 2023, 28, 5580. https://doi.org/10.3390/molecules28145580
Zhang C, Cao L, Jiang Y, Huang Z, Liu G, Wei Y, Xia Q. Molecular Dynamics Simulations on the Adsorbed Monolayers of N-Dodecyl Betaine at the Air–Water Interface. Molecules. 2023; 28(14):5580. https://doi.org/10.3390/molecules28145580
Chicago/Turabian StyleZhang, Chengfeng, Lulu Cao, Yongkang Jiang, Zhiyao Huang, Guokui Liu, Yaoyao Wei, and Qiying Xia. 2023. "Molecular Dynamics Simulations on the Adsorbed Monolayers of N-Dodecyl Betaine at the Air–Water Interface" Molecules 28, no. 14: 5580. https://doi.org/10.3390/molecules28145580
APA StyleZhang, C., Cao, L., Jiang, Y., Huang, Z., Liu, G., Wei, Y., & Xia, Q. (2023). Molecular Dynamics Simulations on the Adsorbed Monolayers of N-Dodecyl Betaine at the Air–Water Interface. Molecules, 28(14), 5580. https://doi.org/10.3390/molecules28145580