
Recent Advances in the Enantioselective Radical Reactions


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
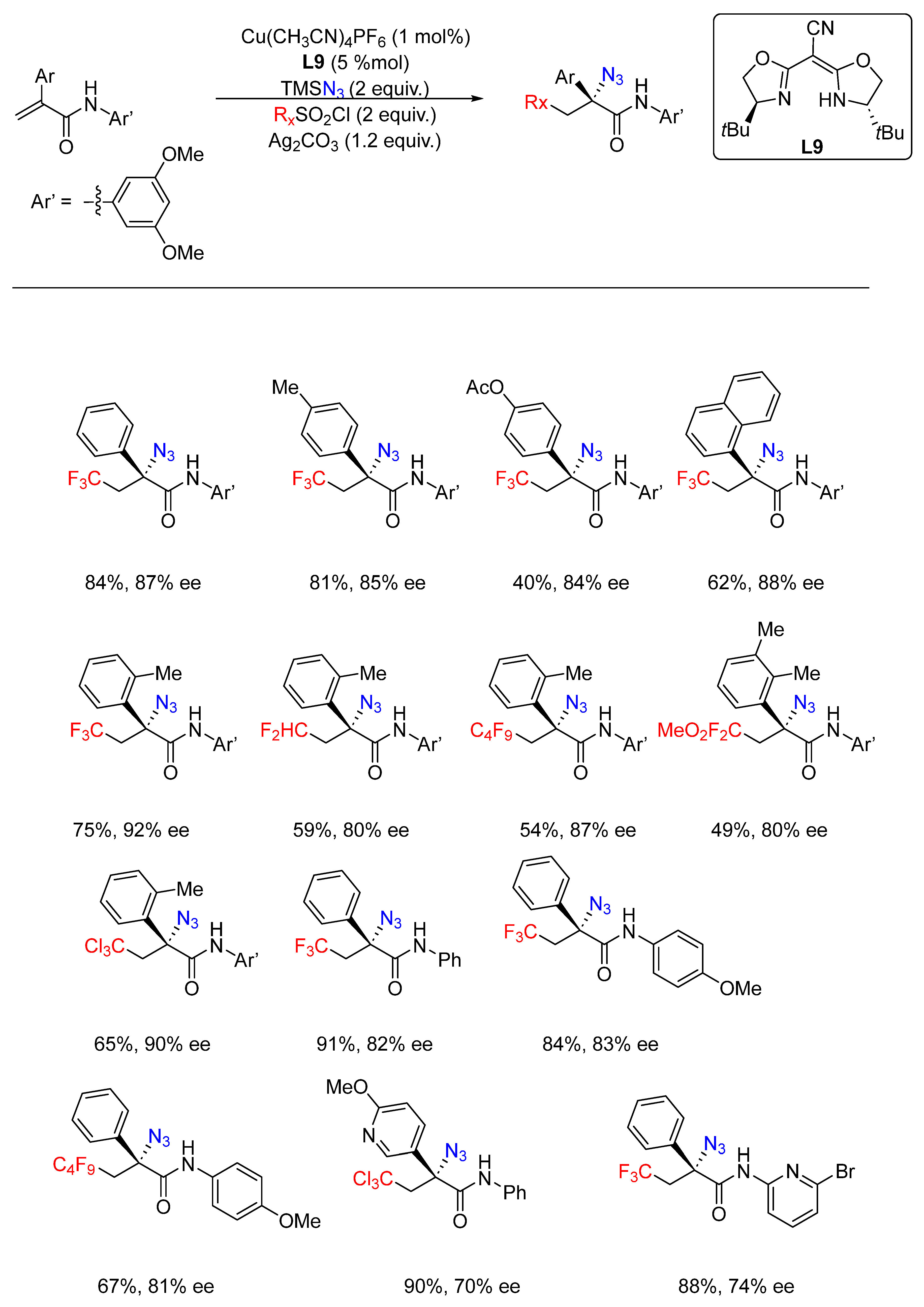{kind=link}
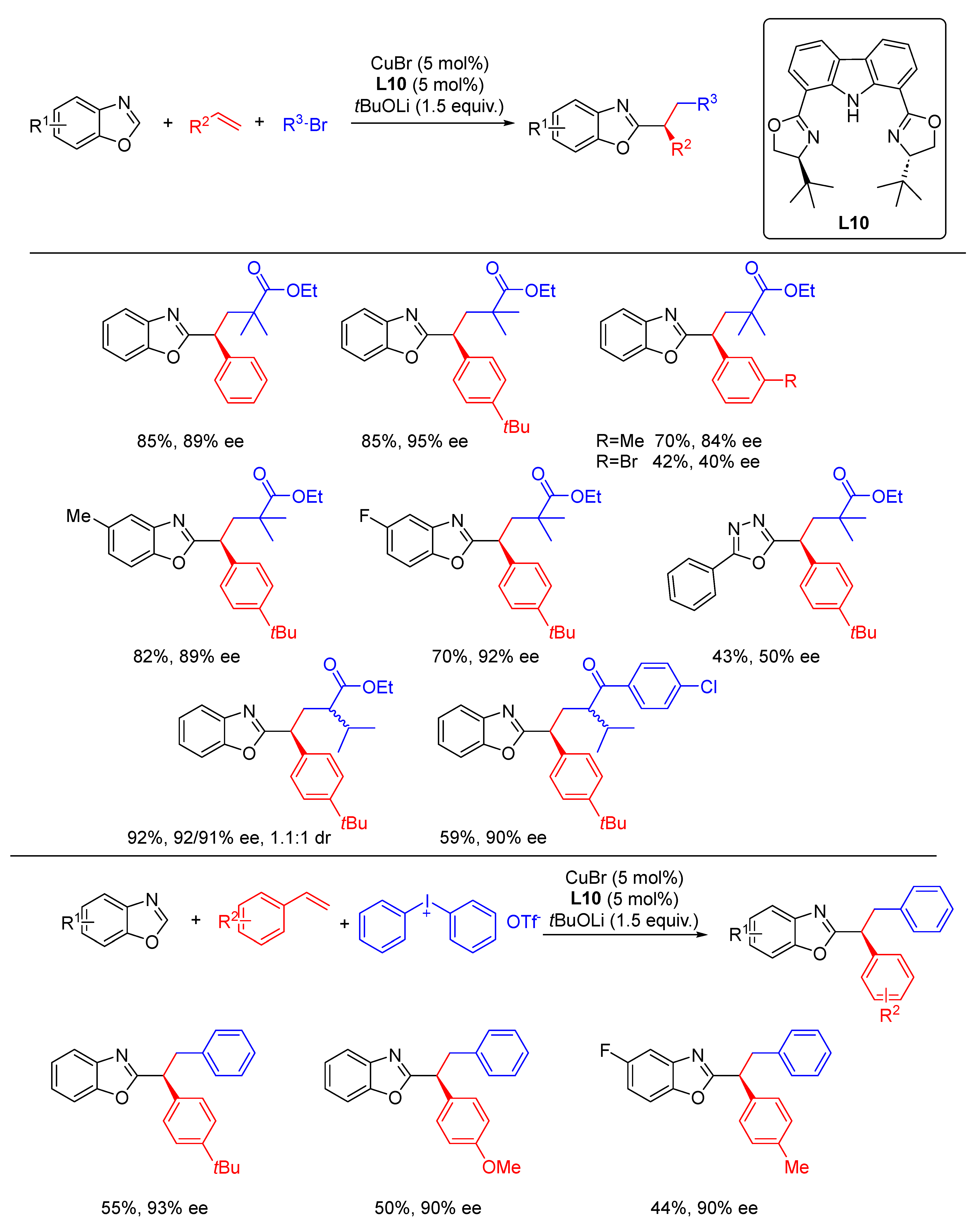{kind=link}
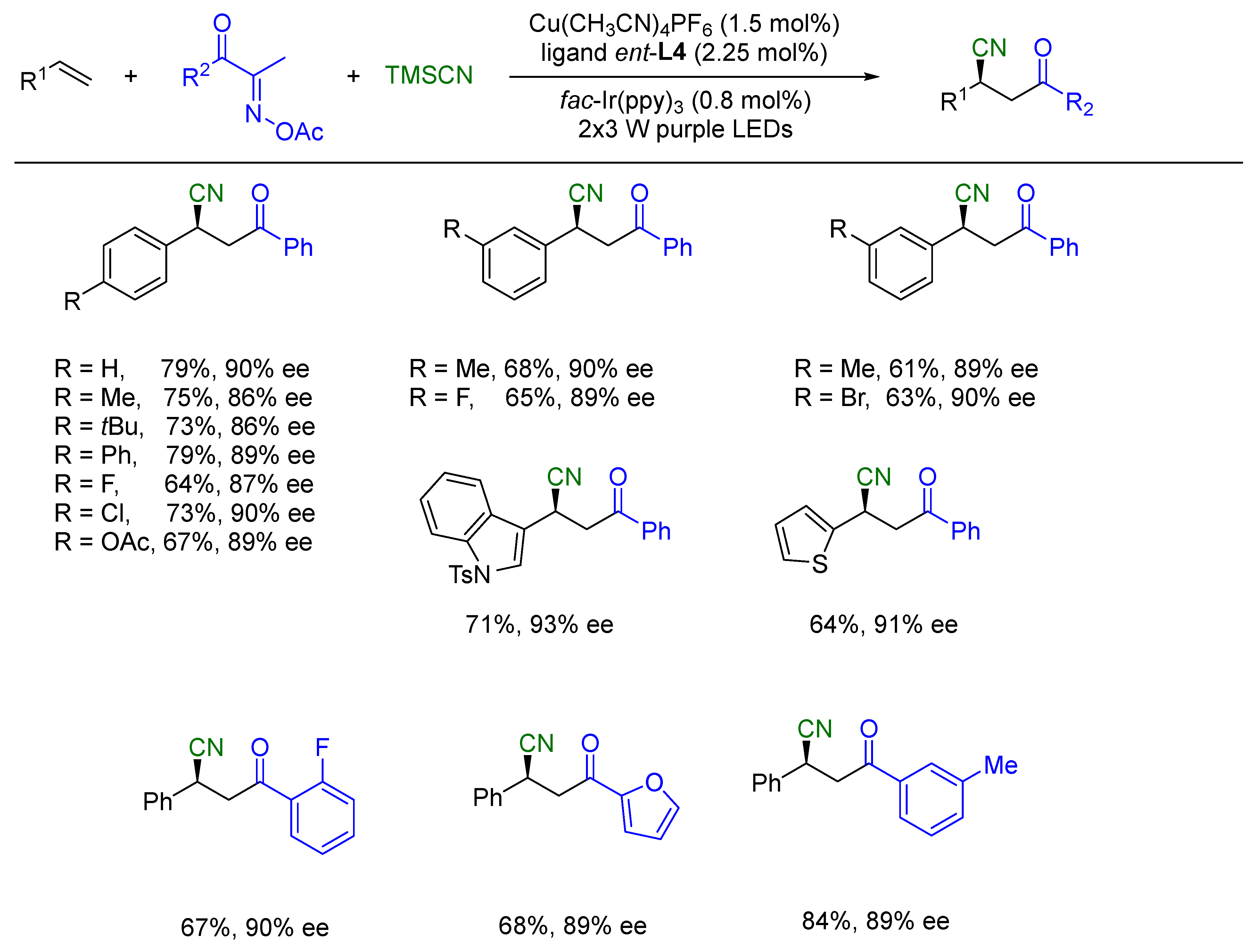{kind=link}
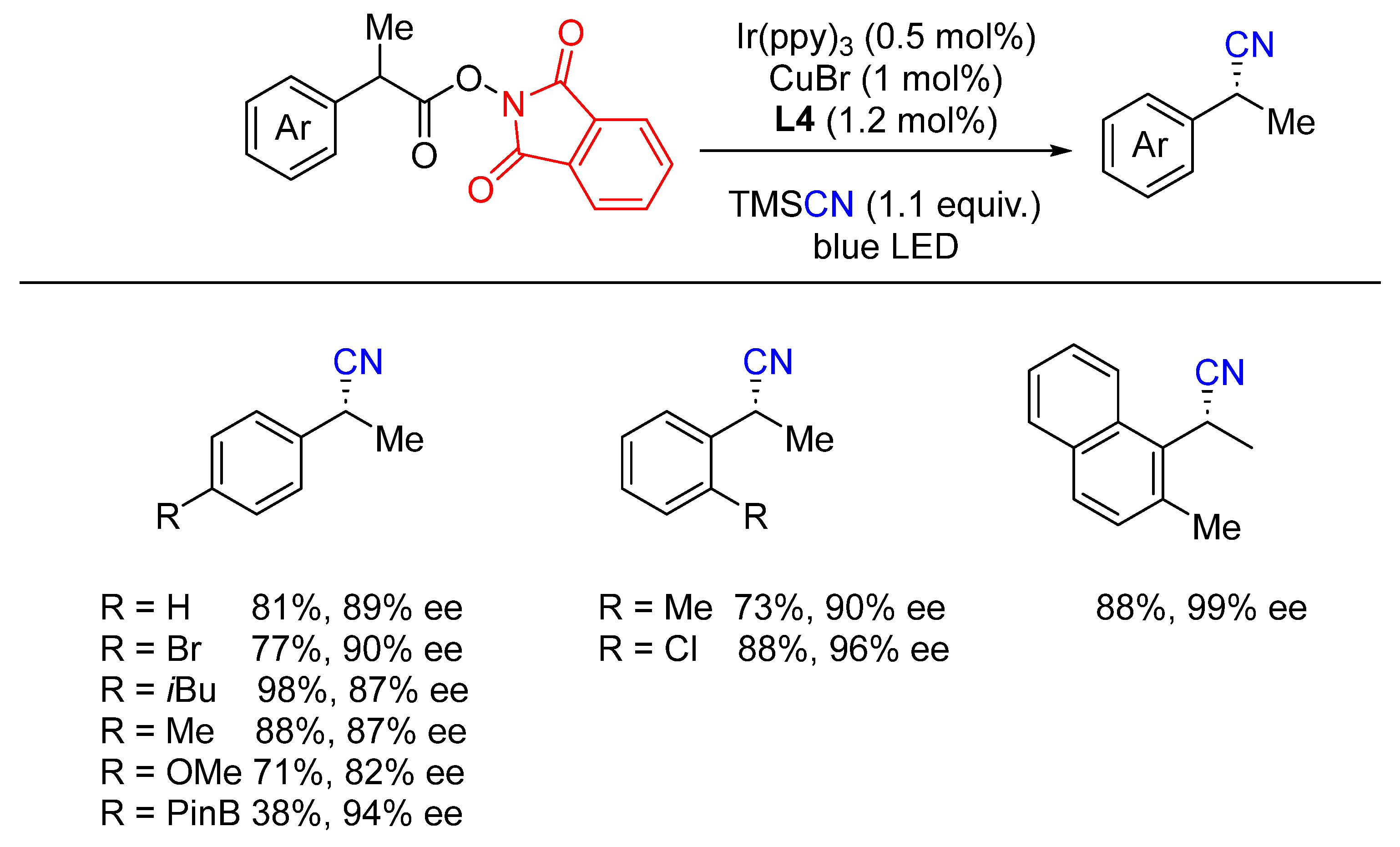{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
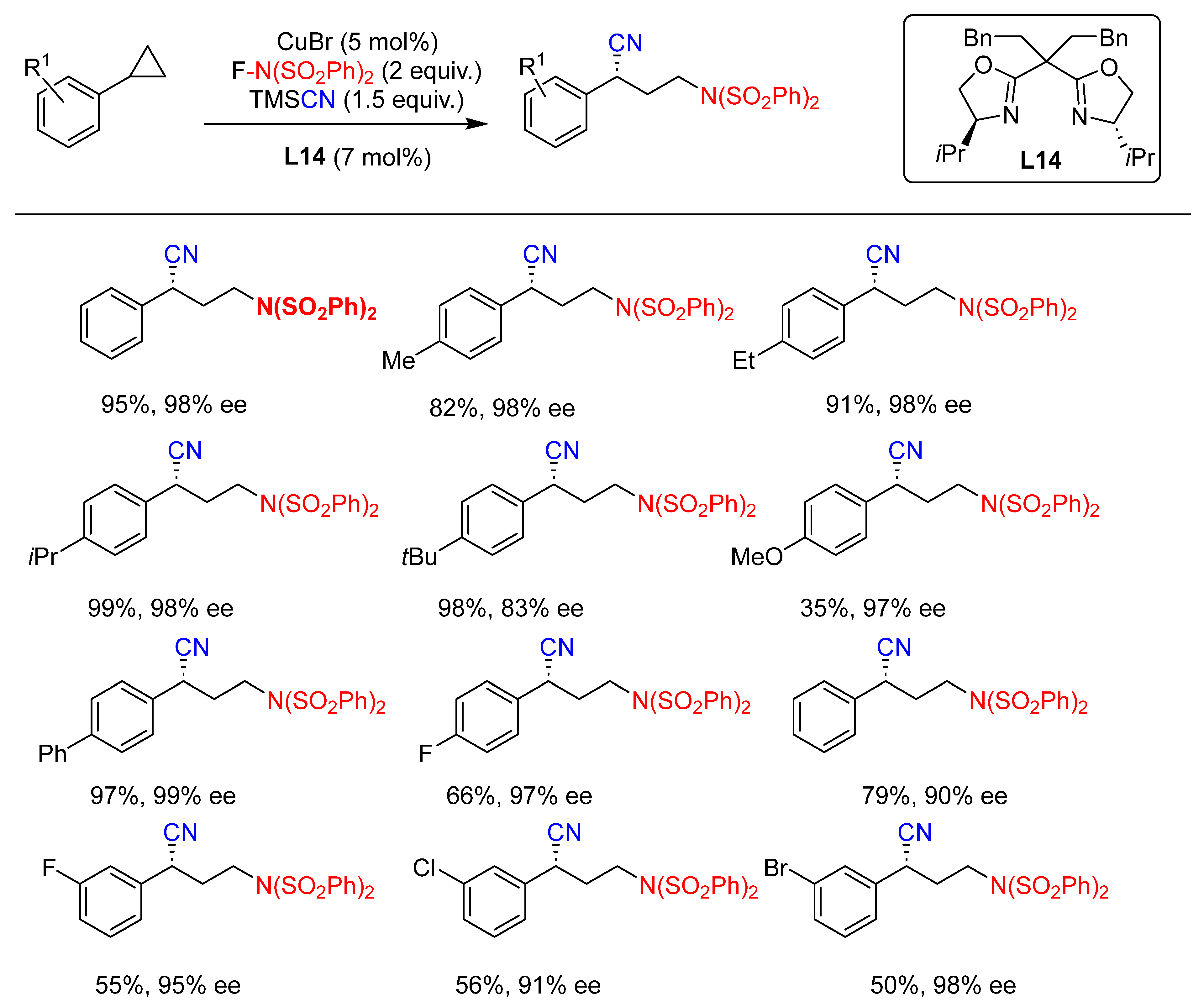{kind=link}
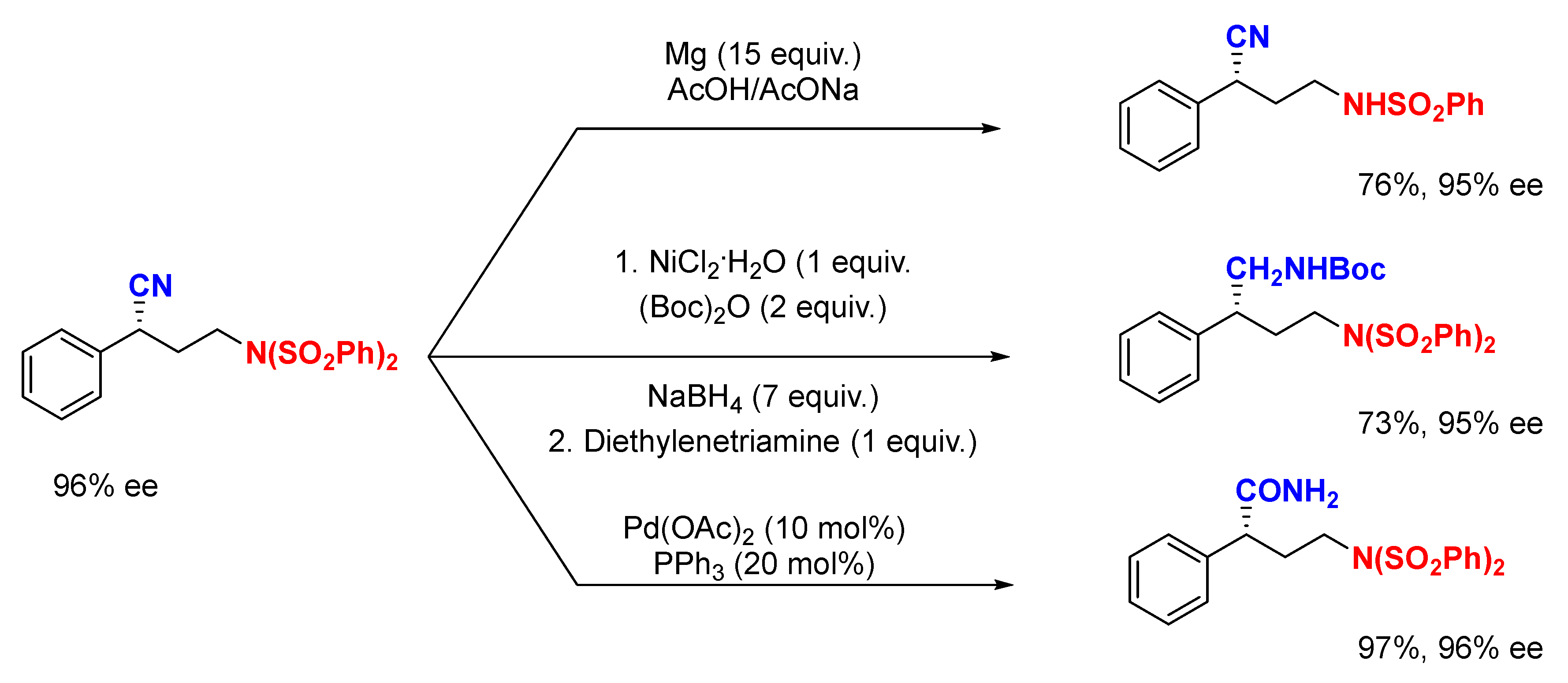{kind=link}
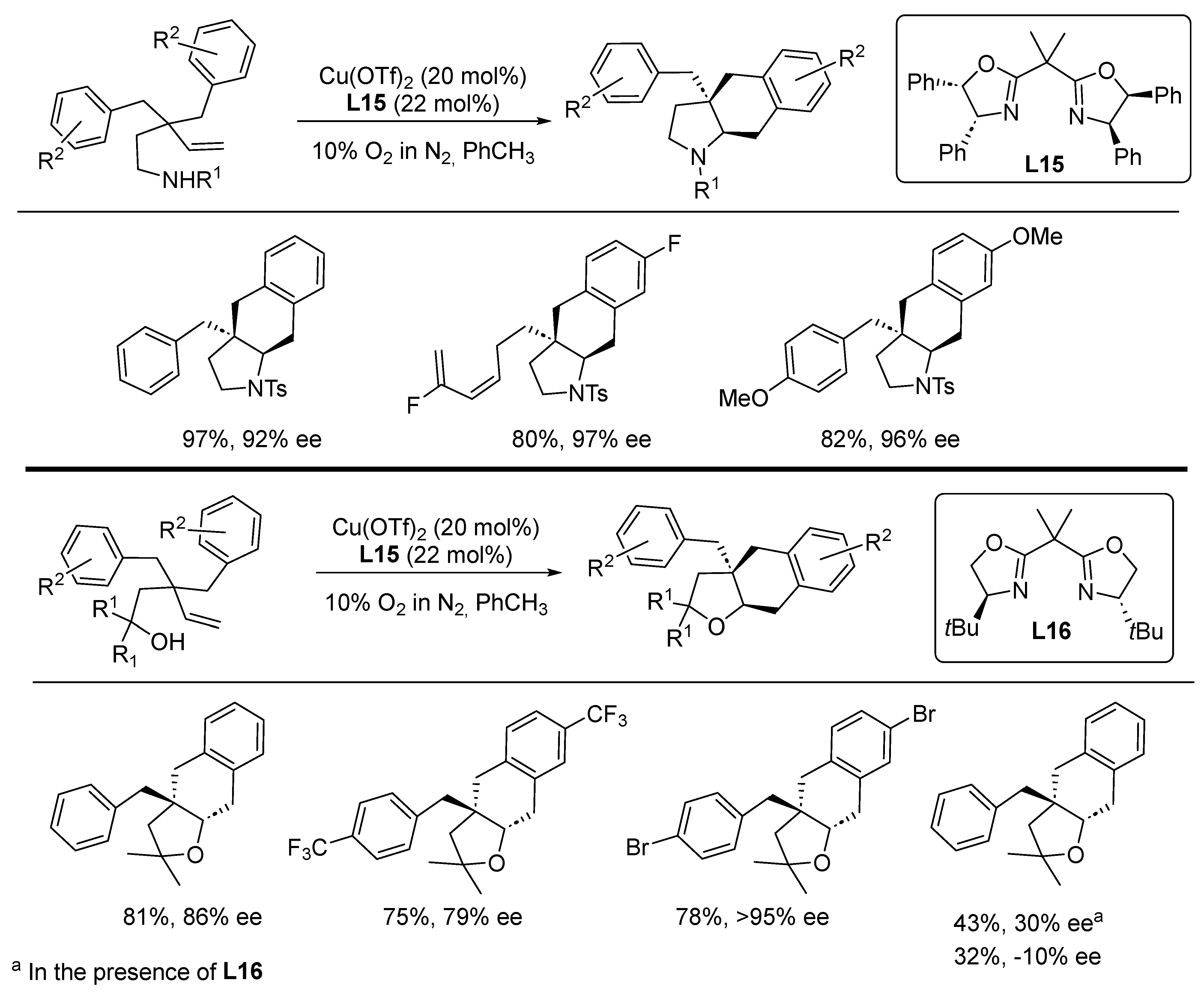{kind=link}
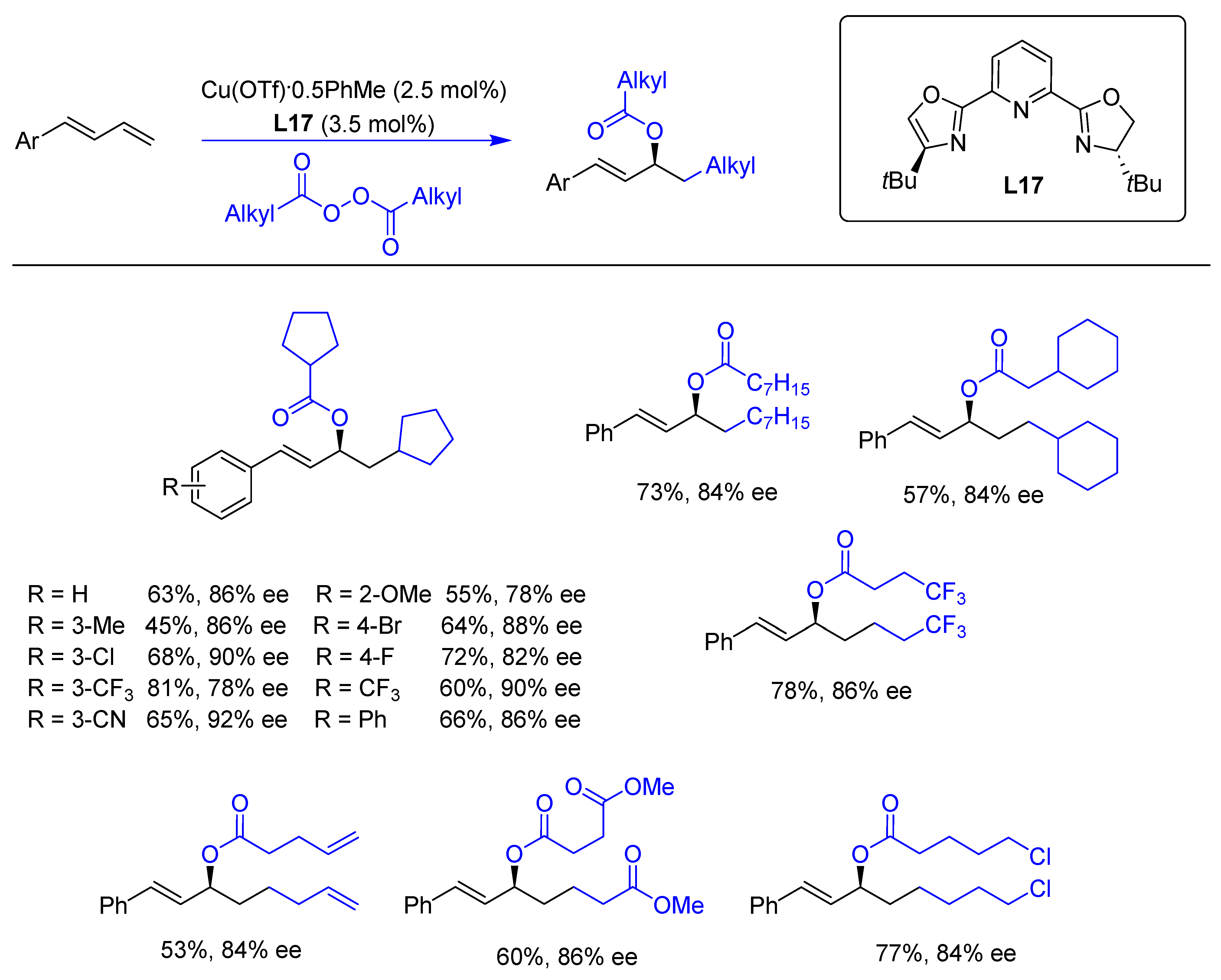{kind=link}
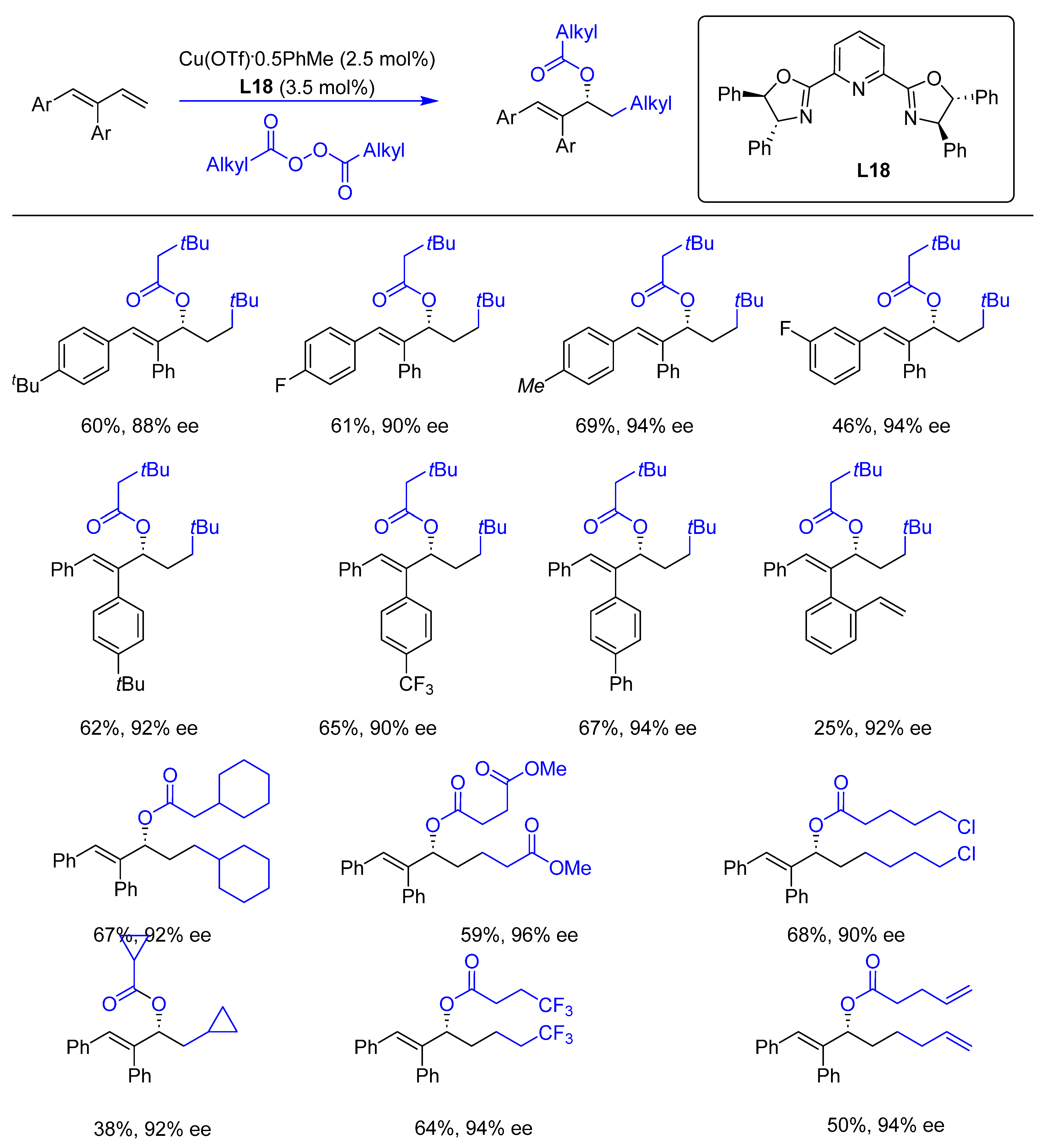{kind=link}
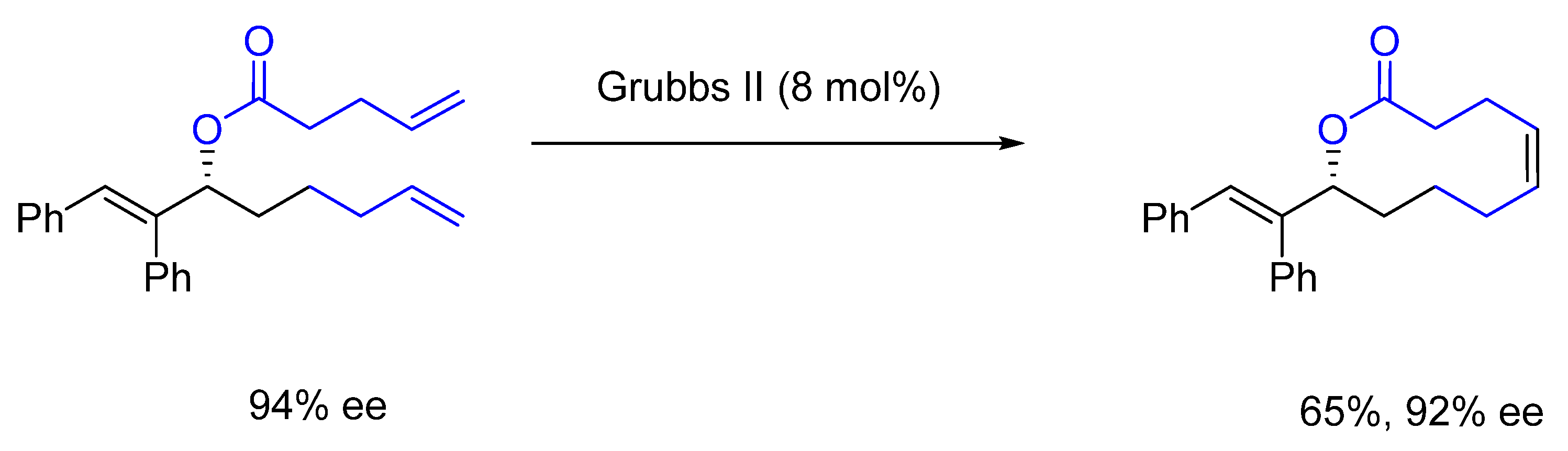{kind=link}
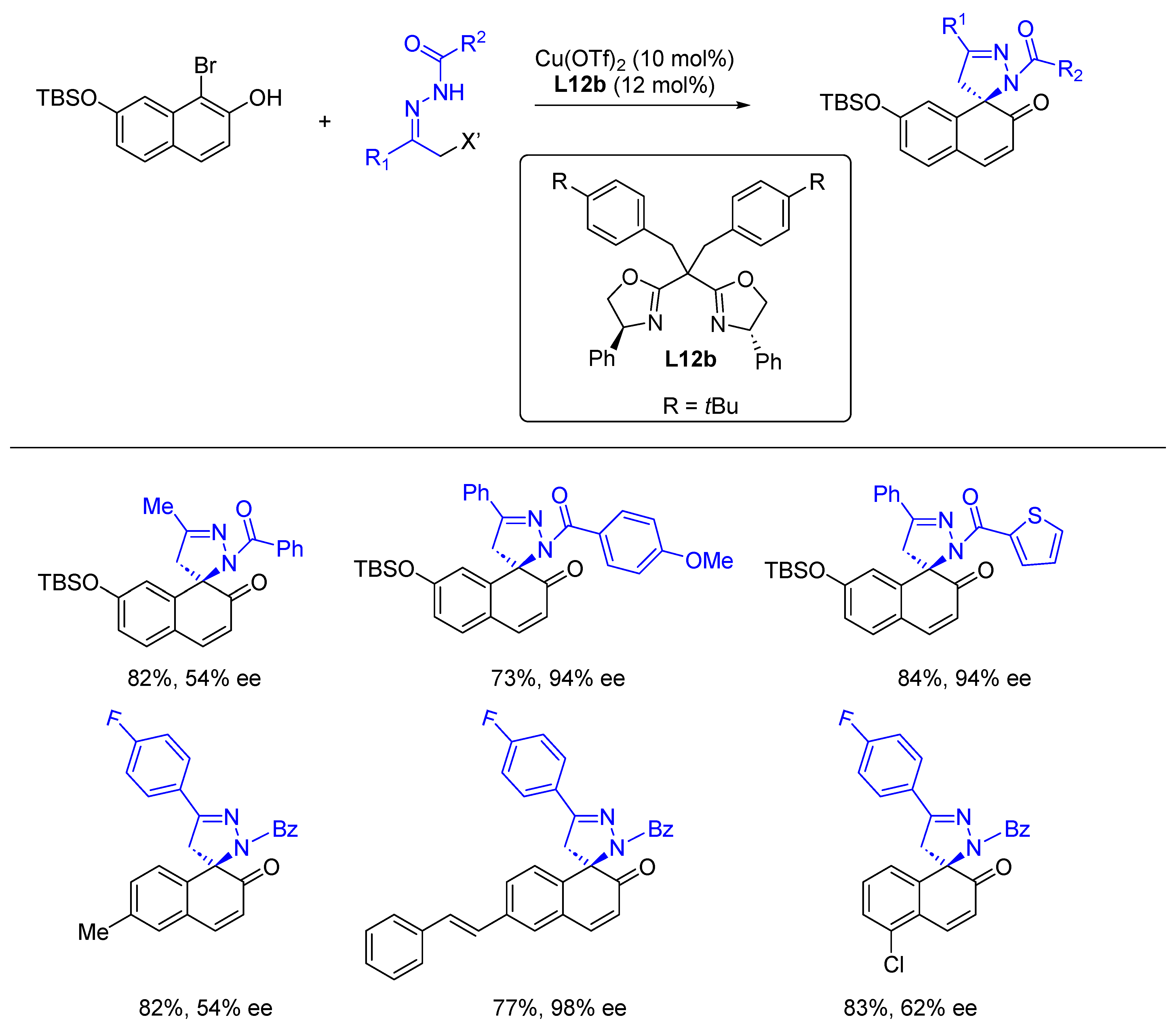{kind=link}
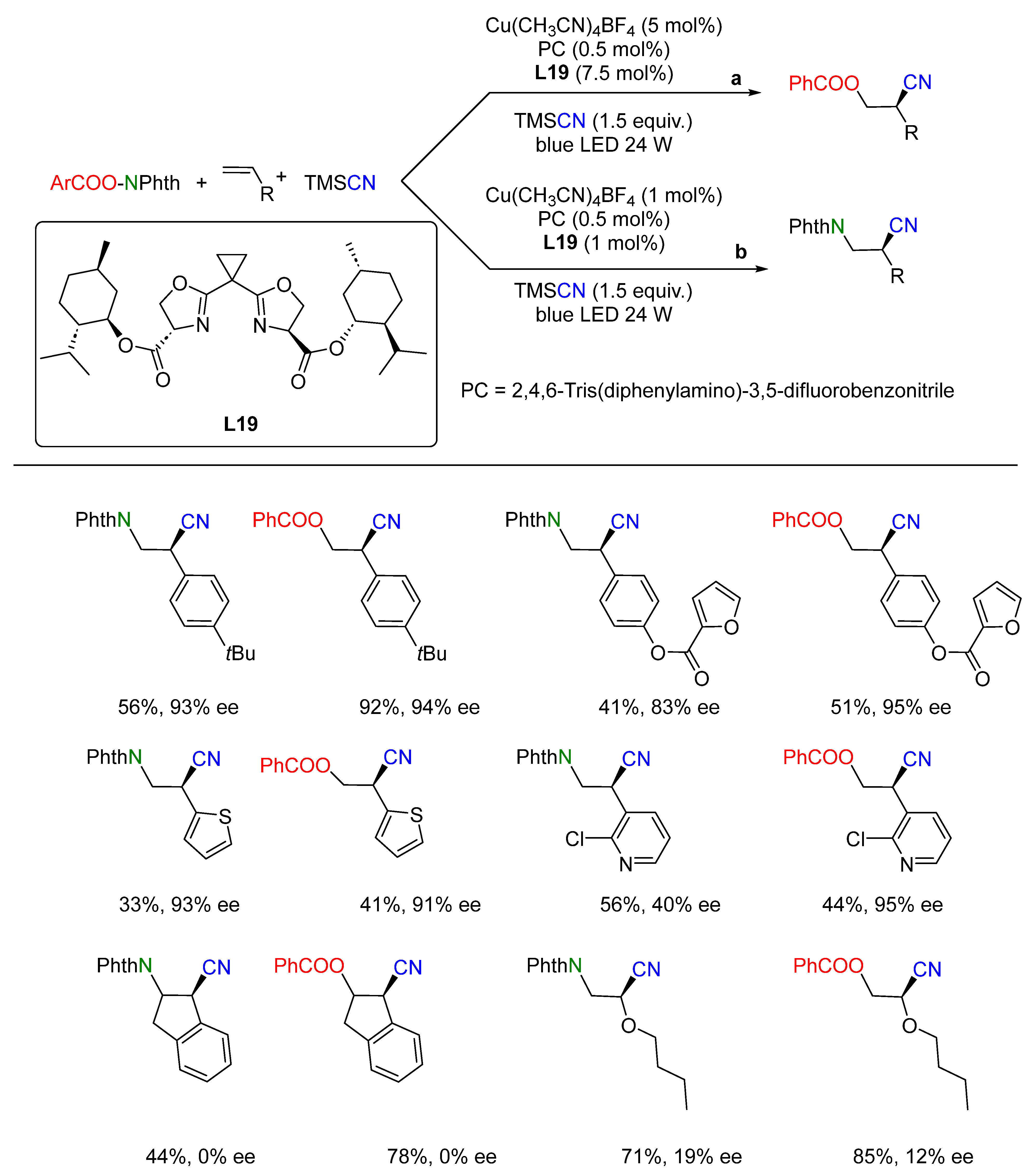{kind=link}
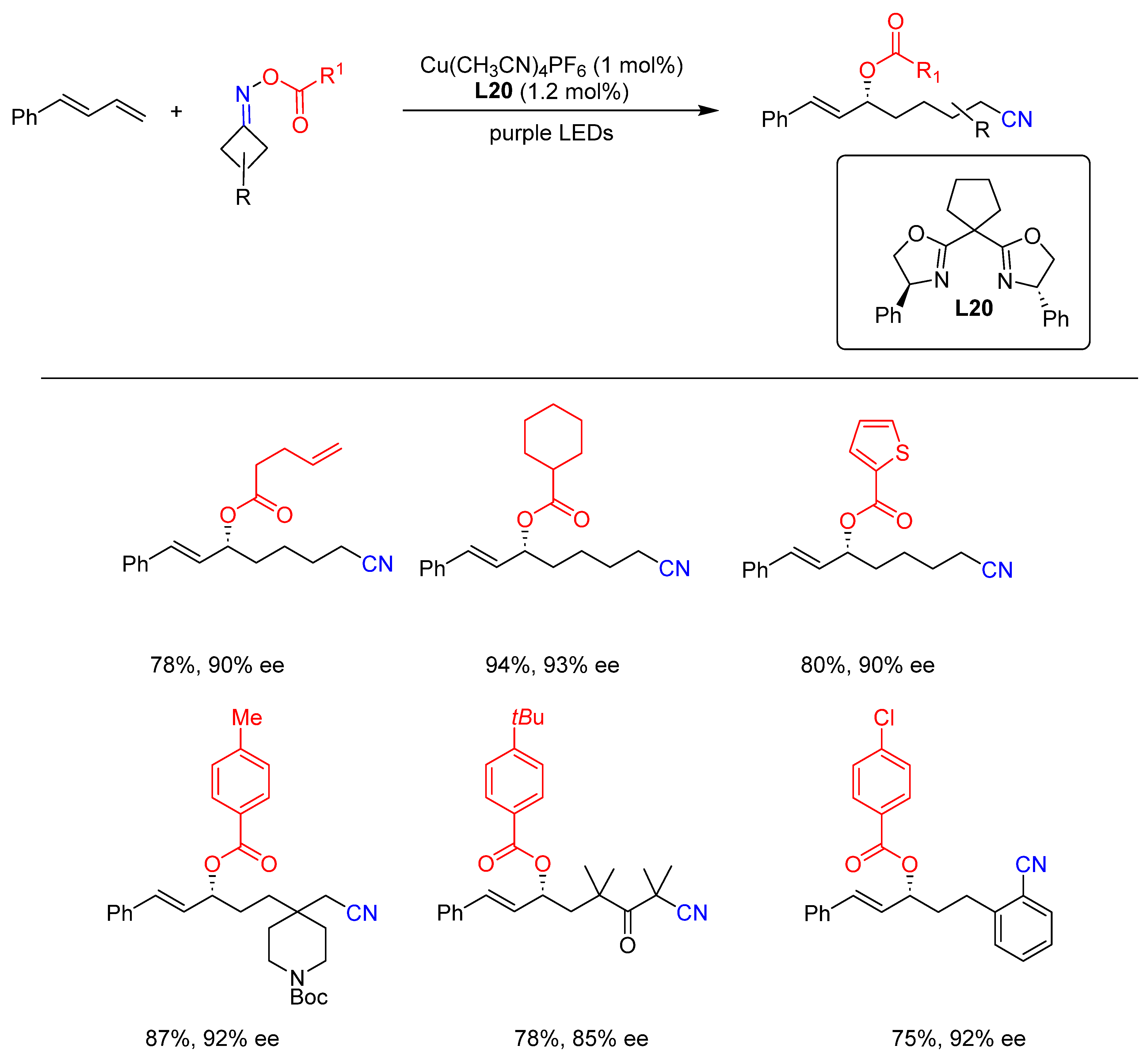{kind=link}
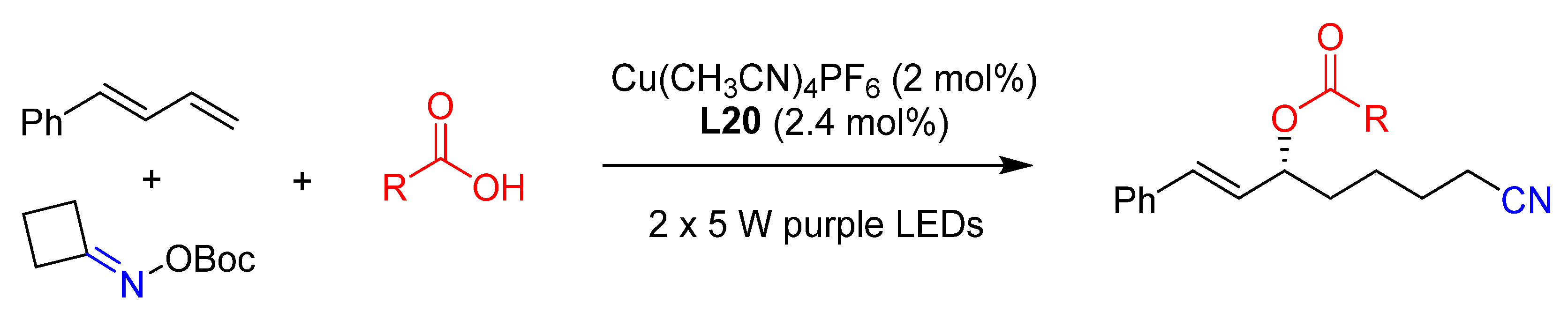{kind=link}
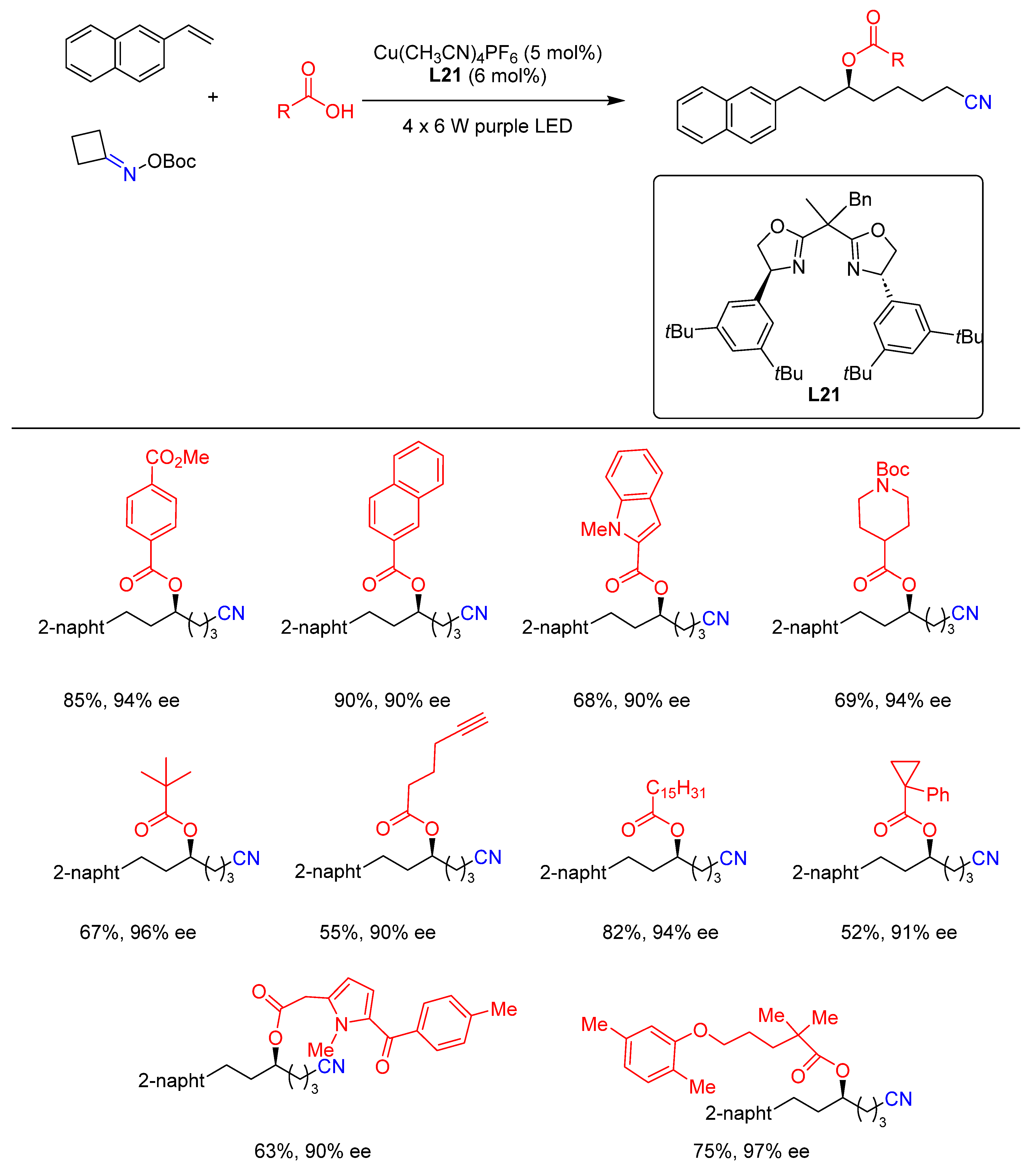{kind=link}
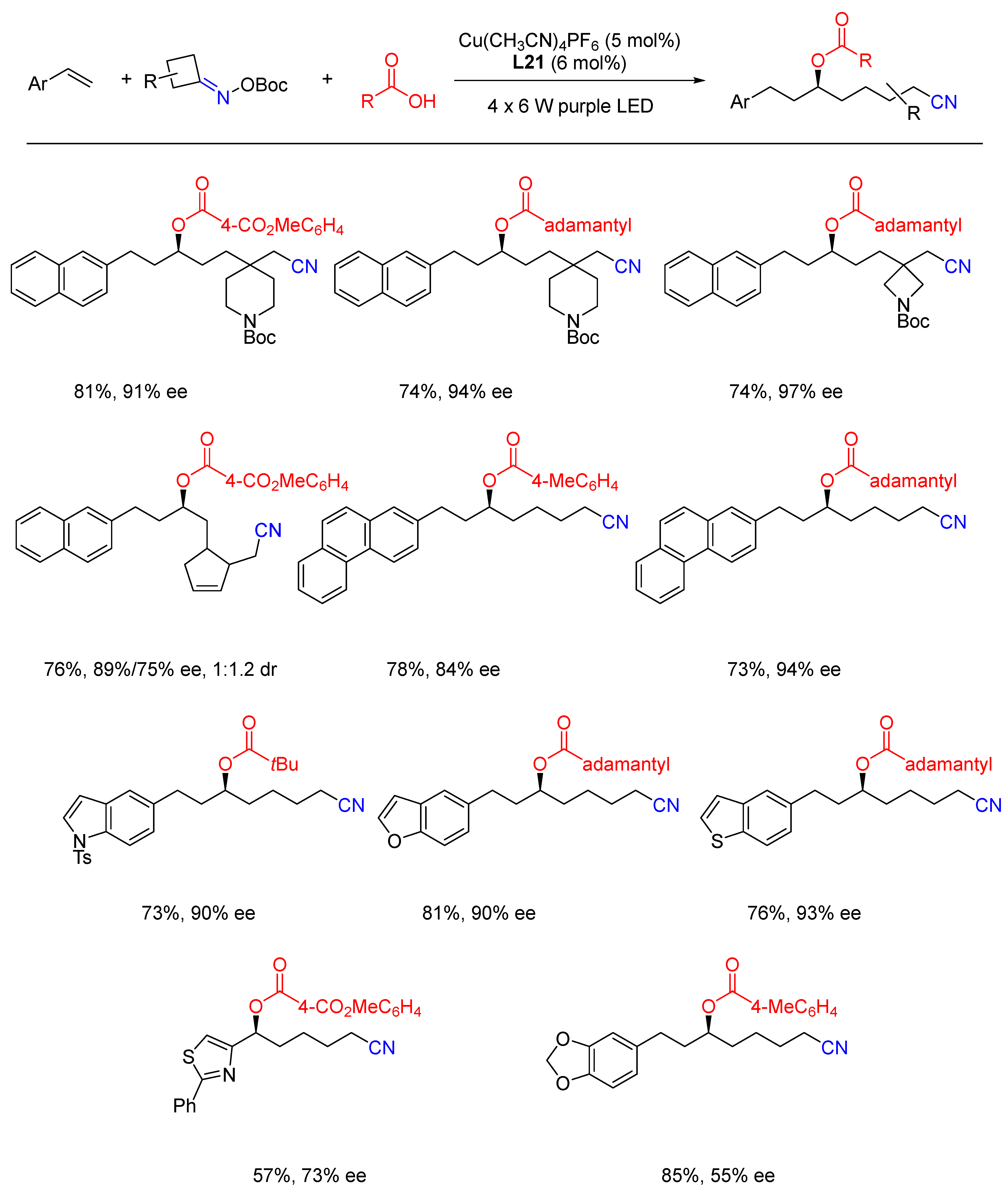{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
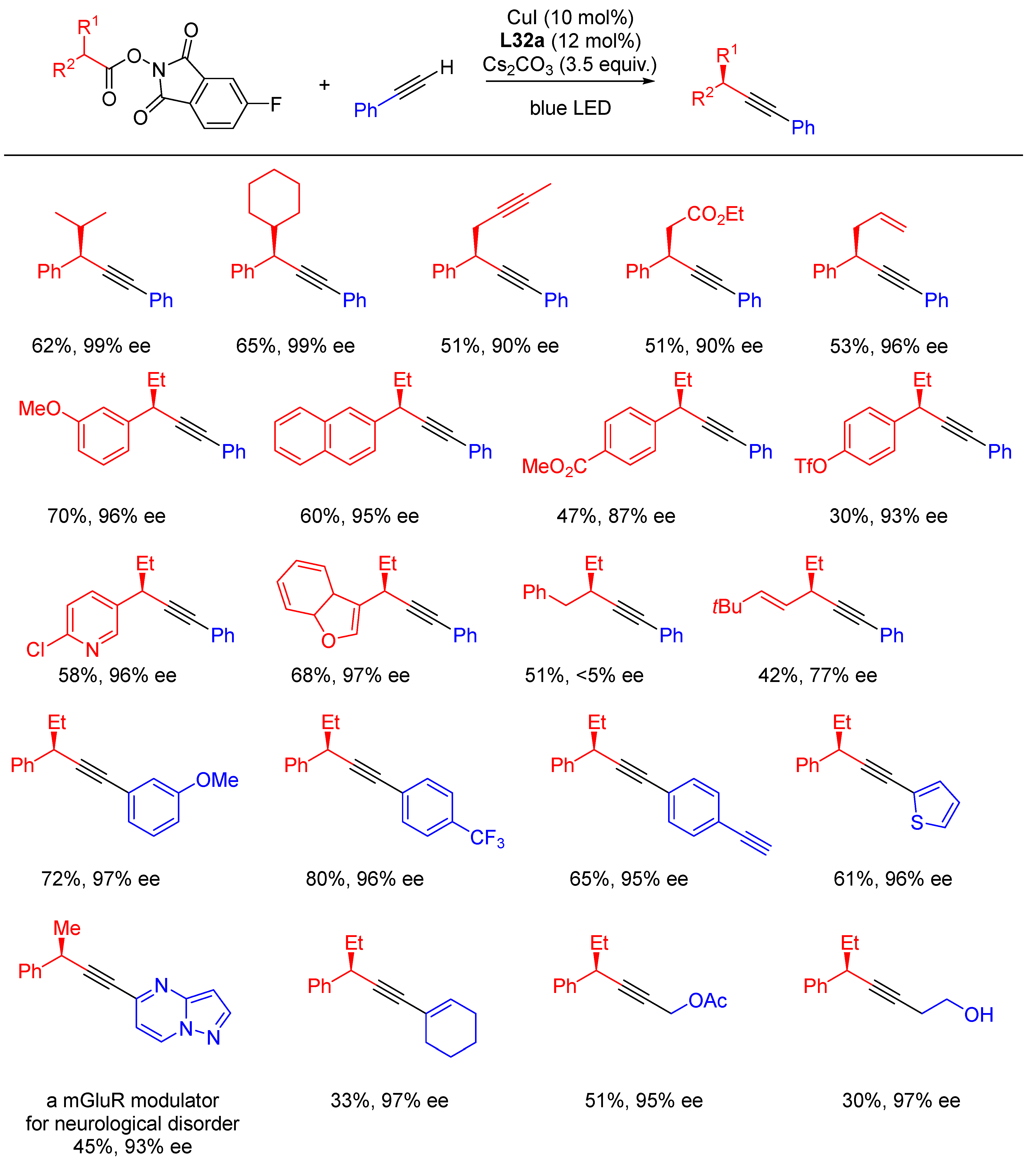{kind=link}
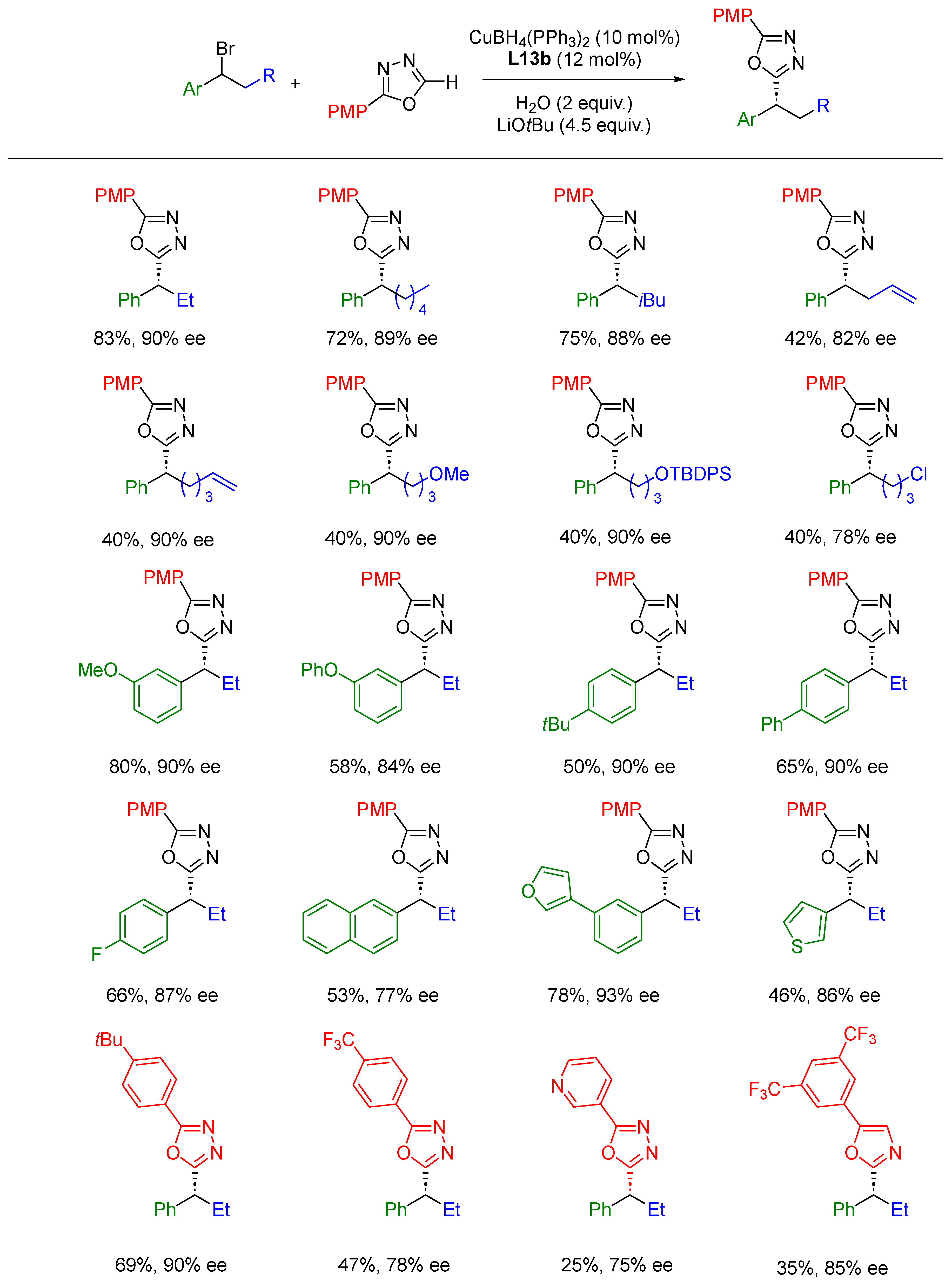{kind=link}
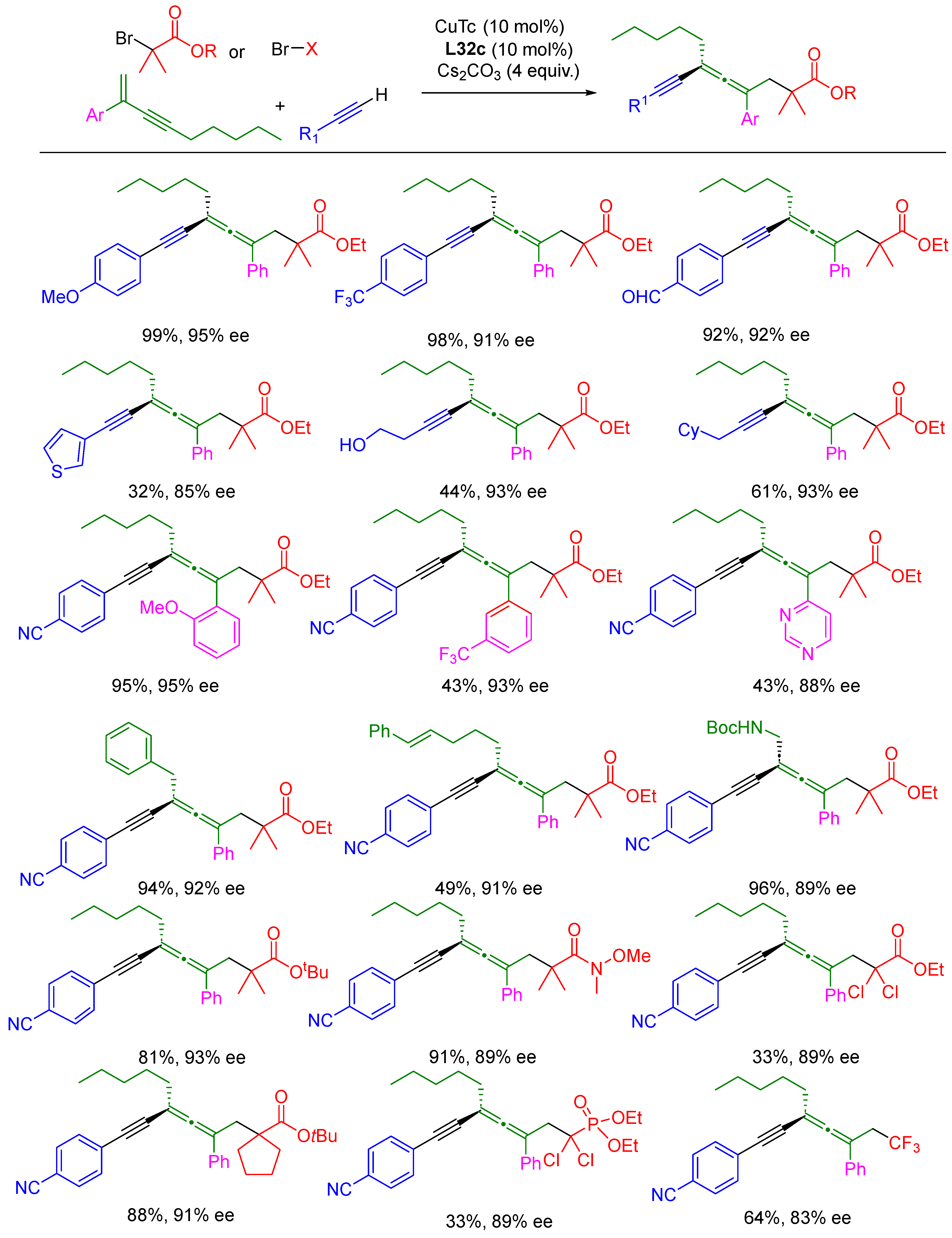{kind=link}
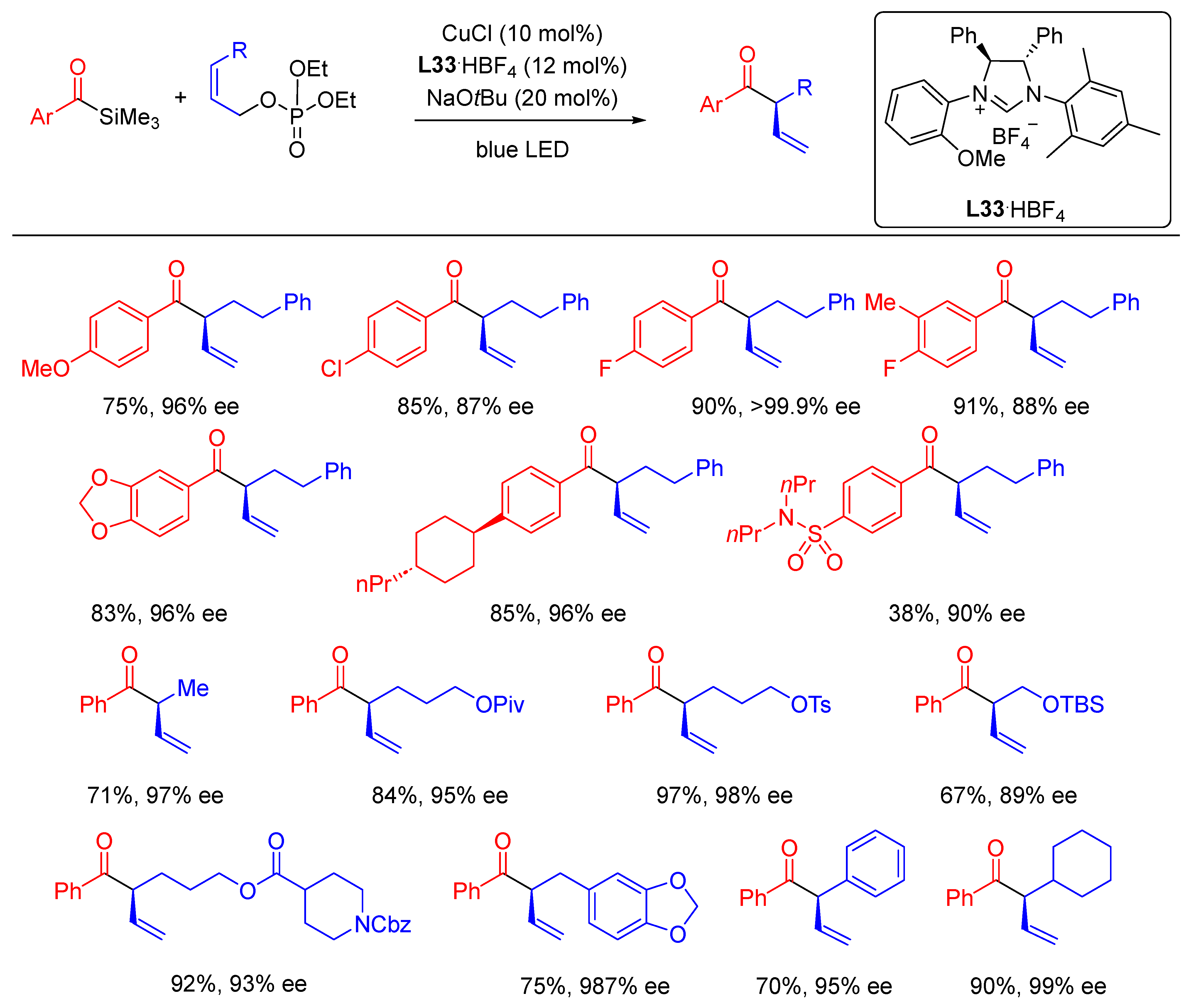{kind=link}
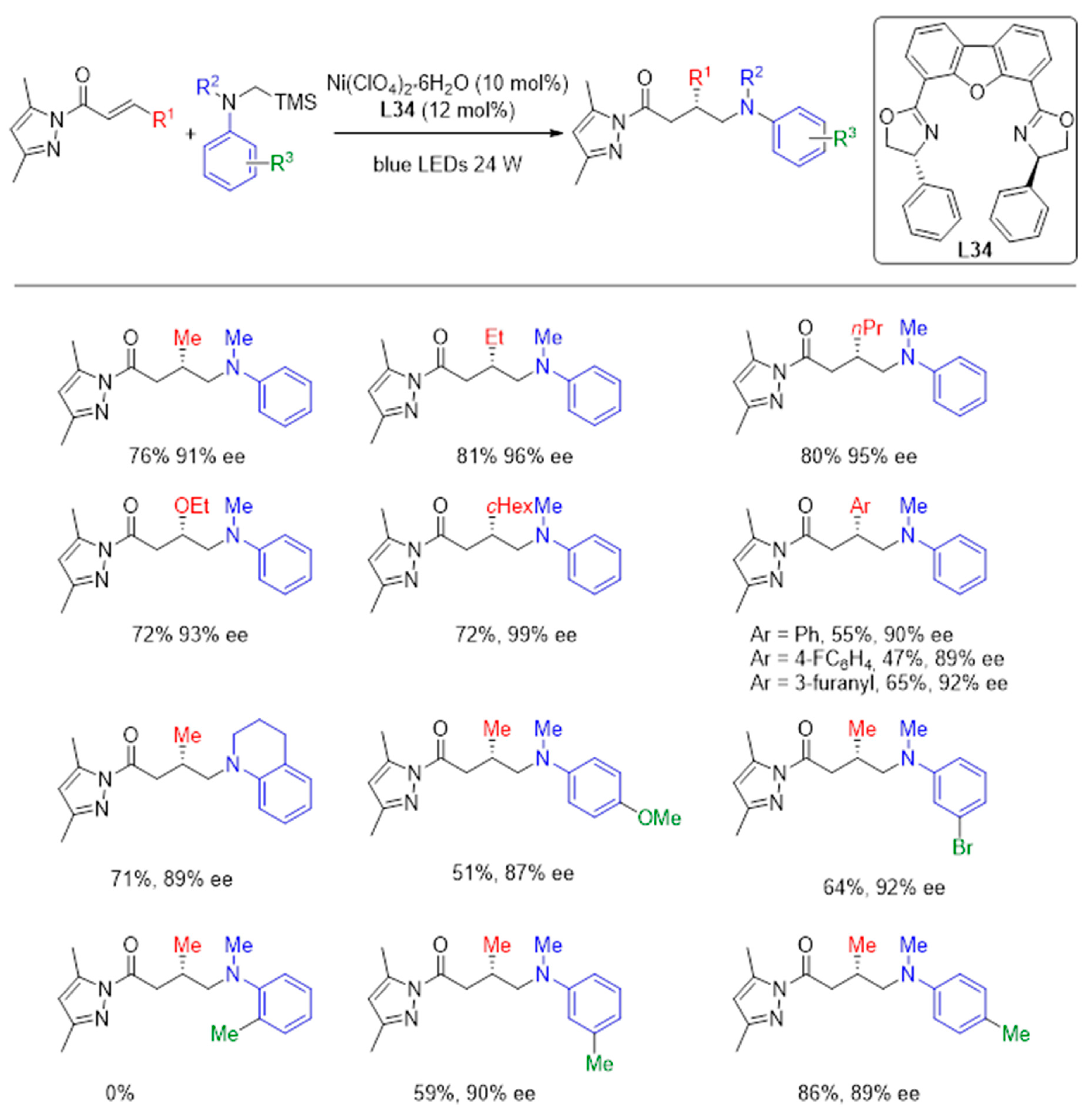{kind=link}
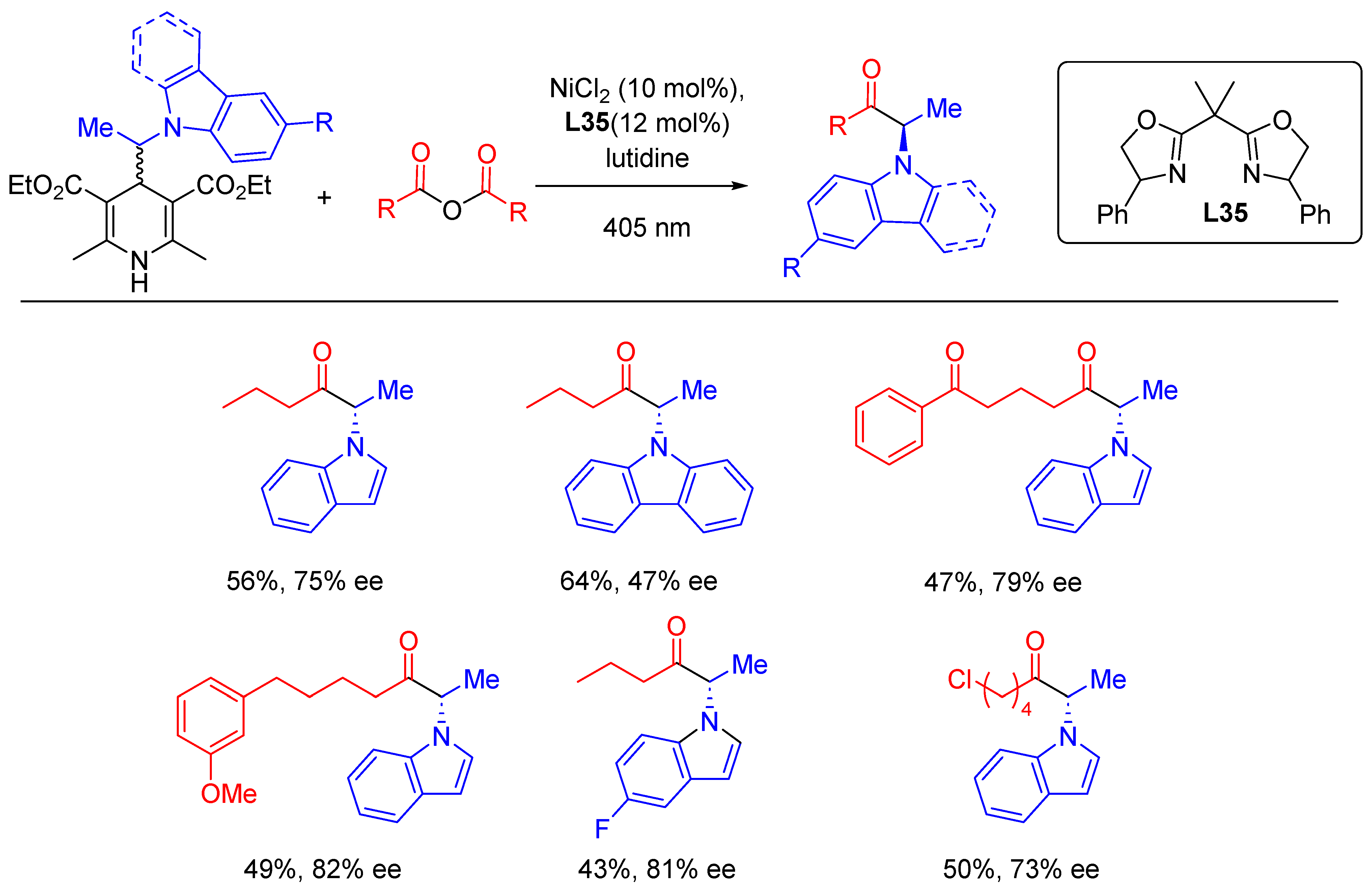{kind=link}
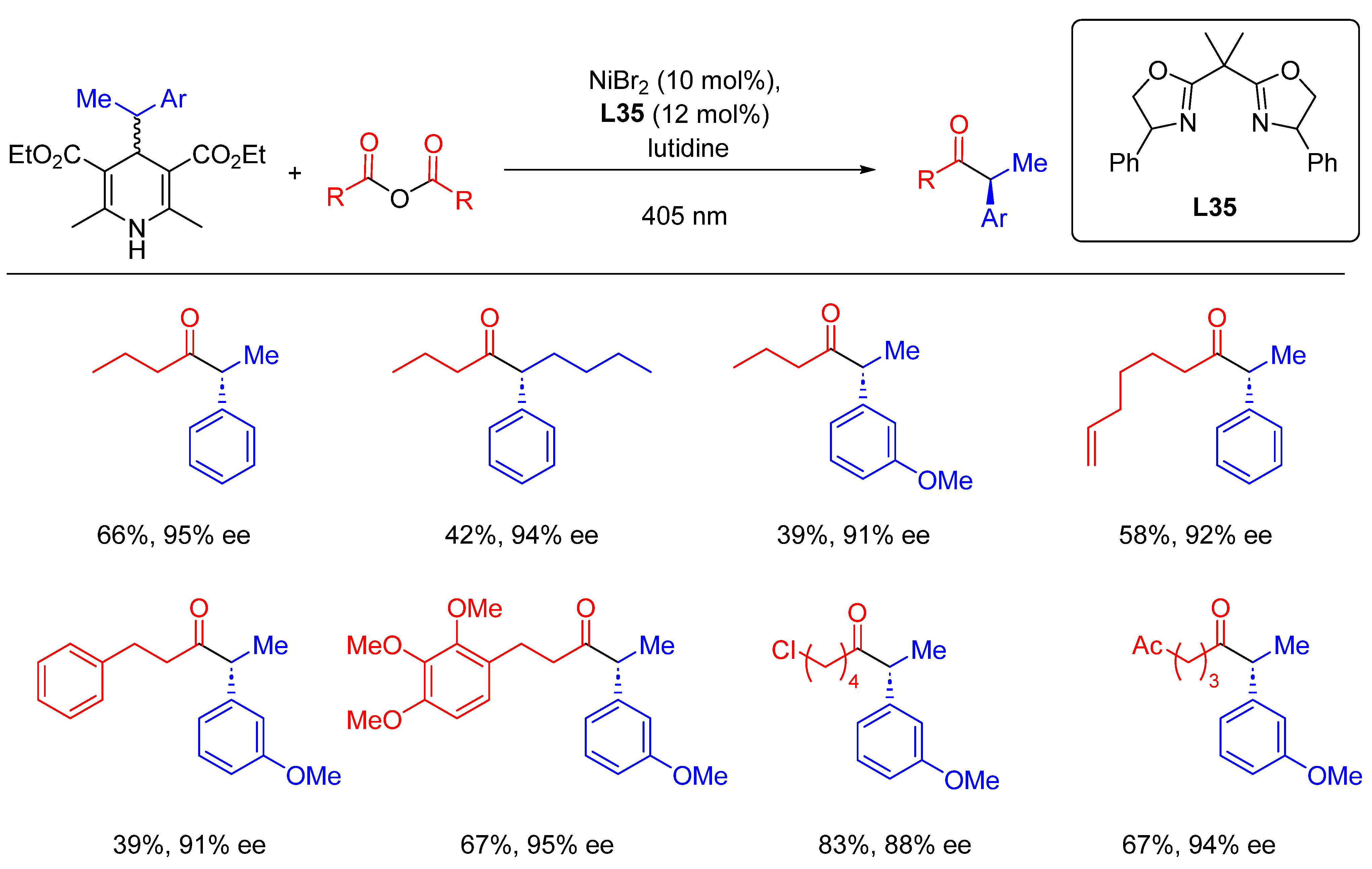{kind=link}
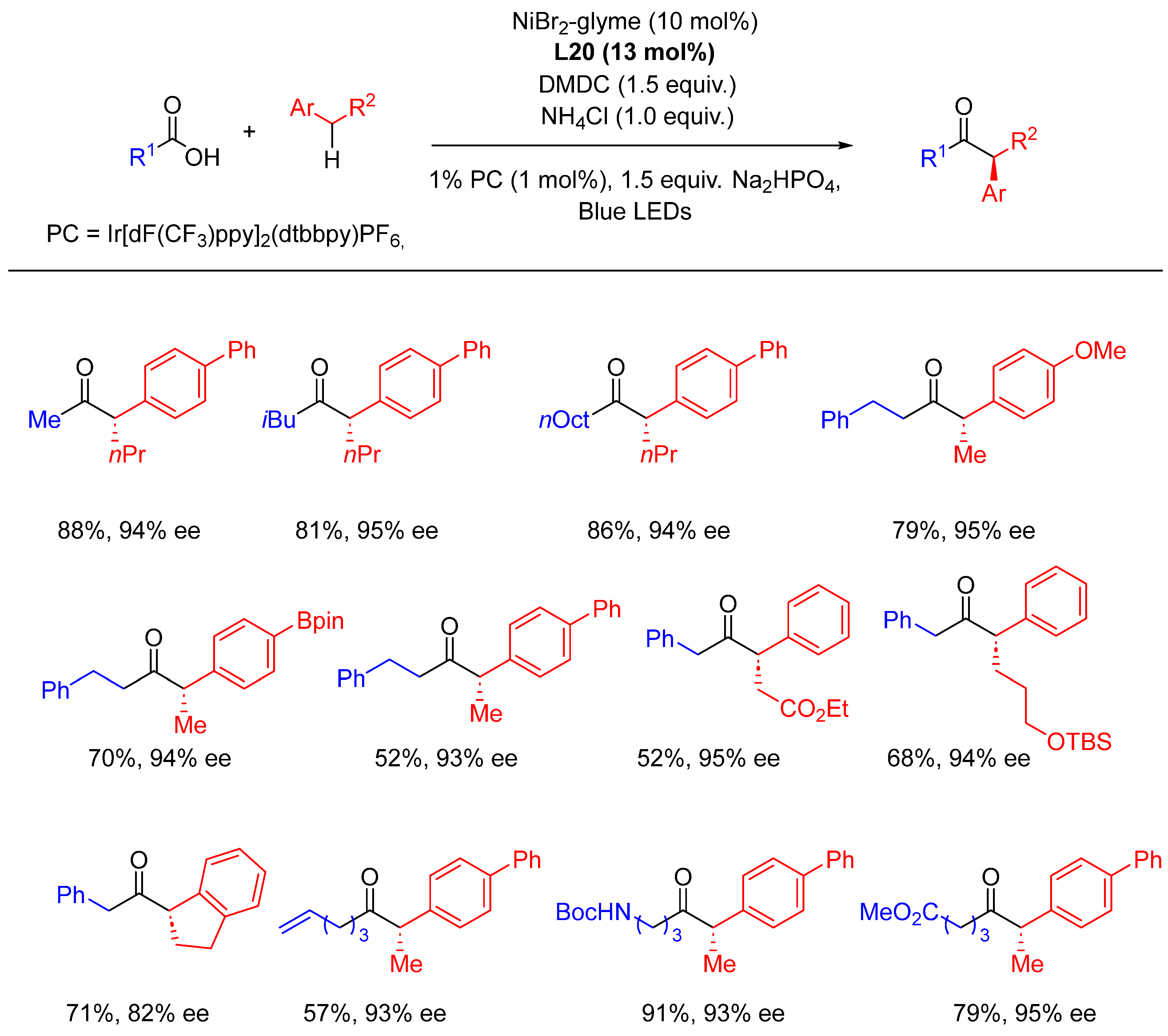{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
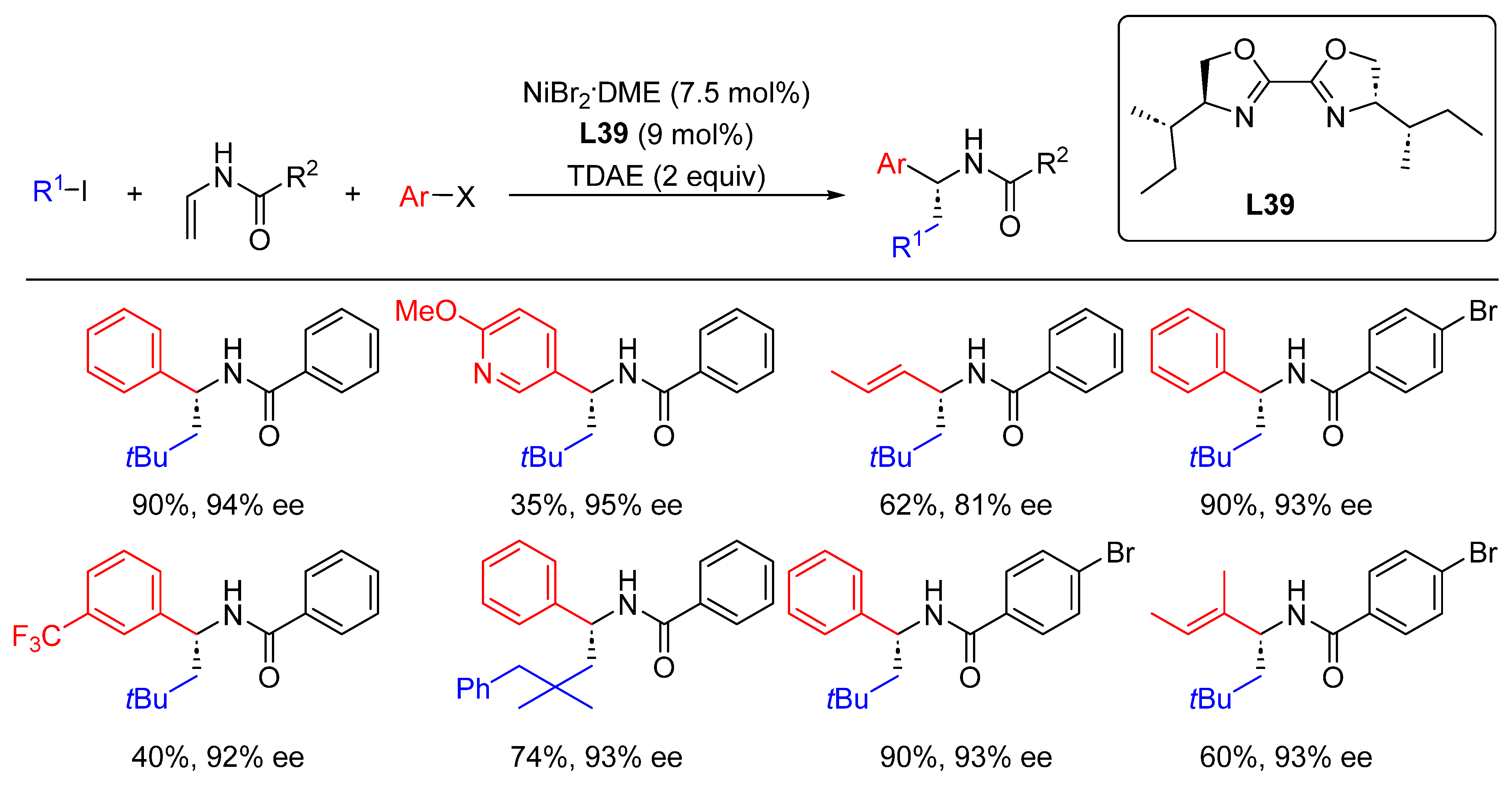{kind=link}
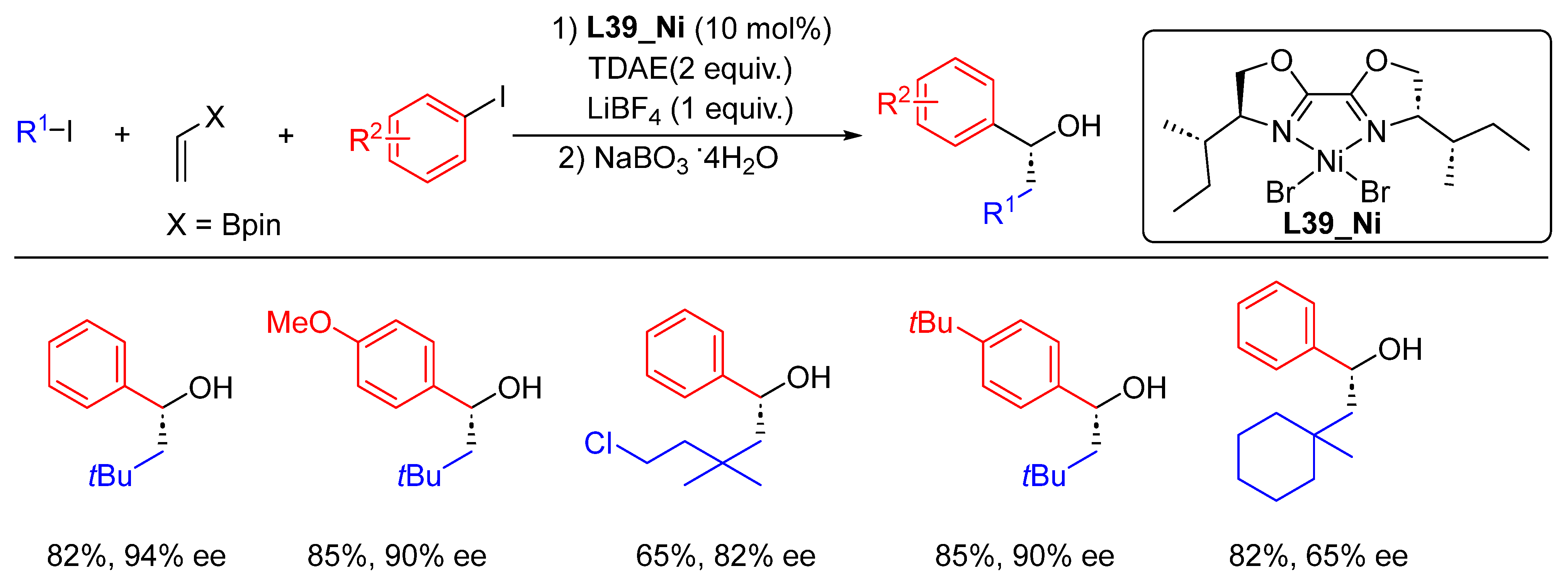{kind=link}
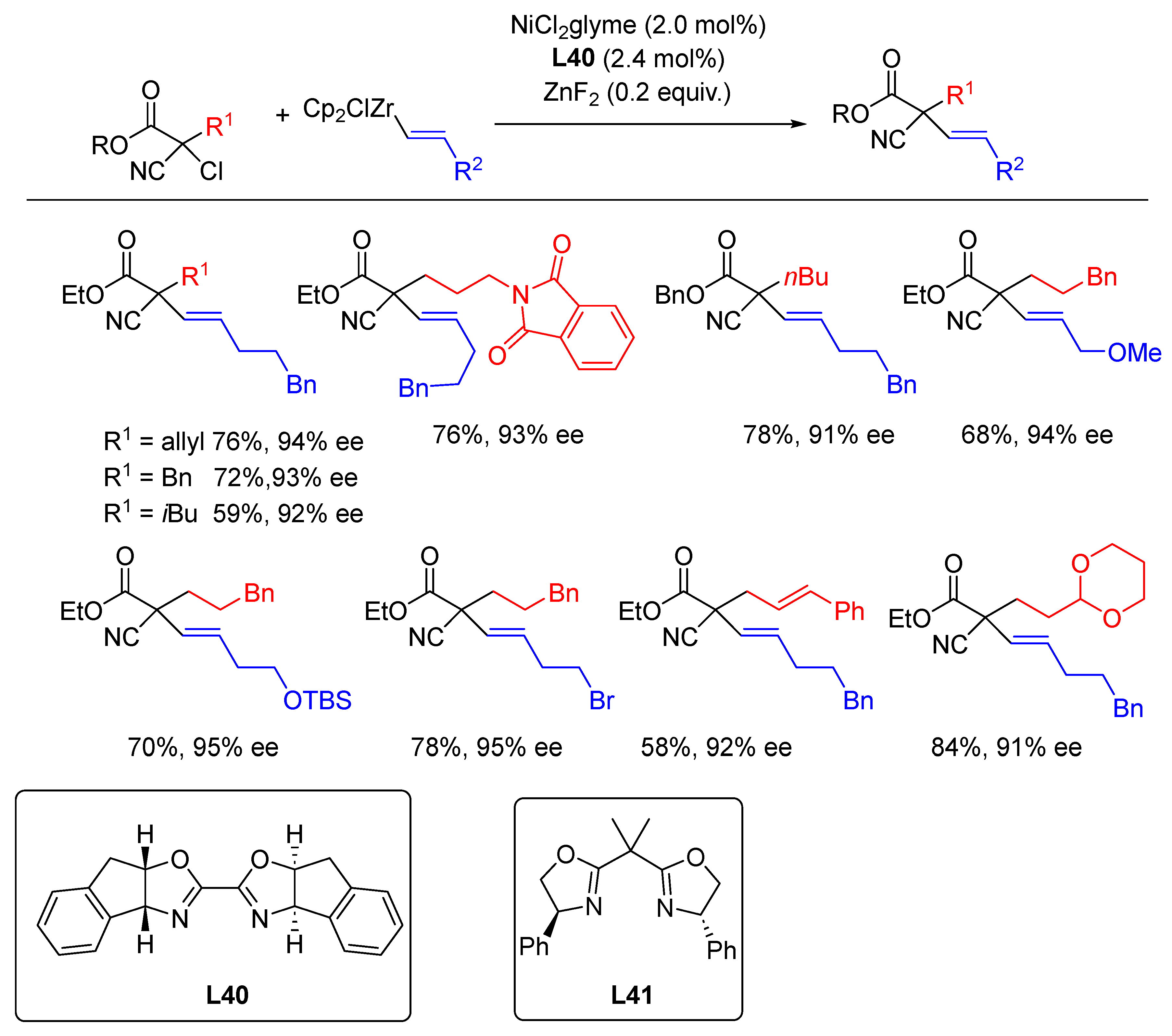{kind=link}
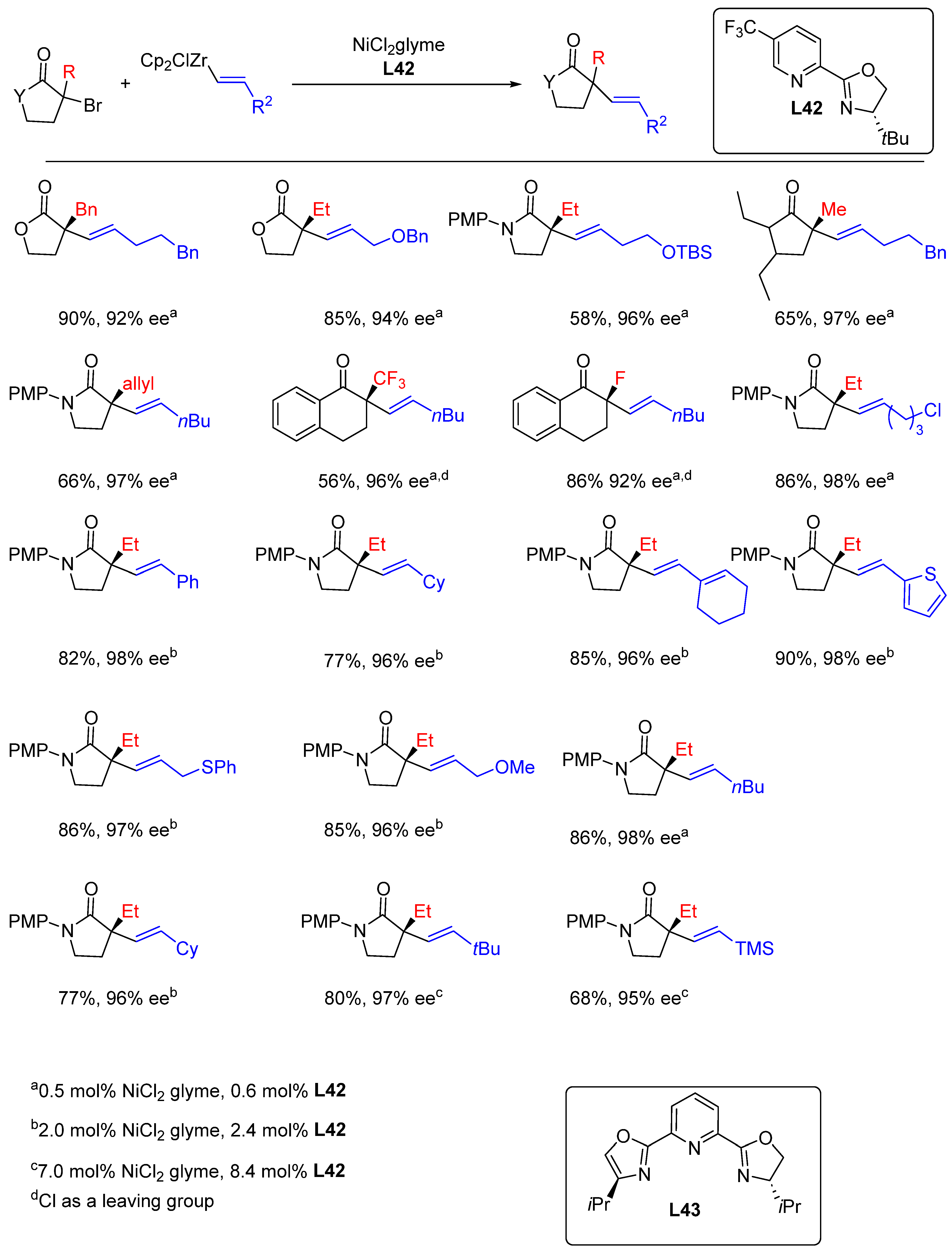{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
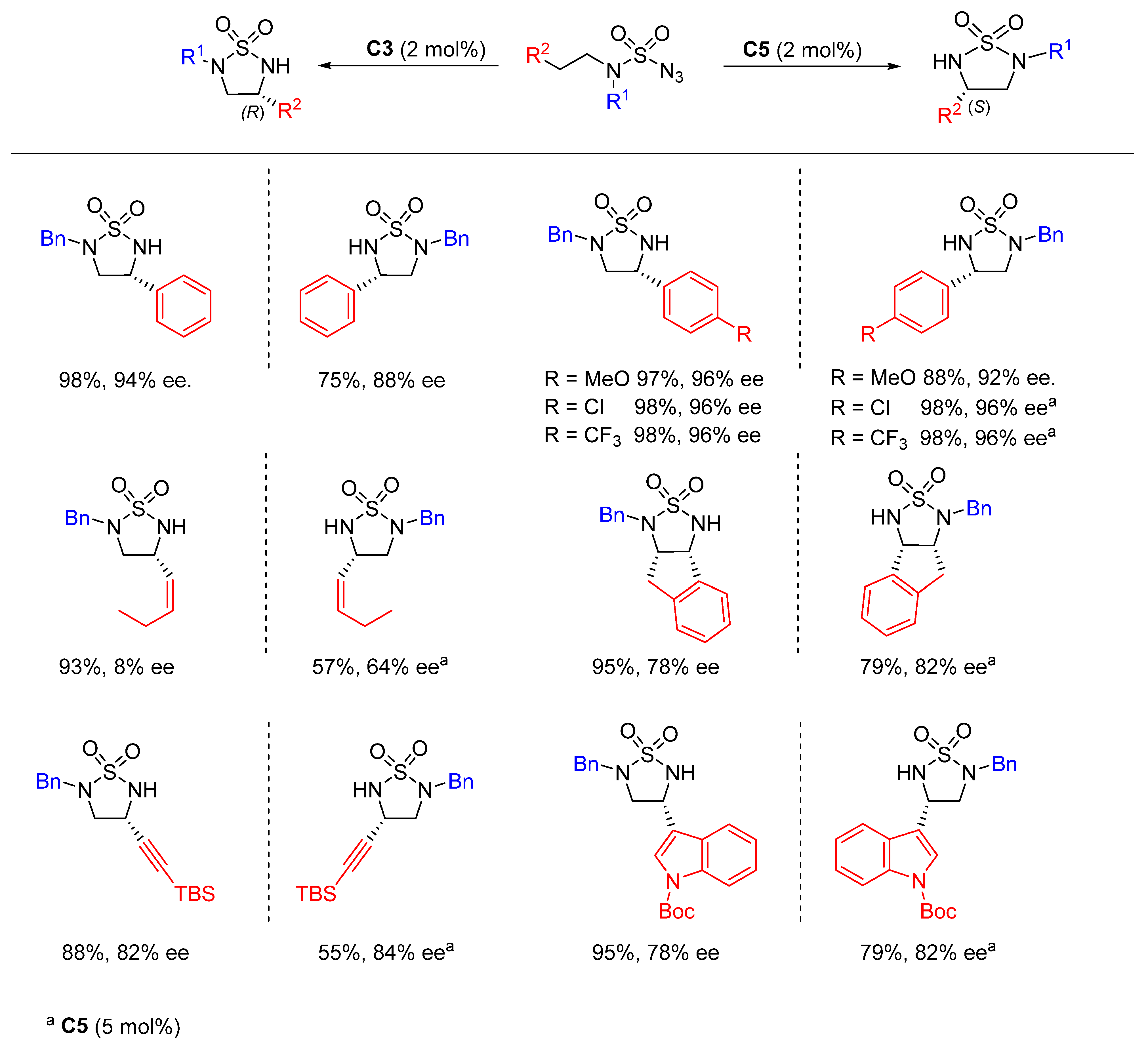{kind=link}
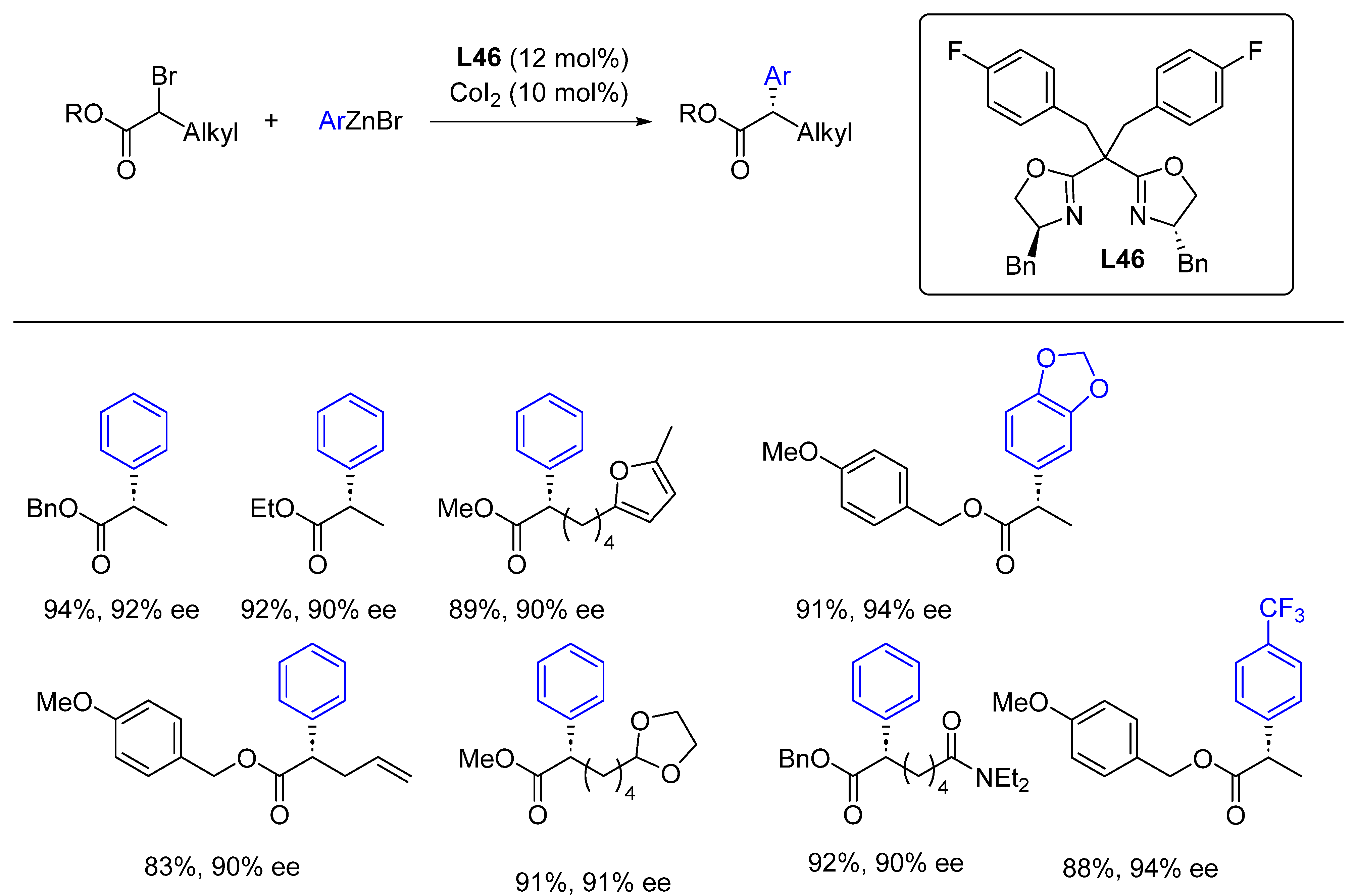{kind=link}
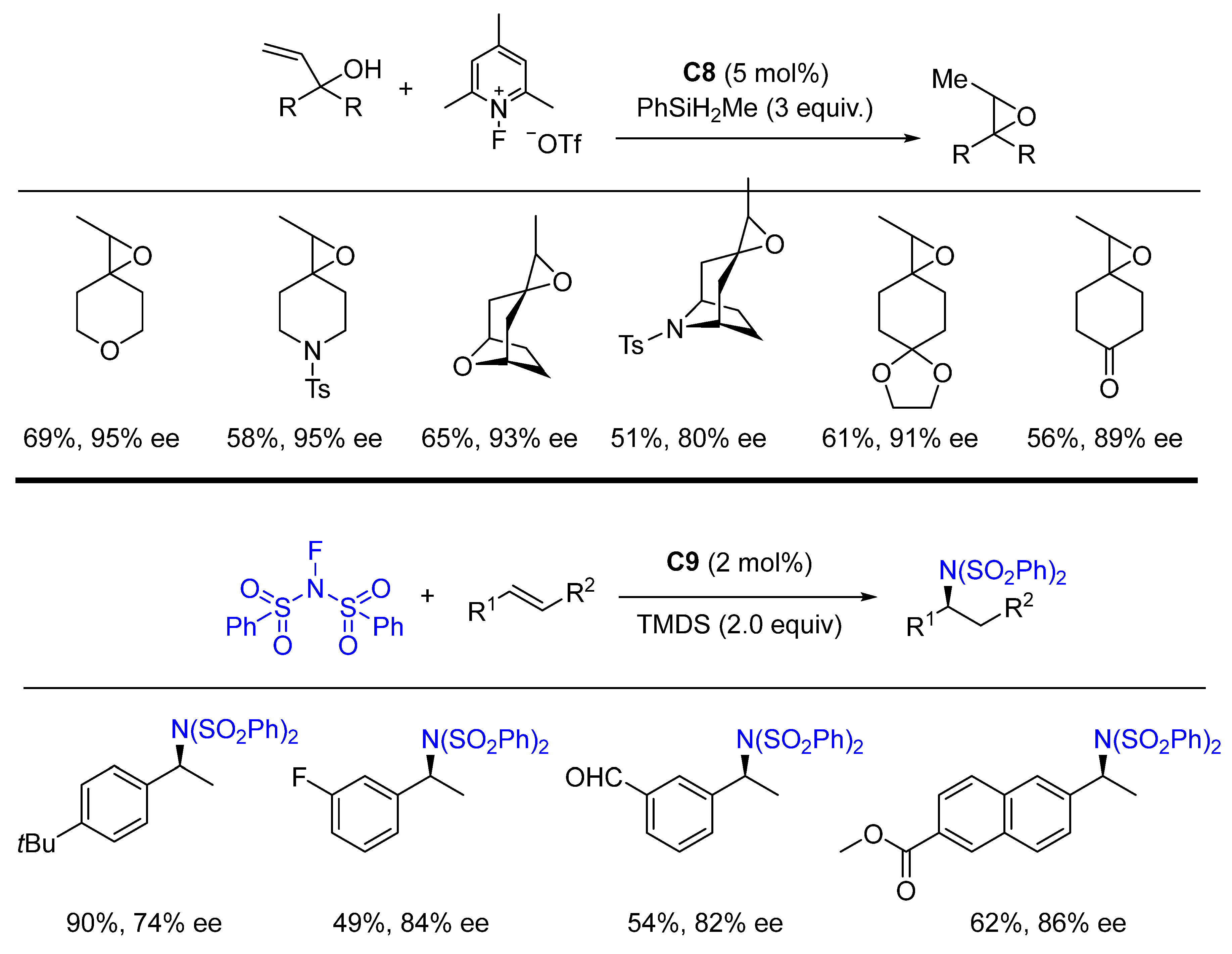{kind=link}
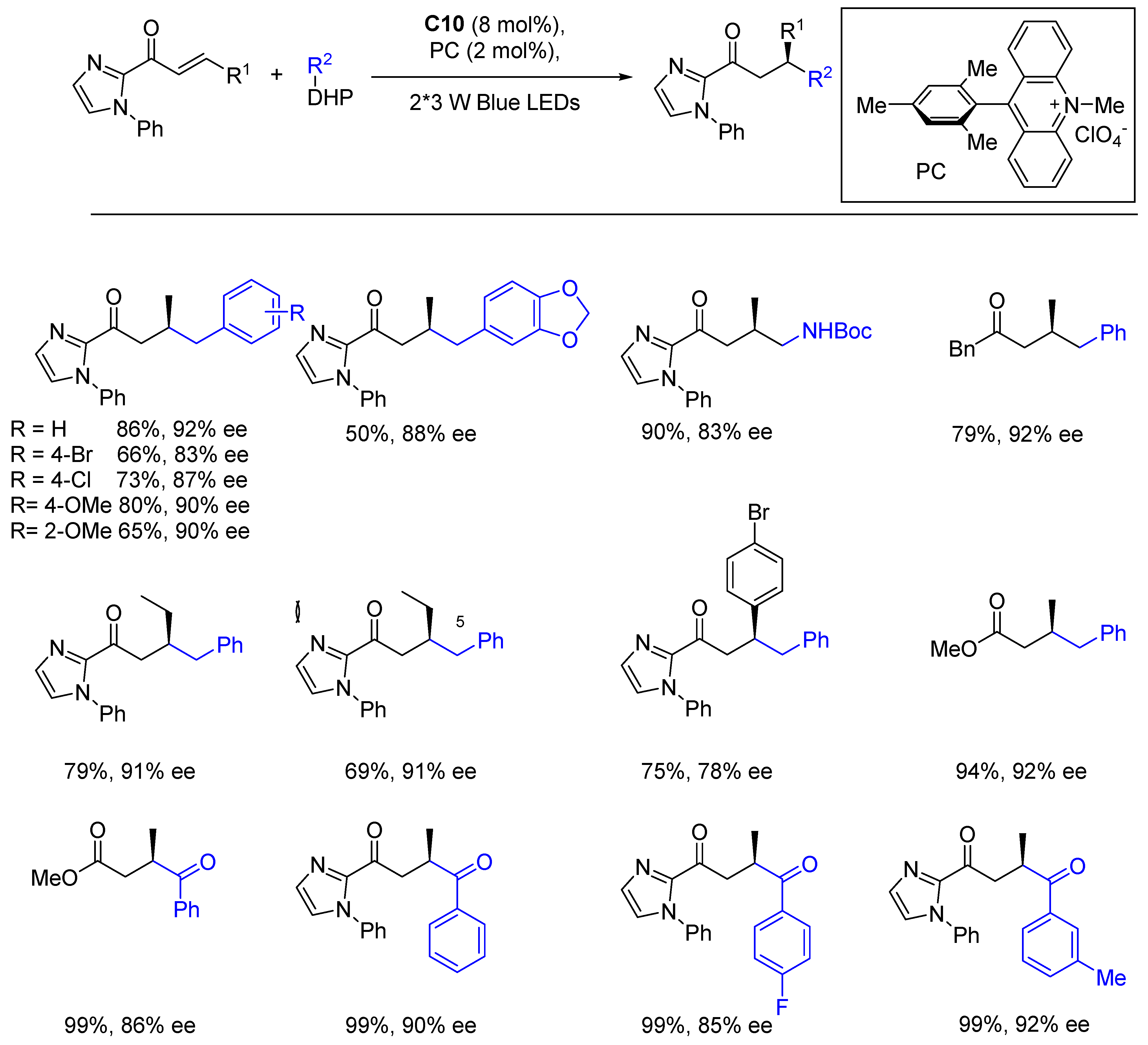{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
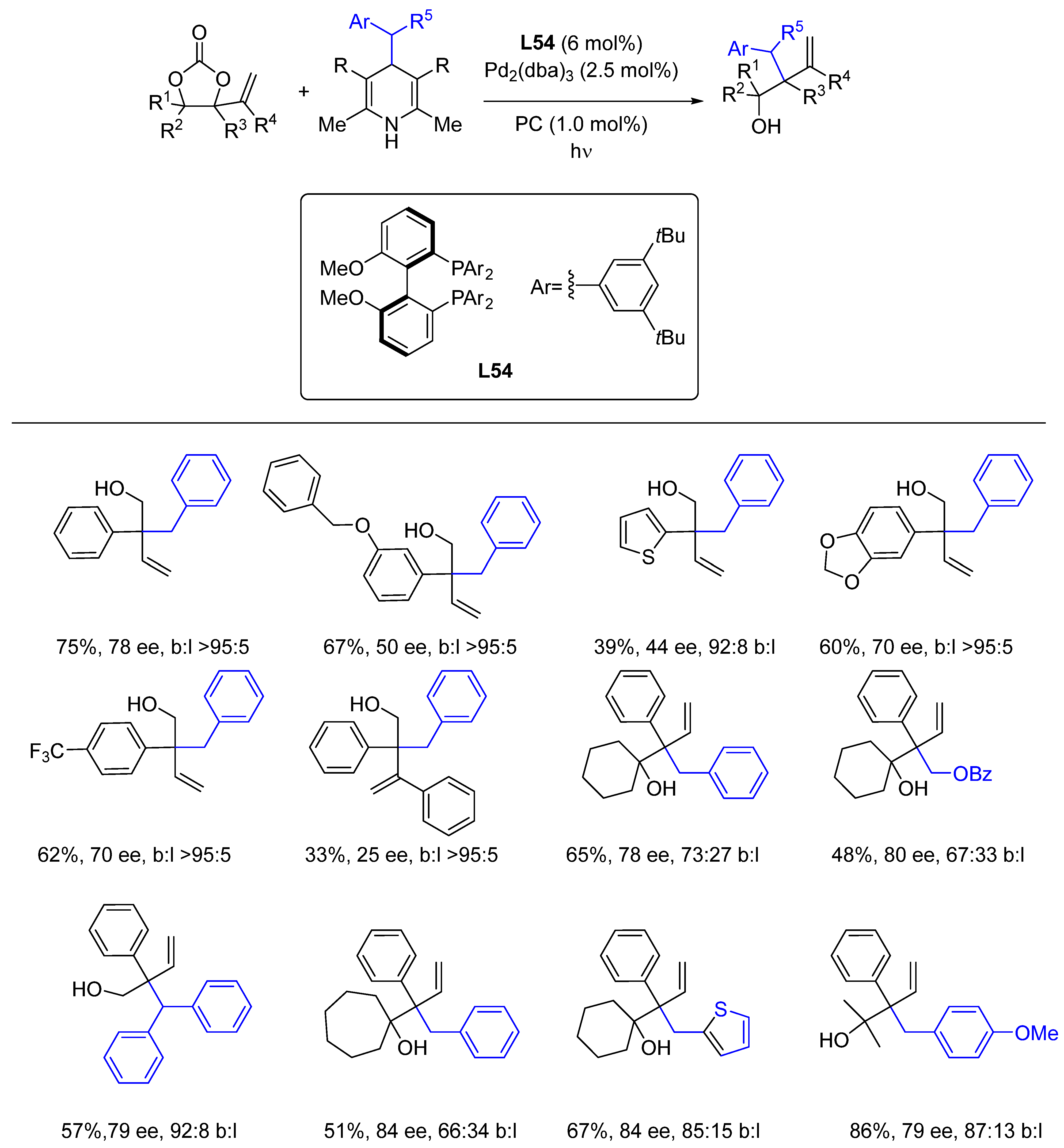{kind=link}
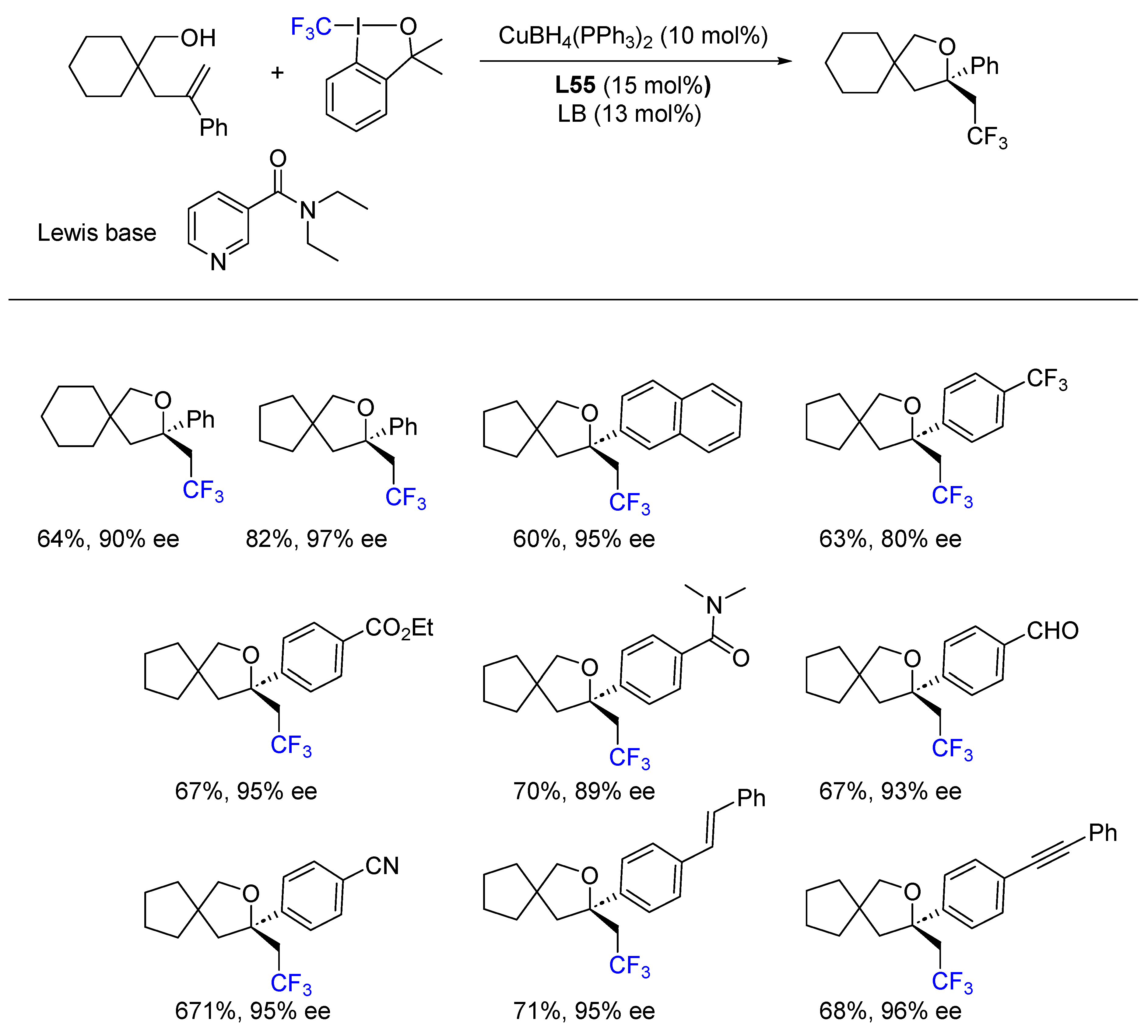{kind=link}
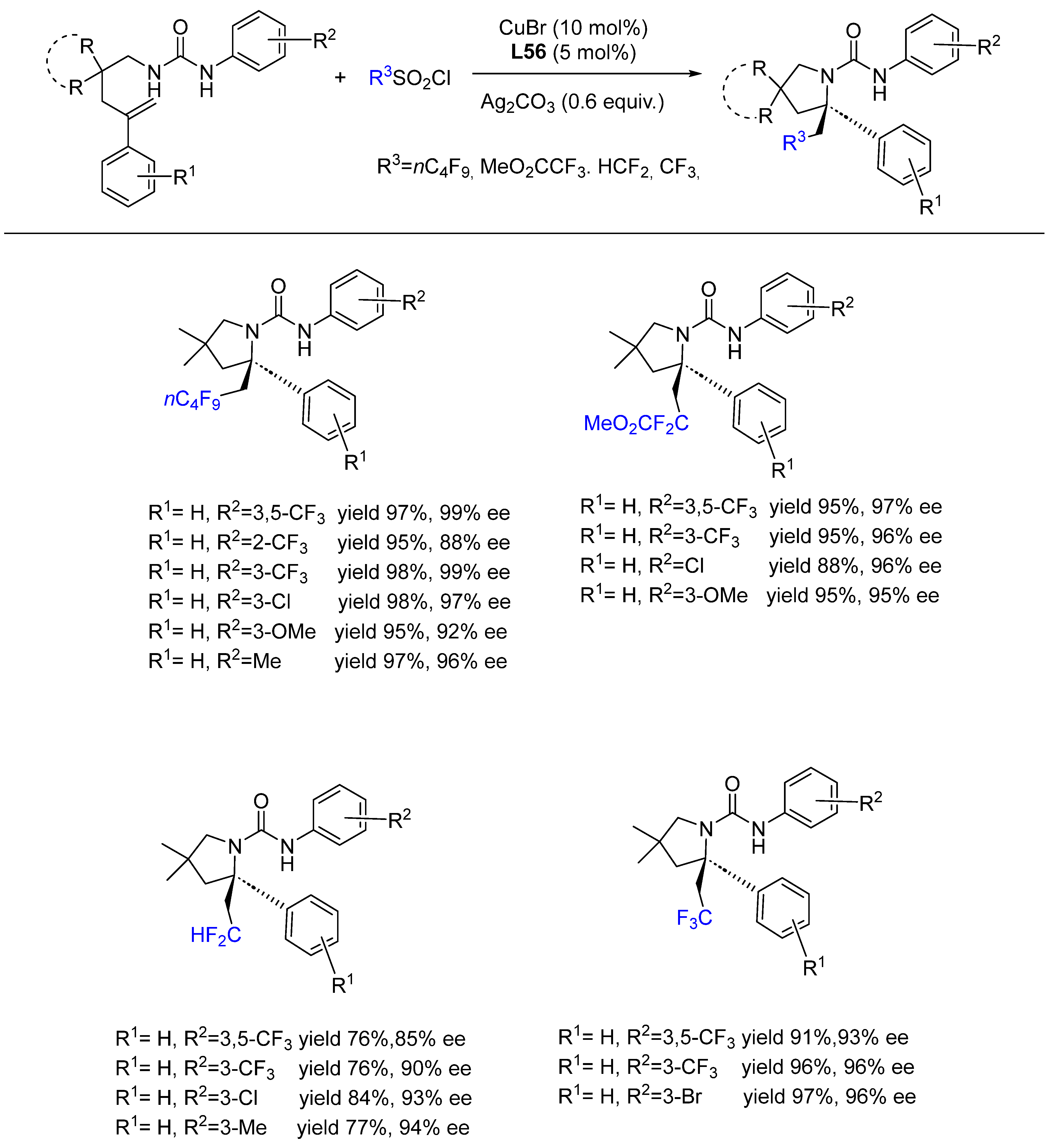{kind=link}
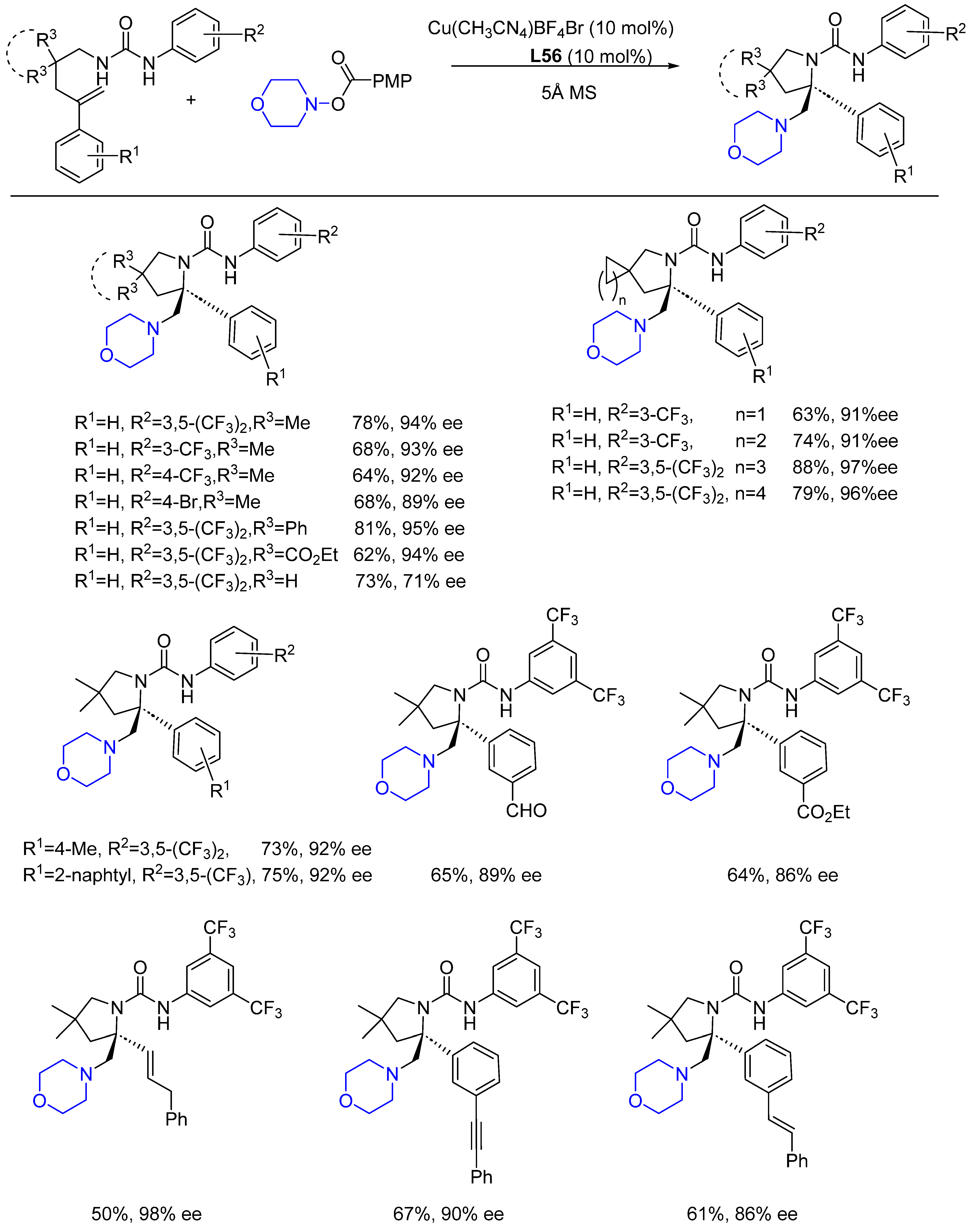{kind=link}
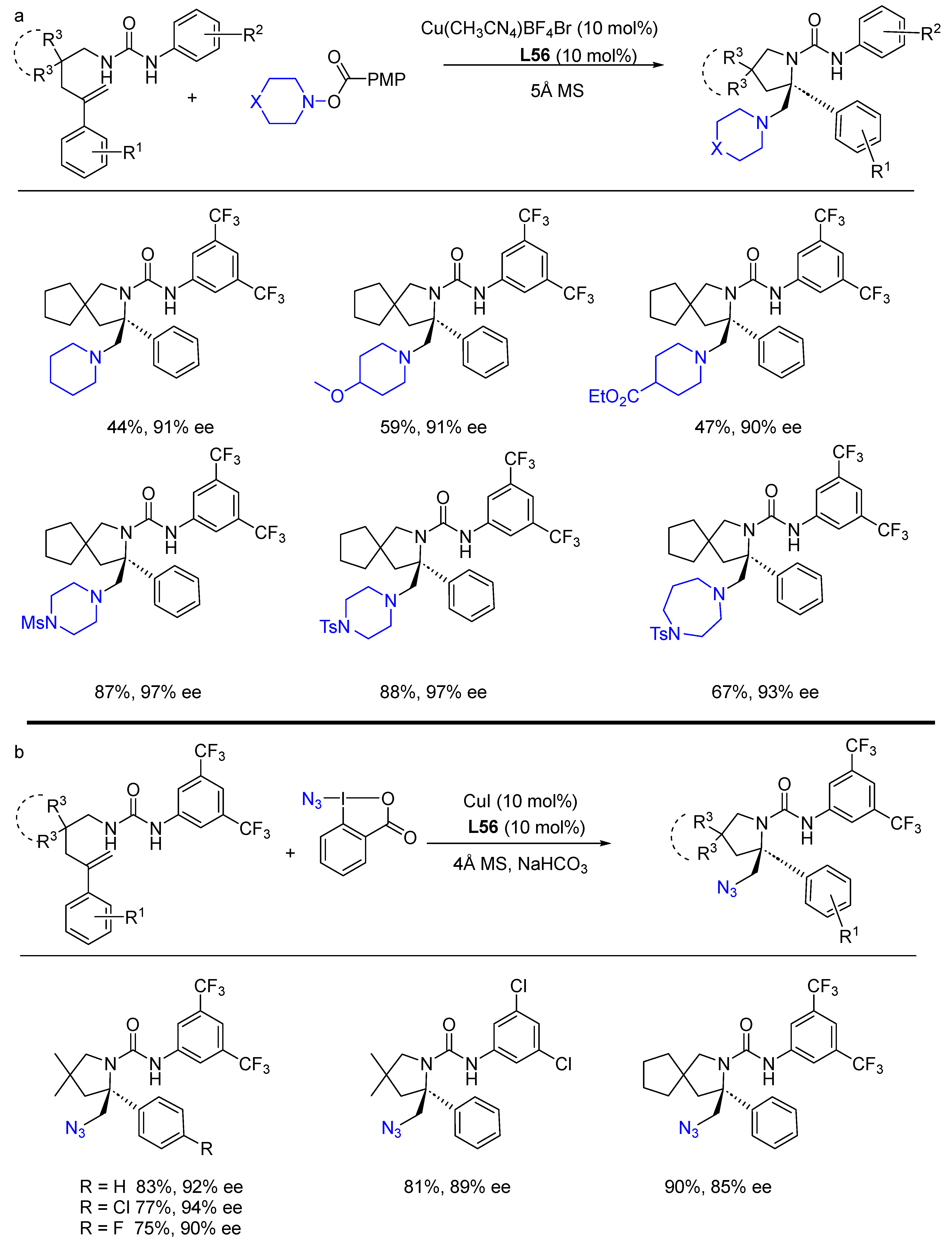{kind=link}
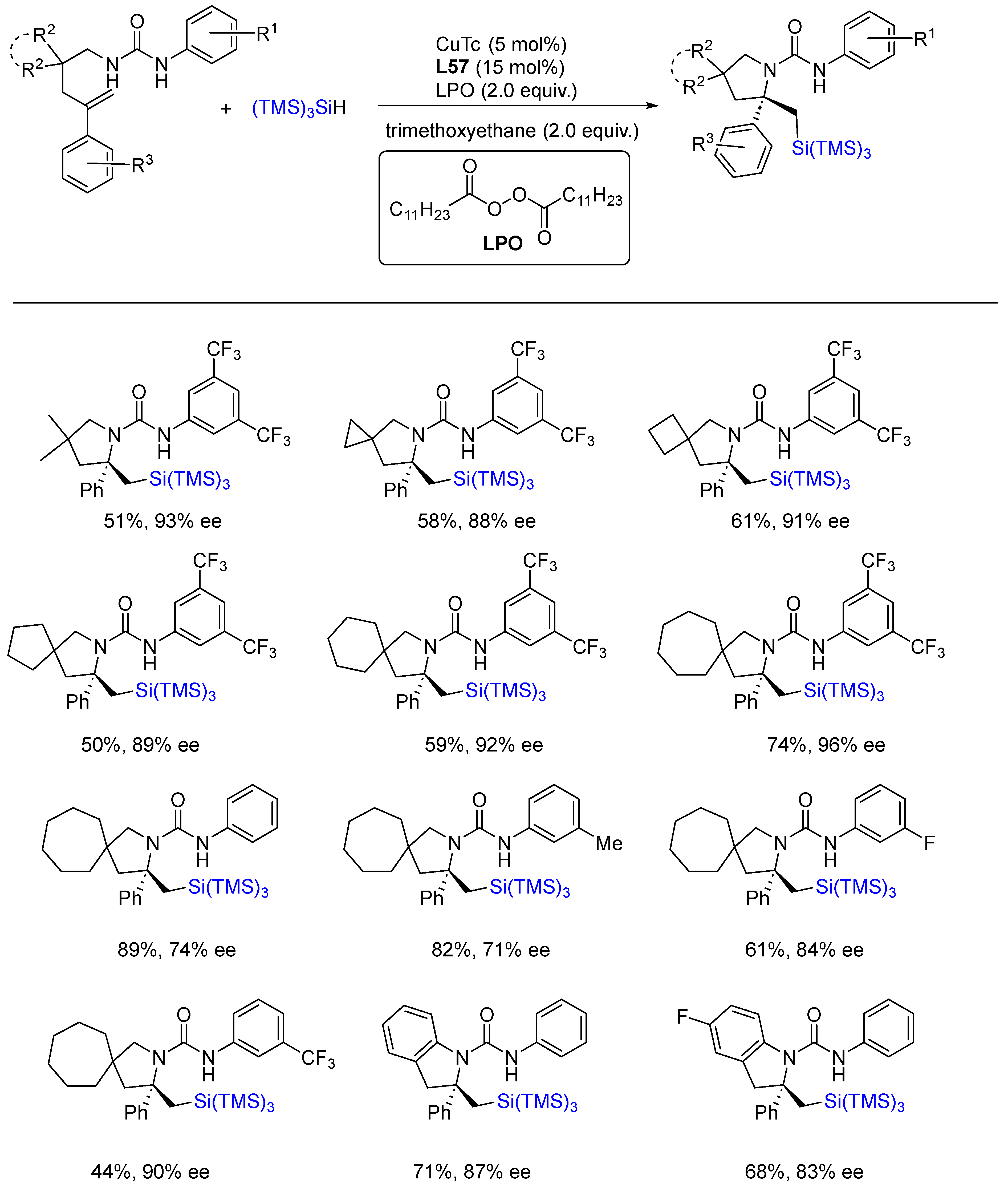{kind=link}
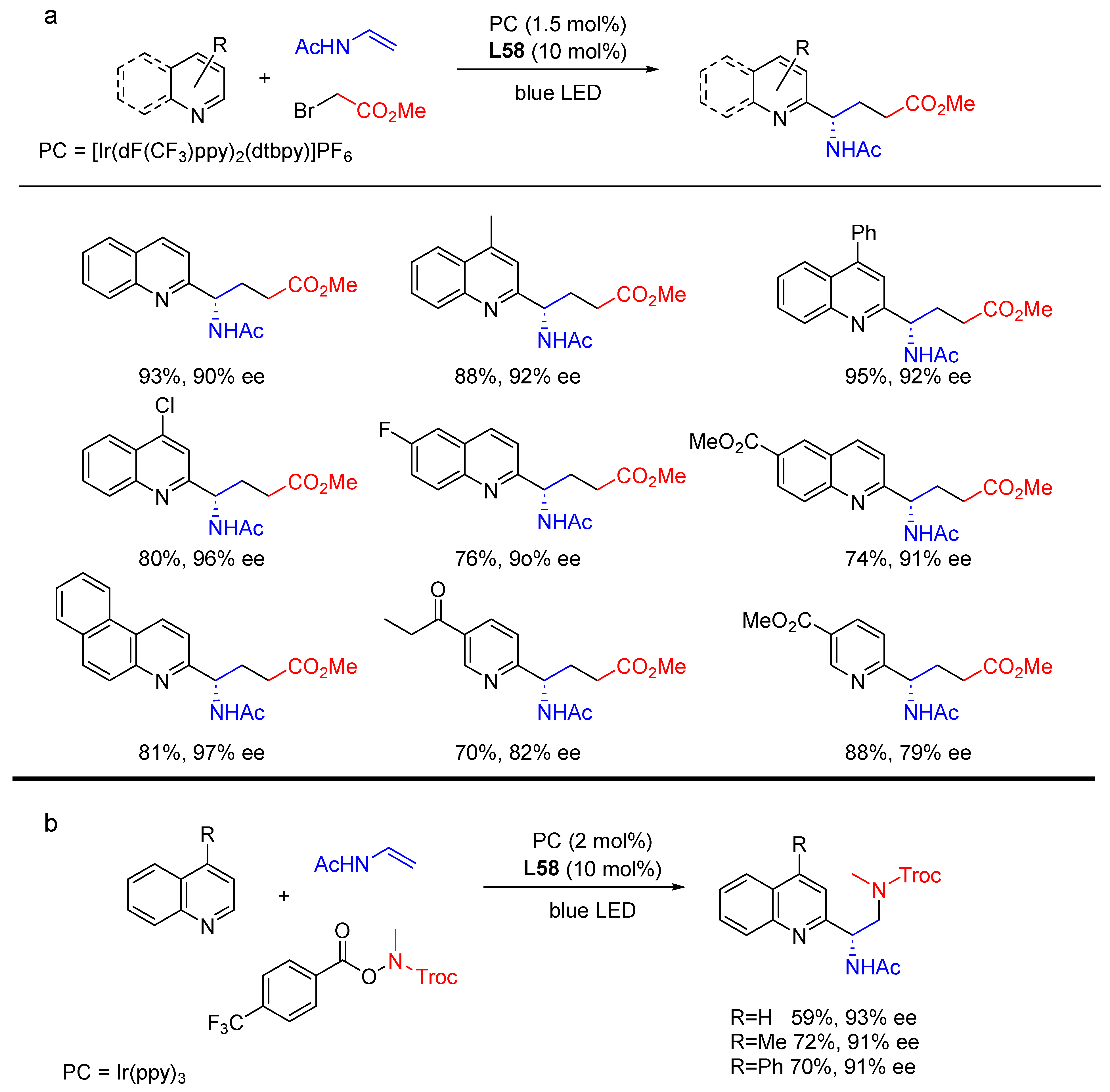{kind=link}
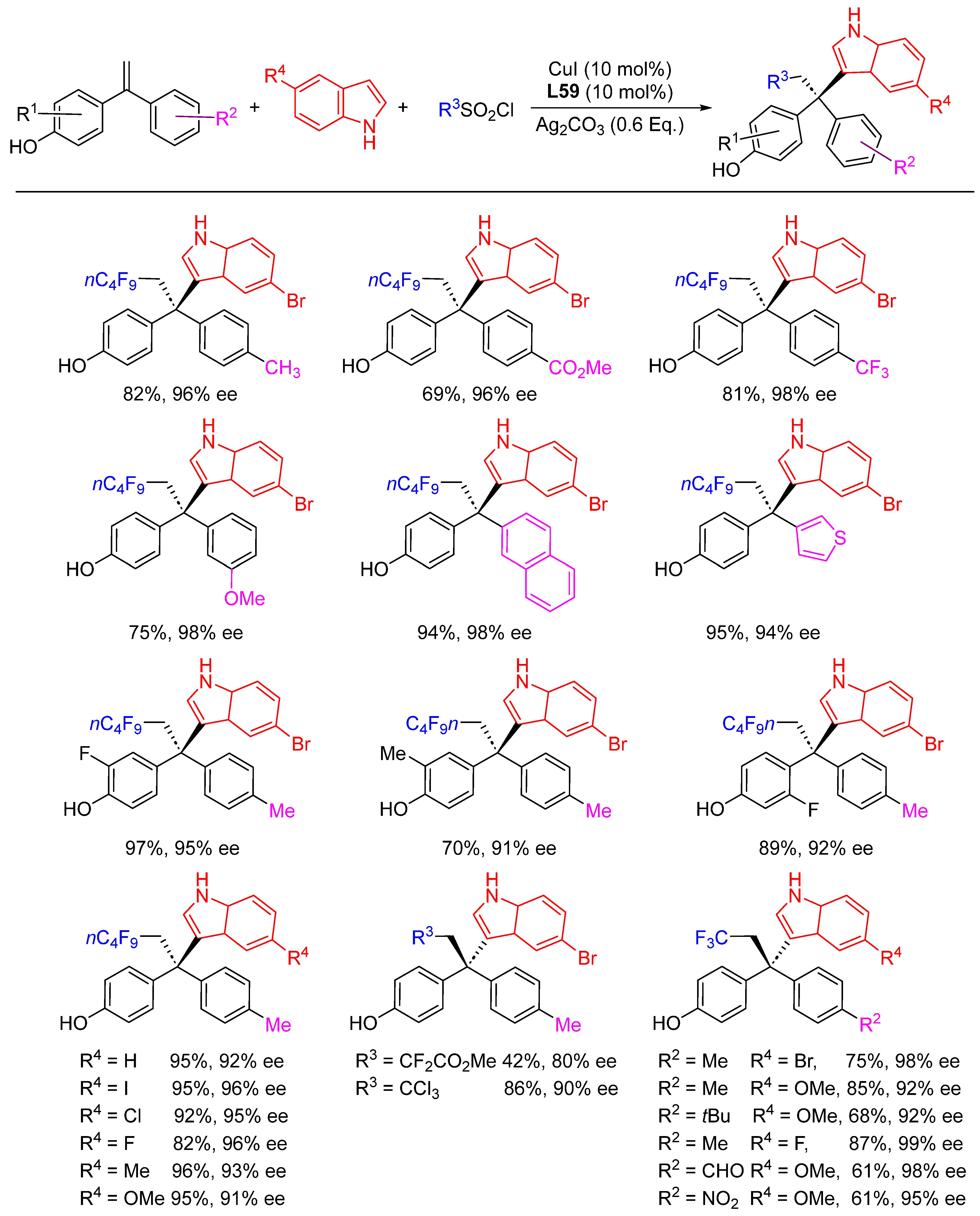{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
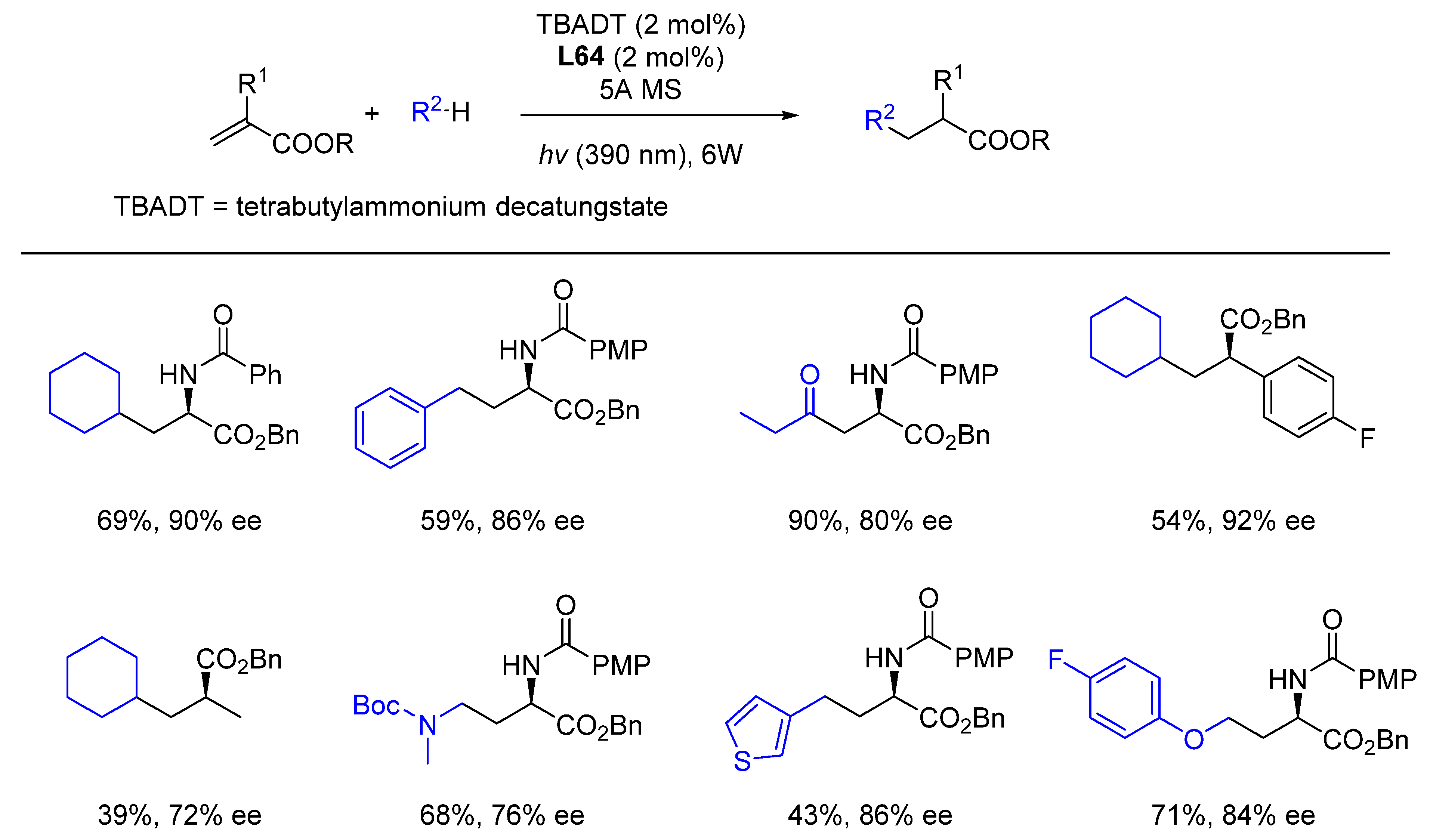{kind=link}
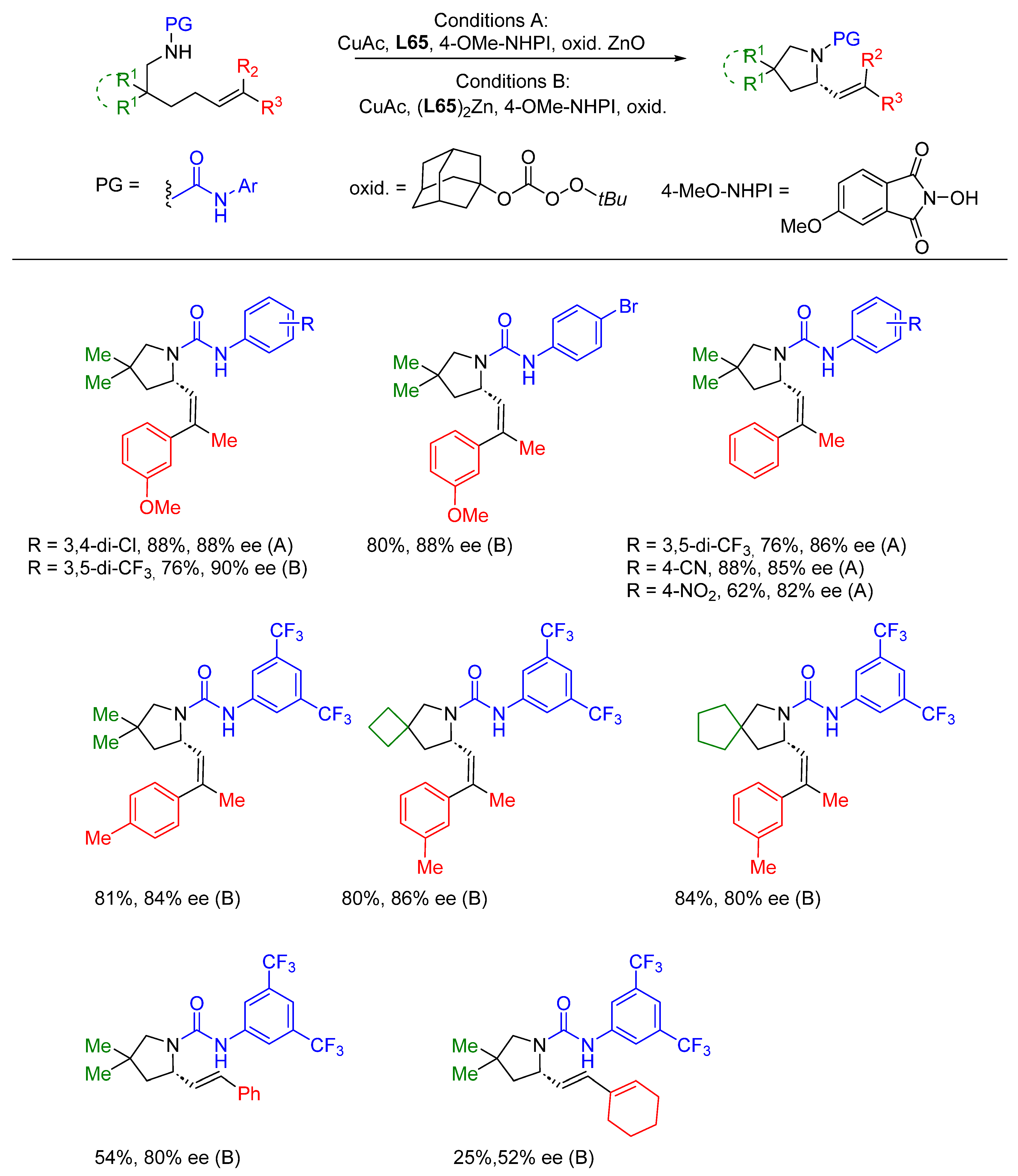{kind=link}
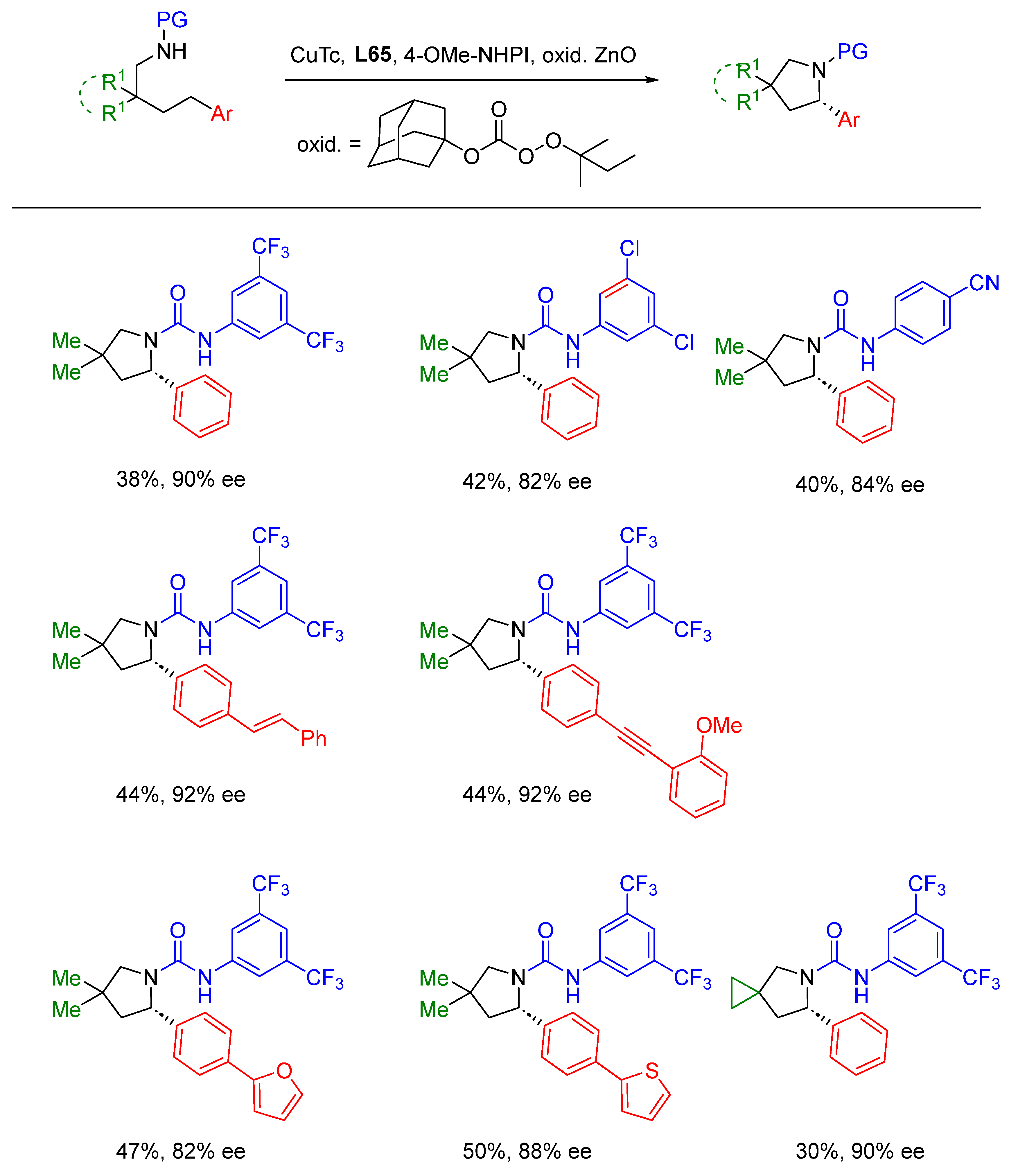{kind=link}
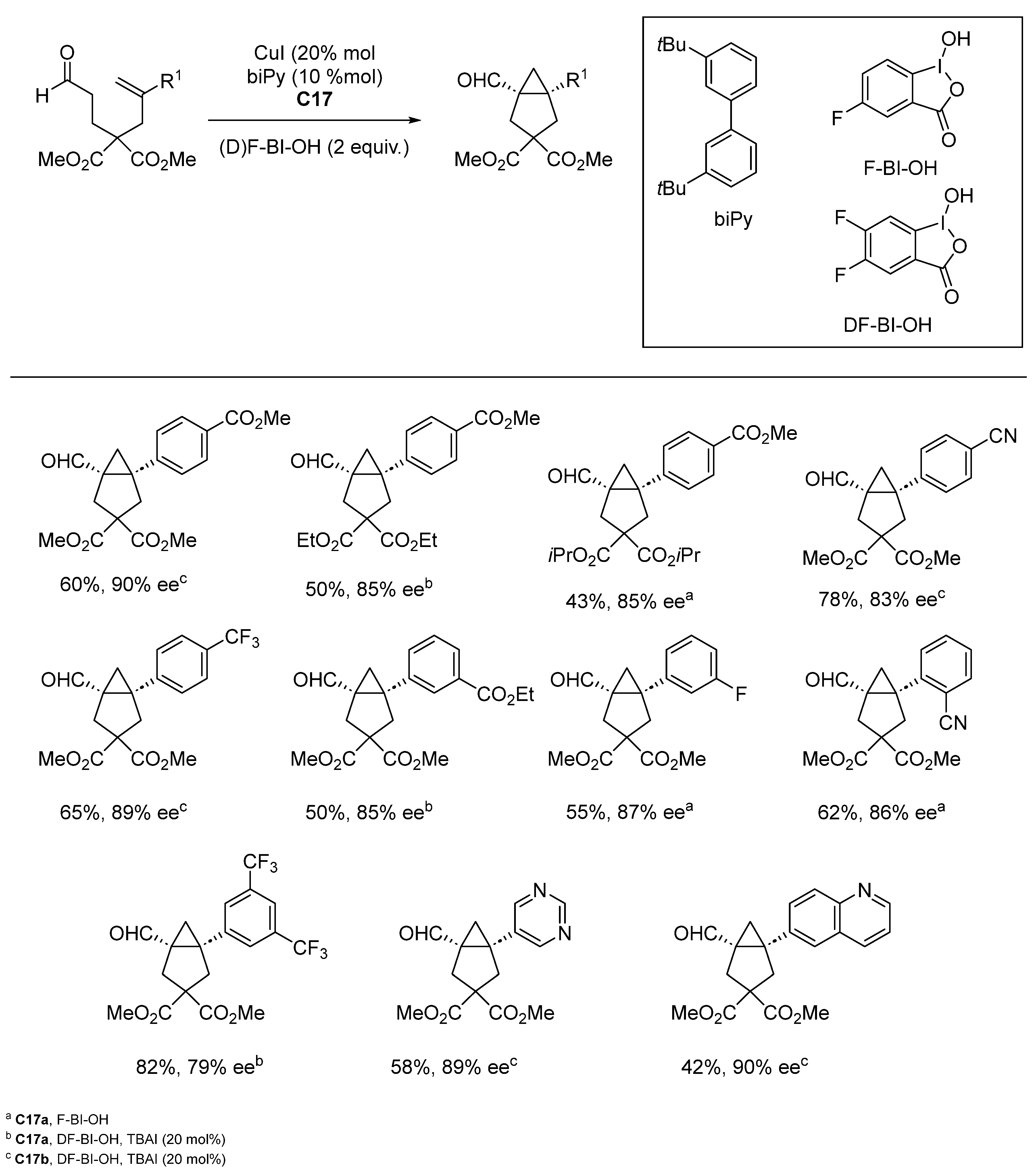{kind=link}
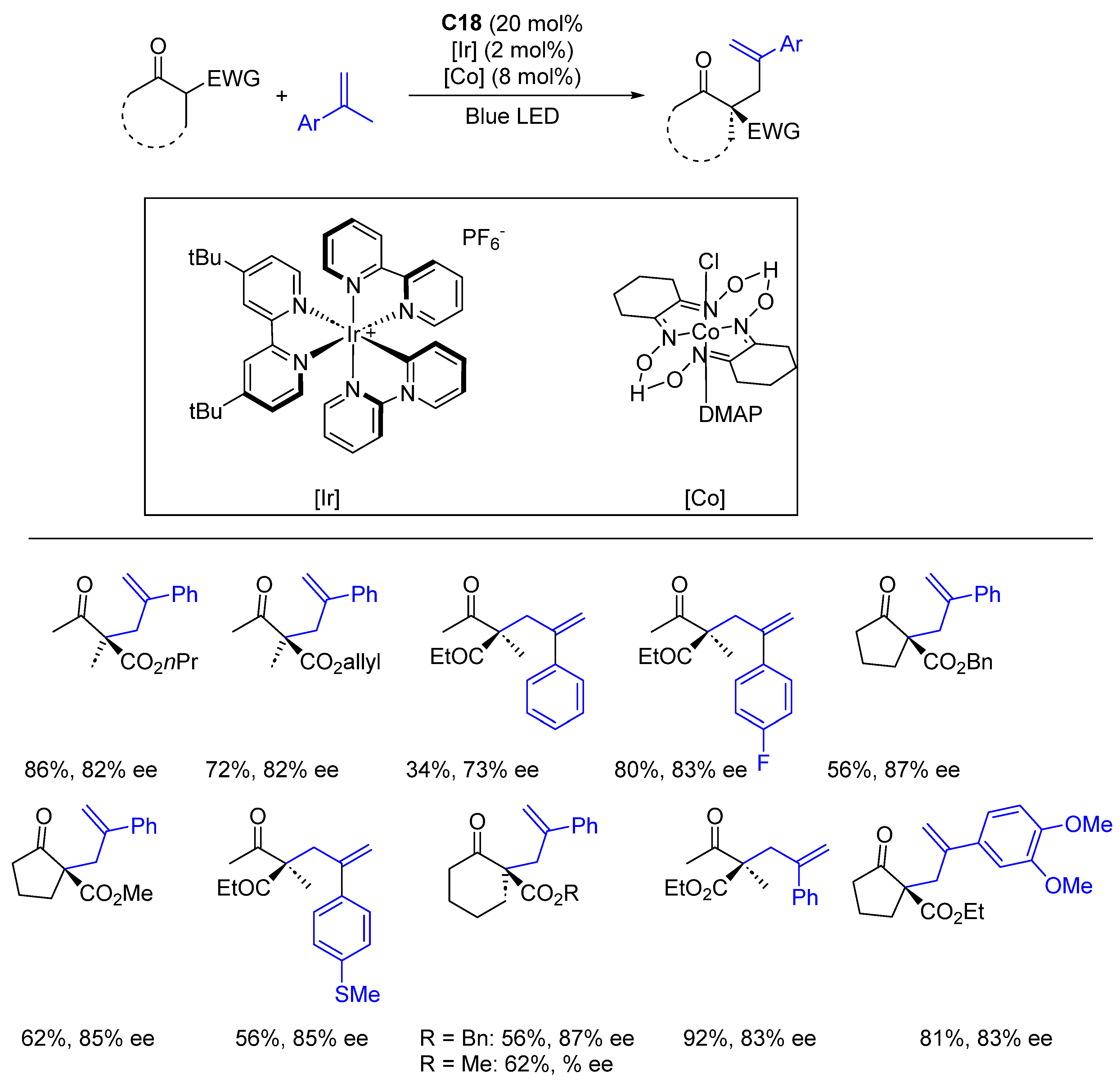{kind=link}
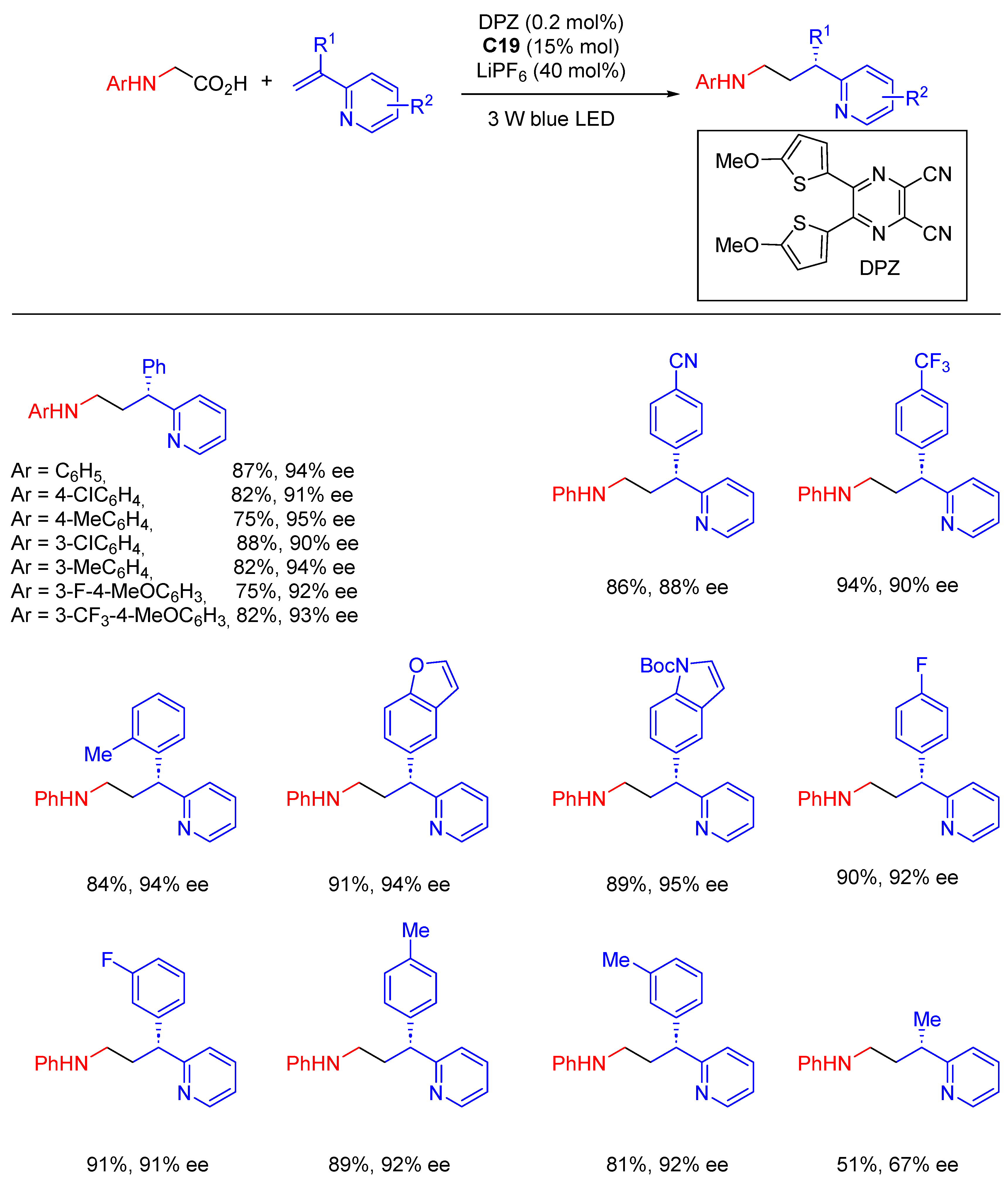{kind=link}
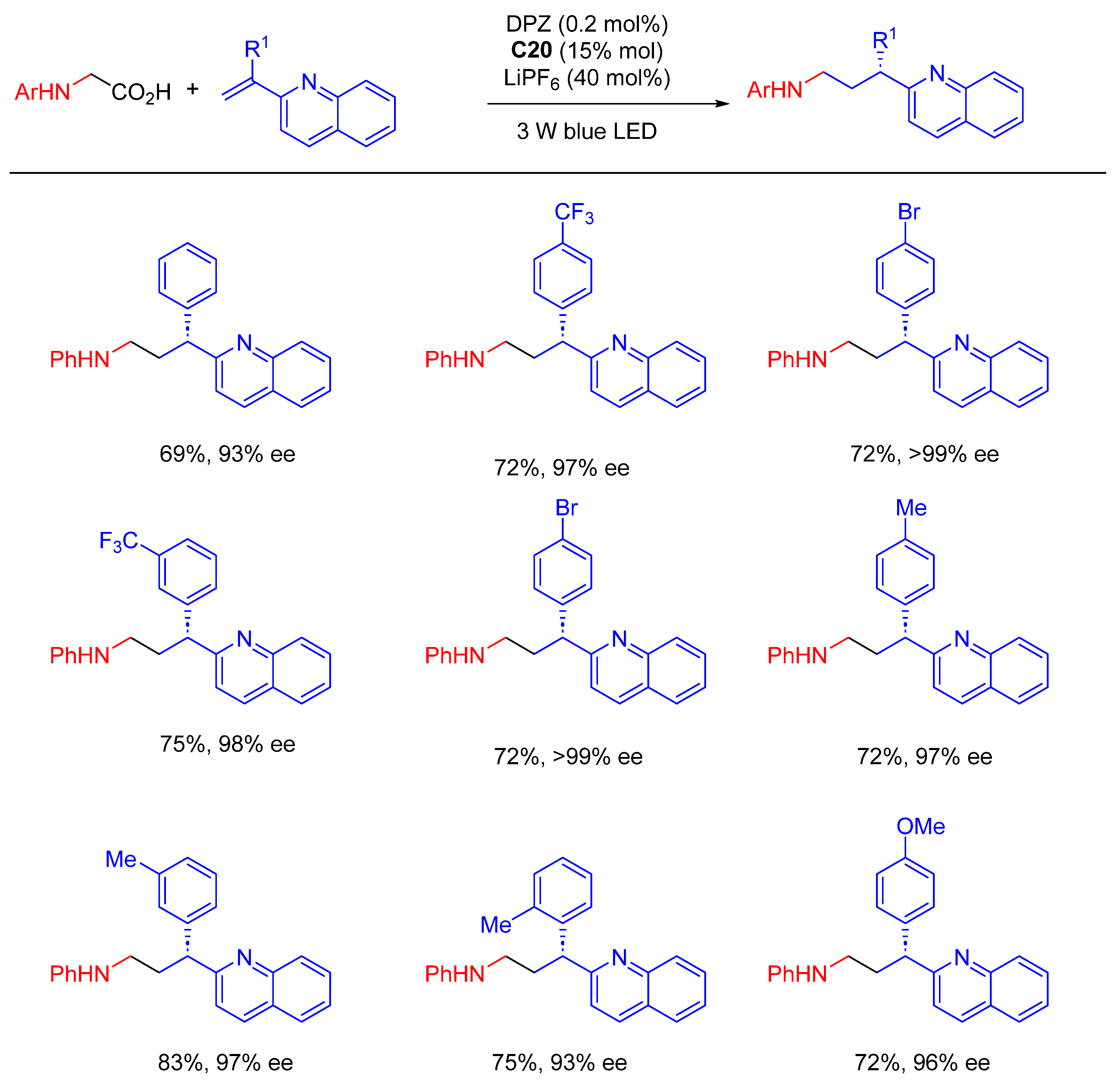{kind=link}
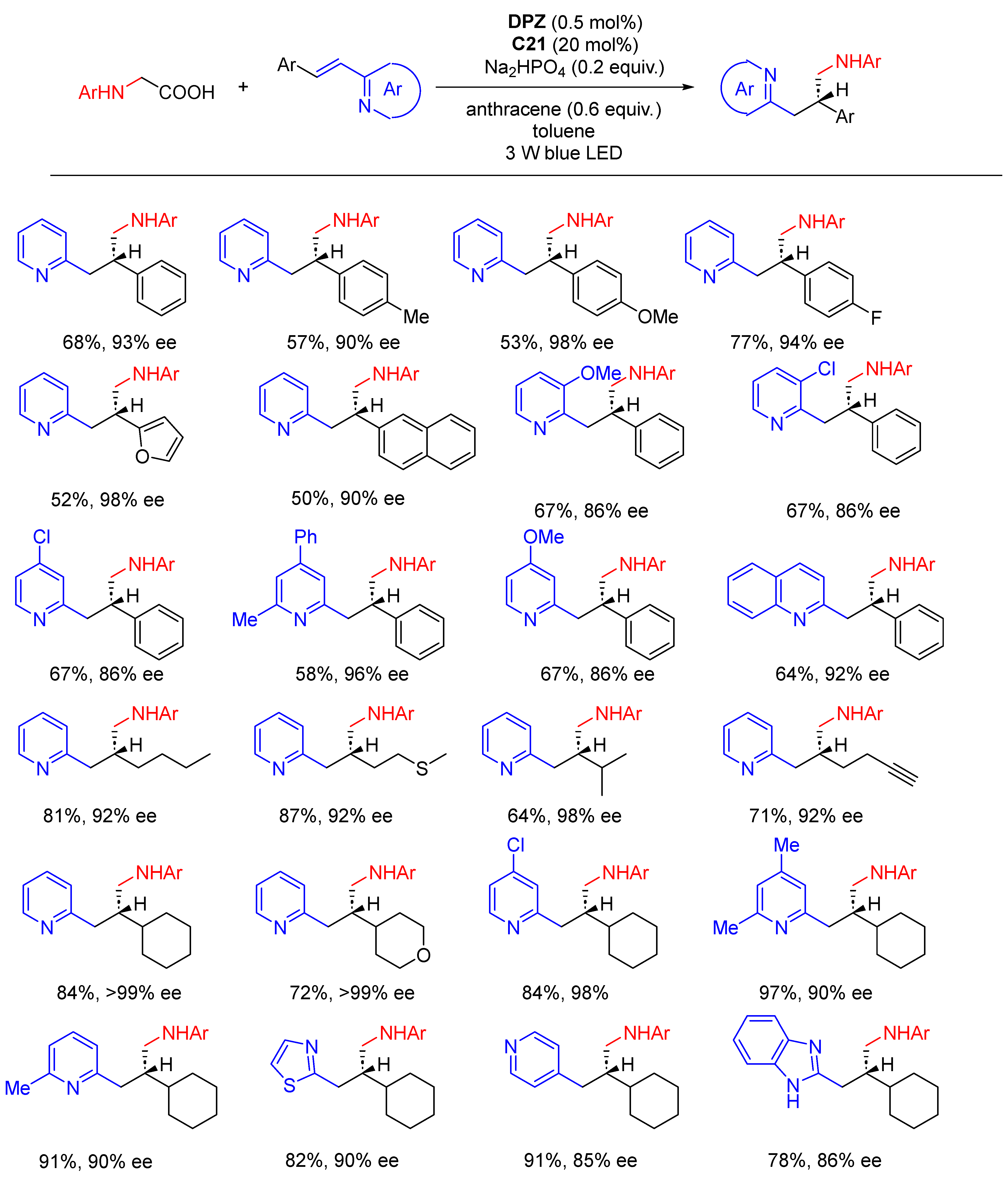{kind=link}
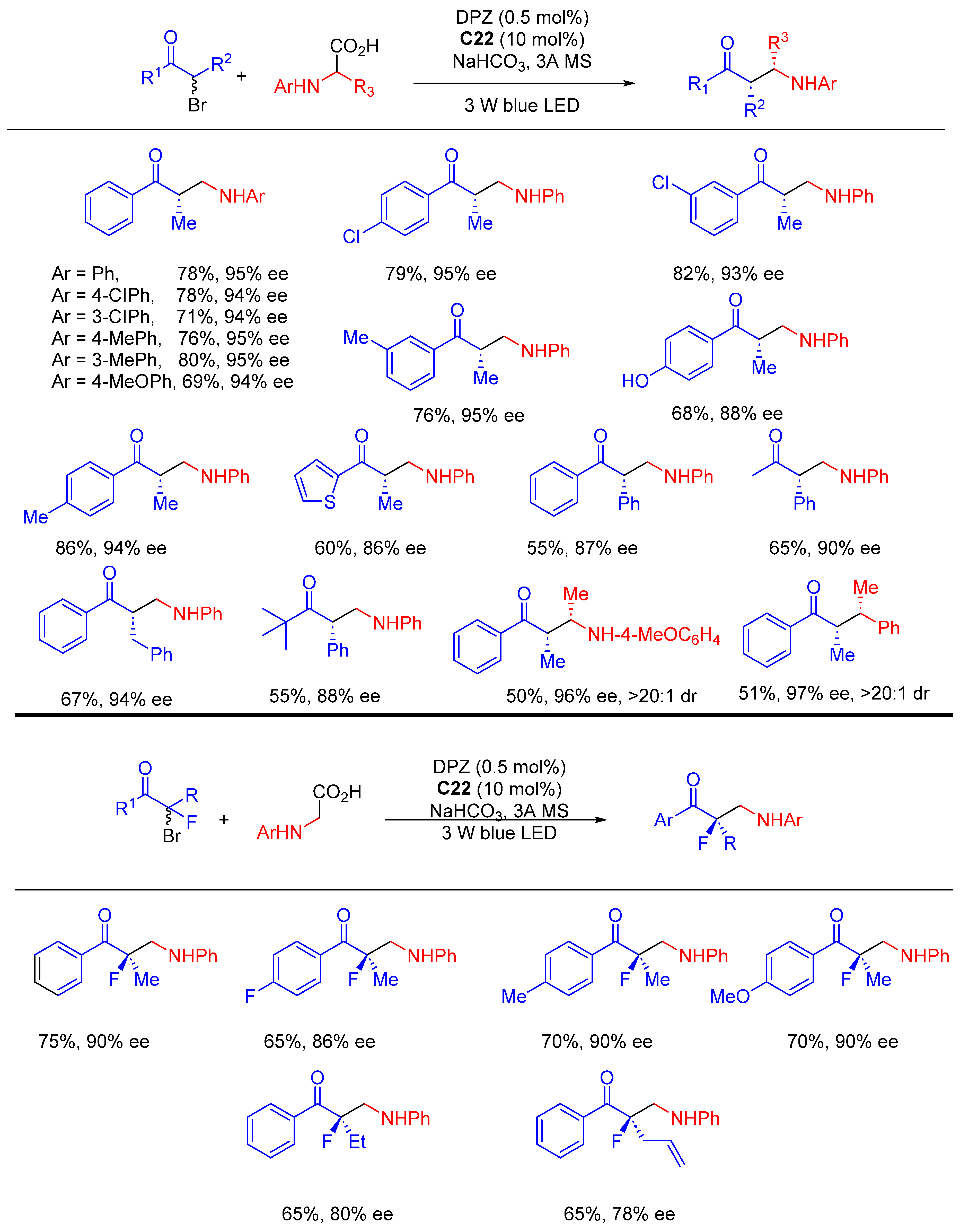{kind=link}
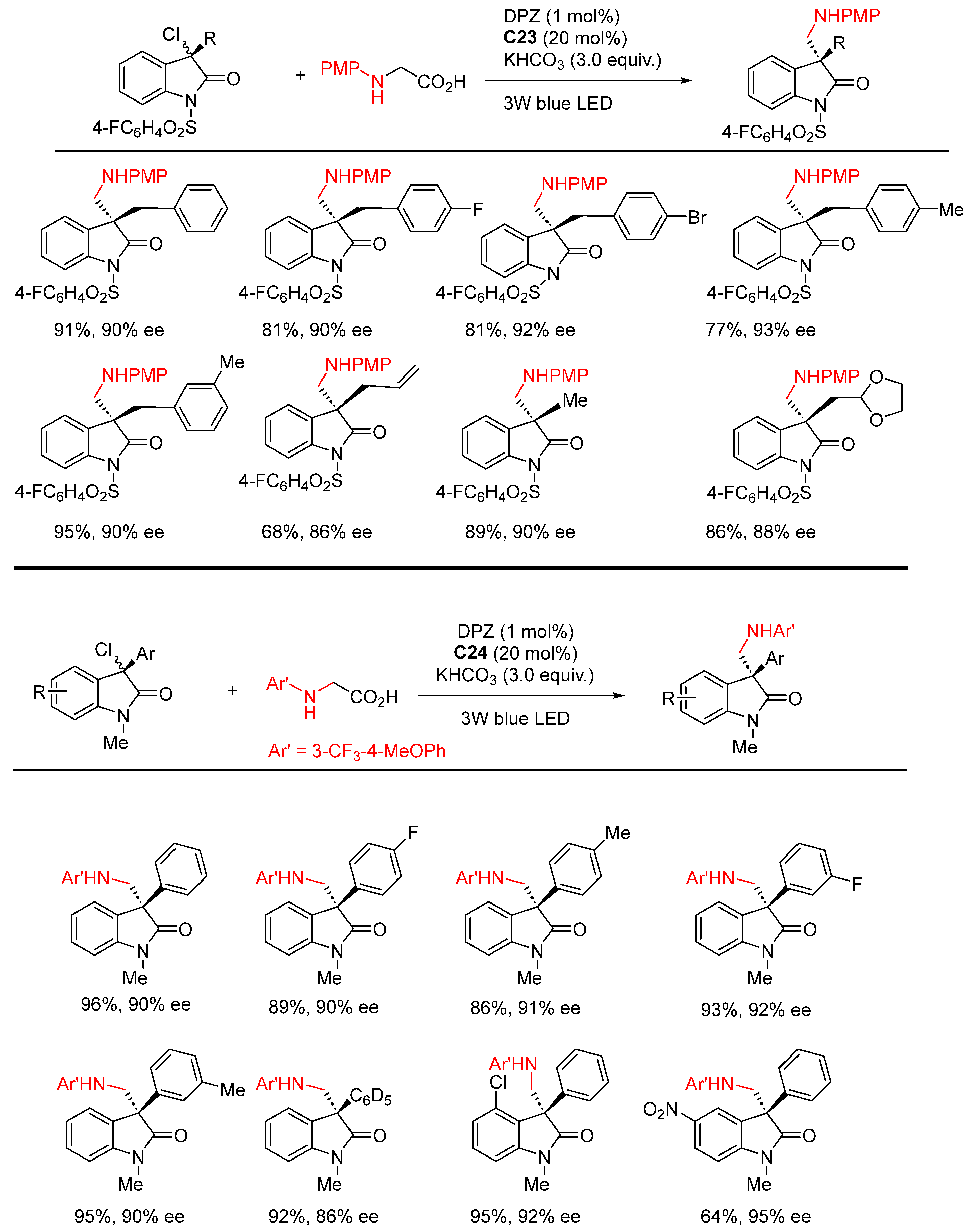{kind=link}
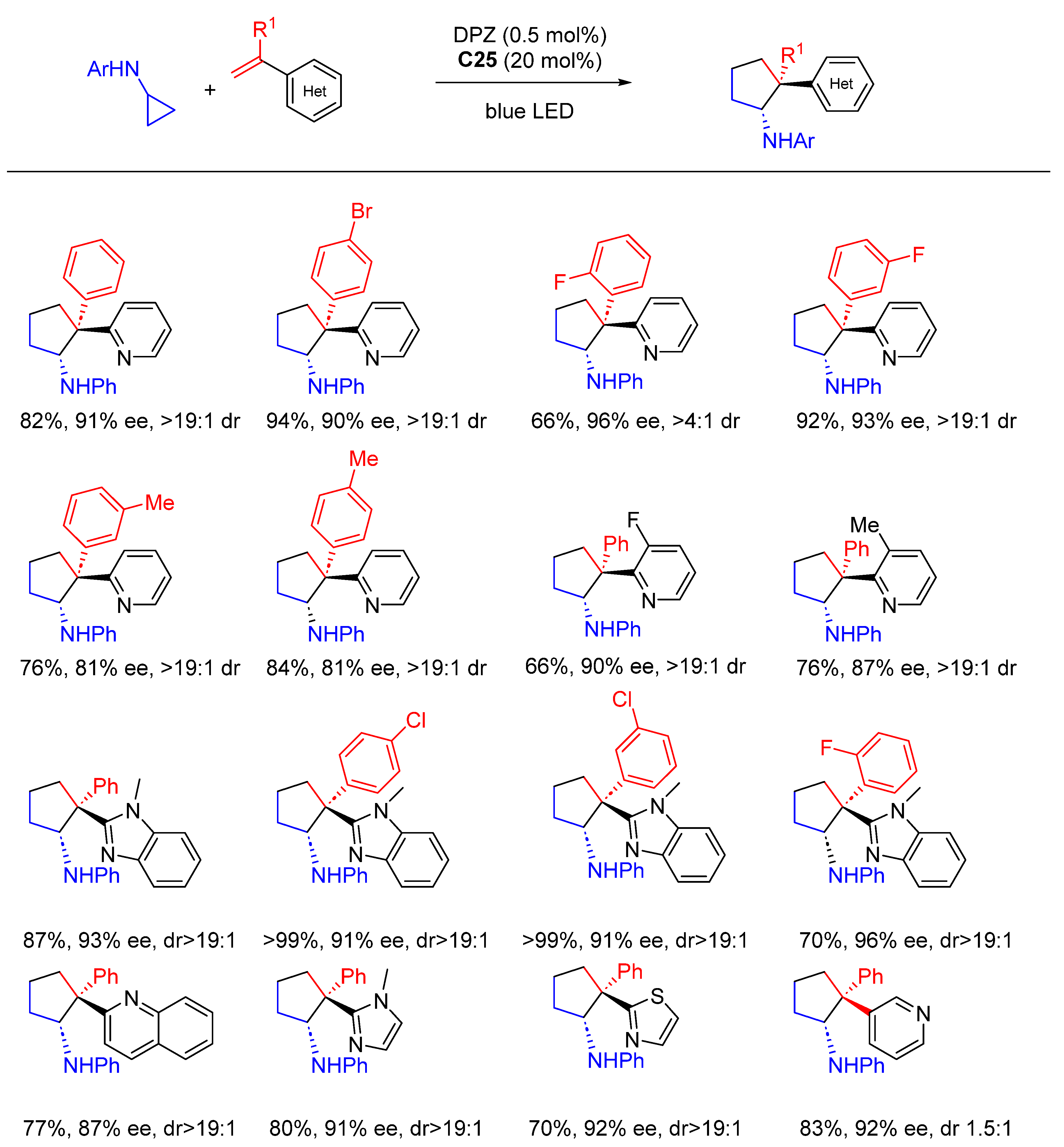{kind=link}
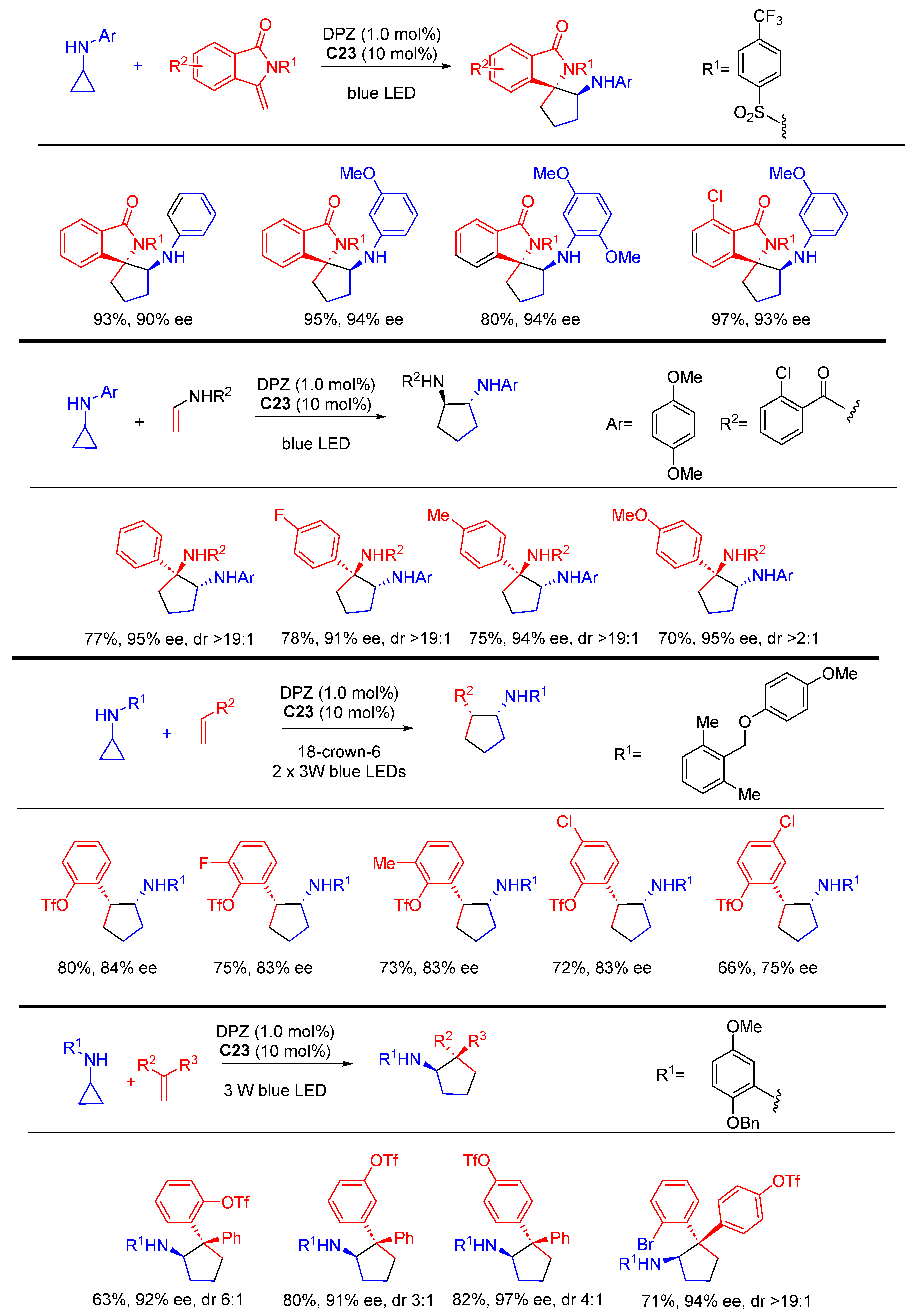{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
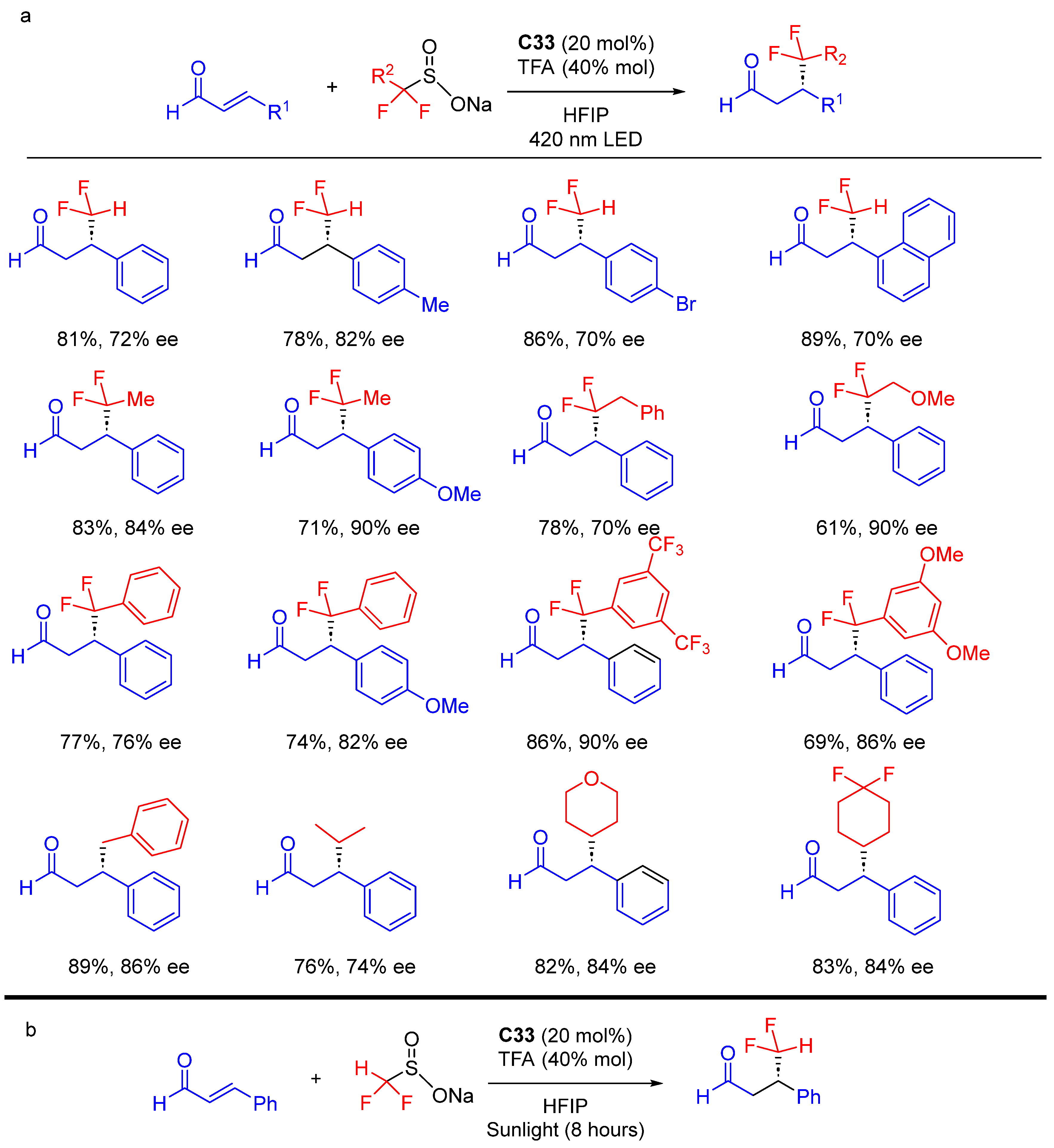{kind=link}
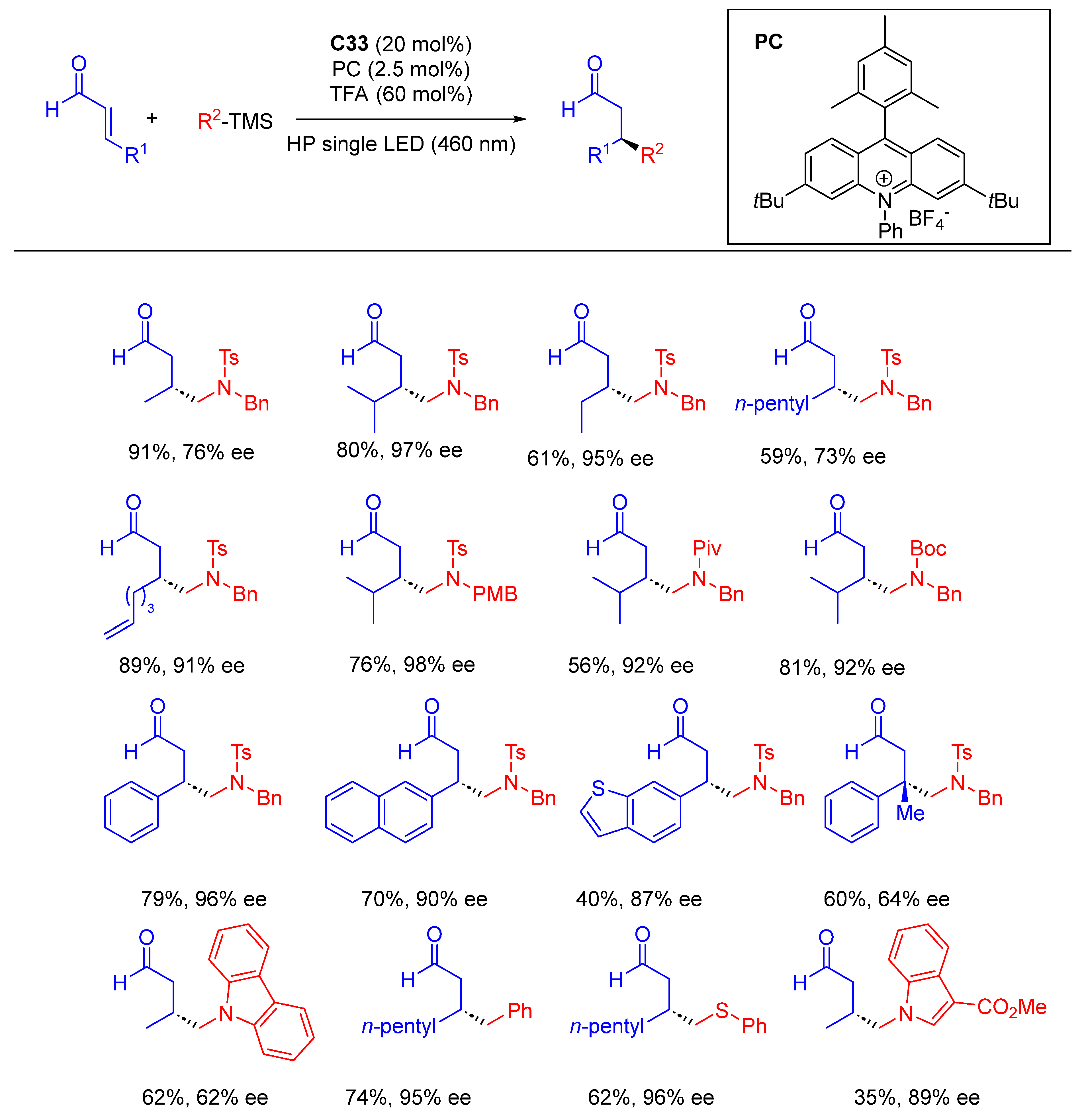{kind=link}
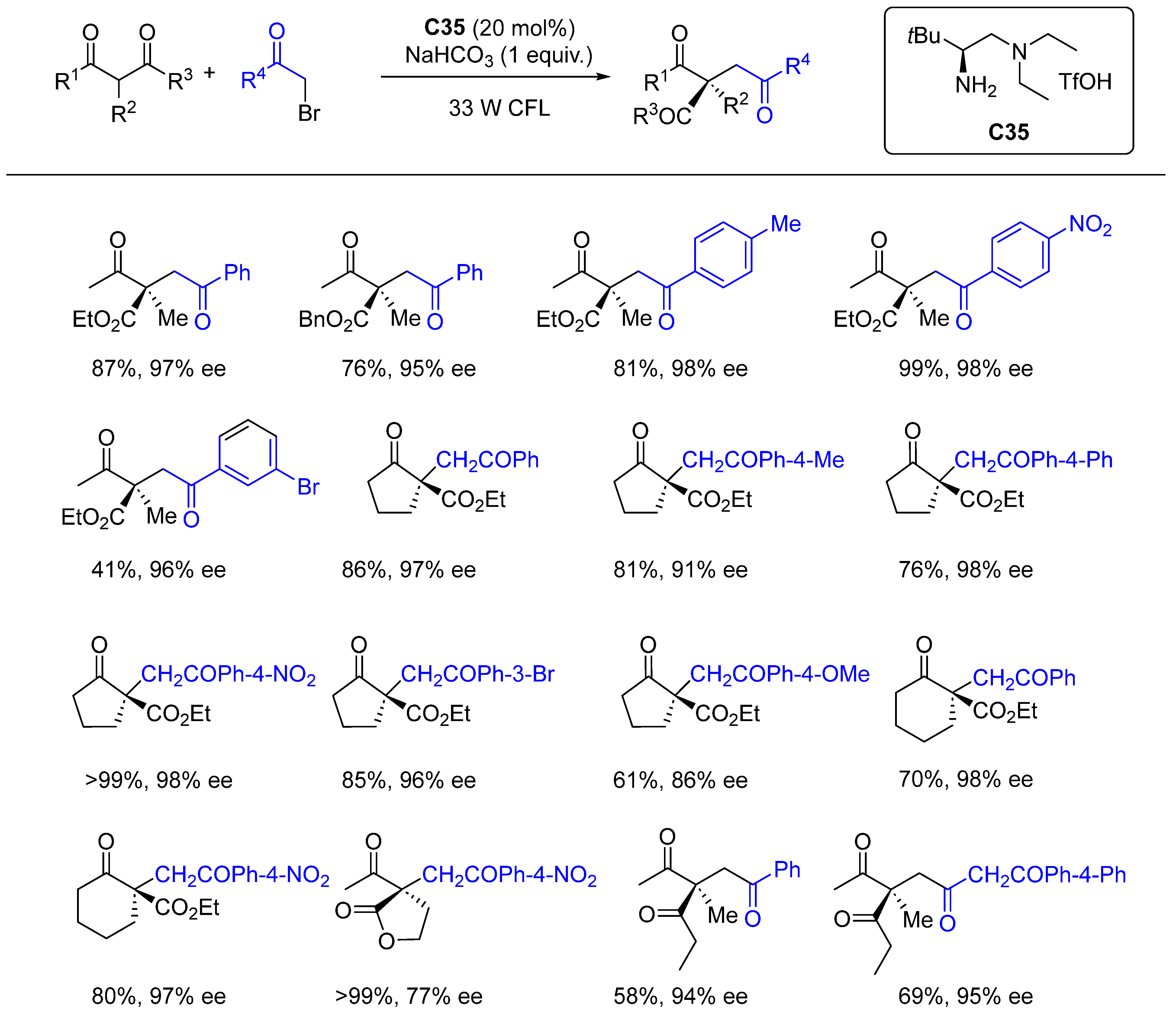{kind=link}
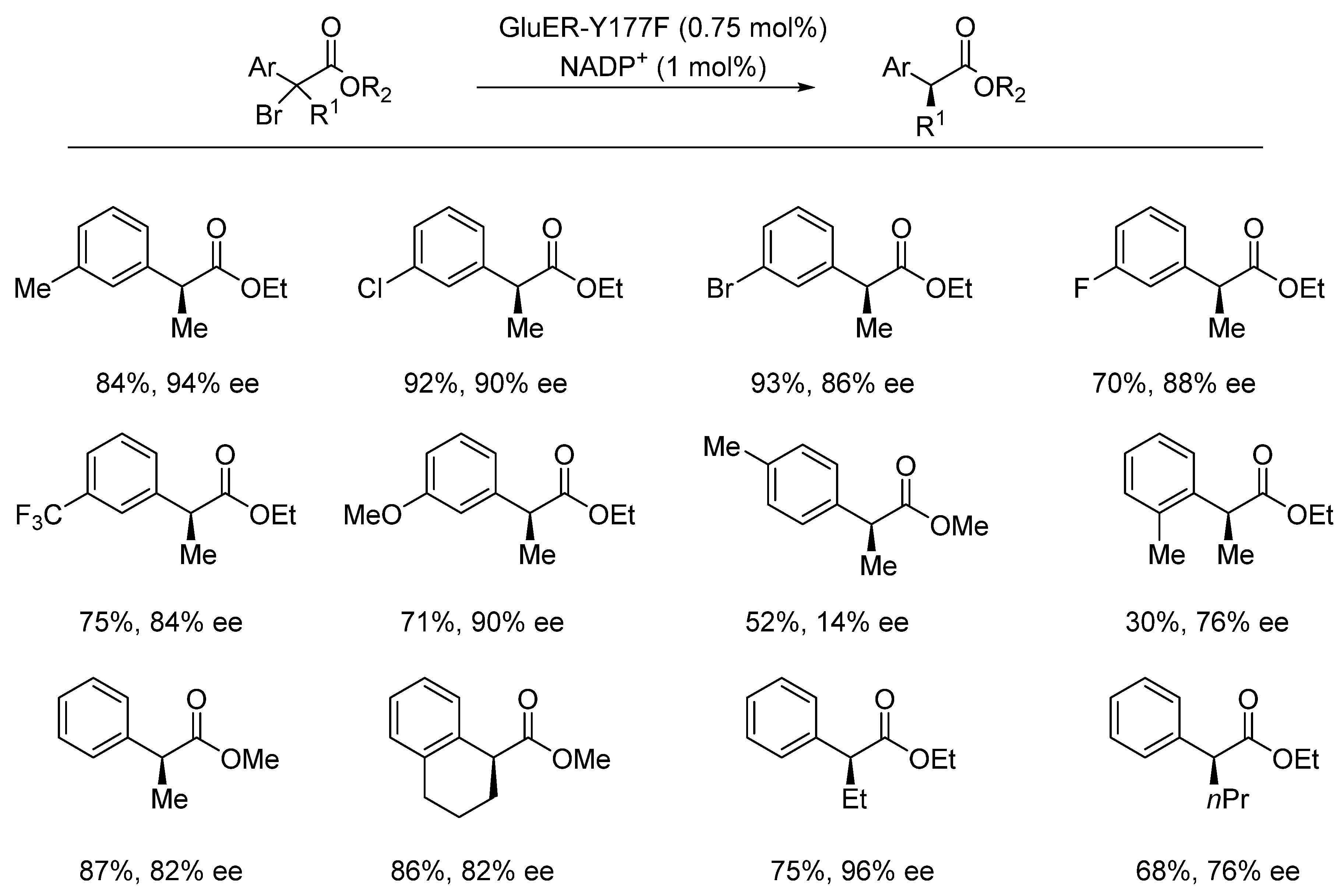{kind=link}
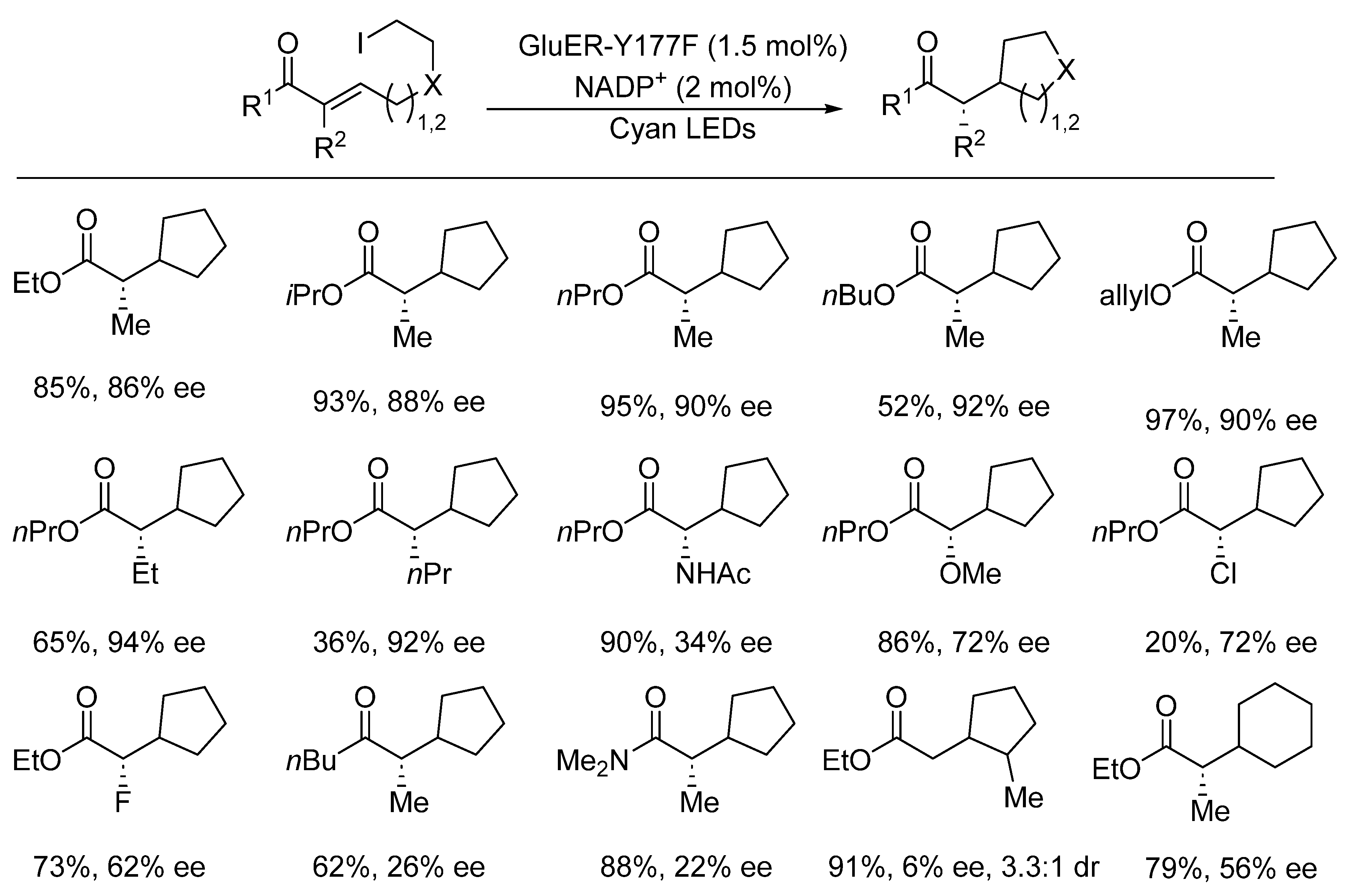{kind=link}
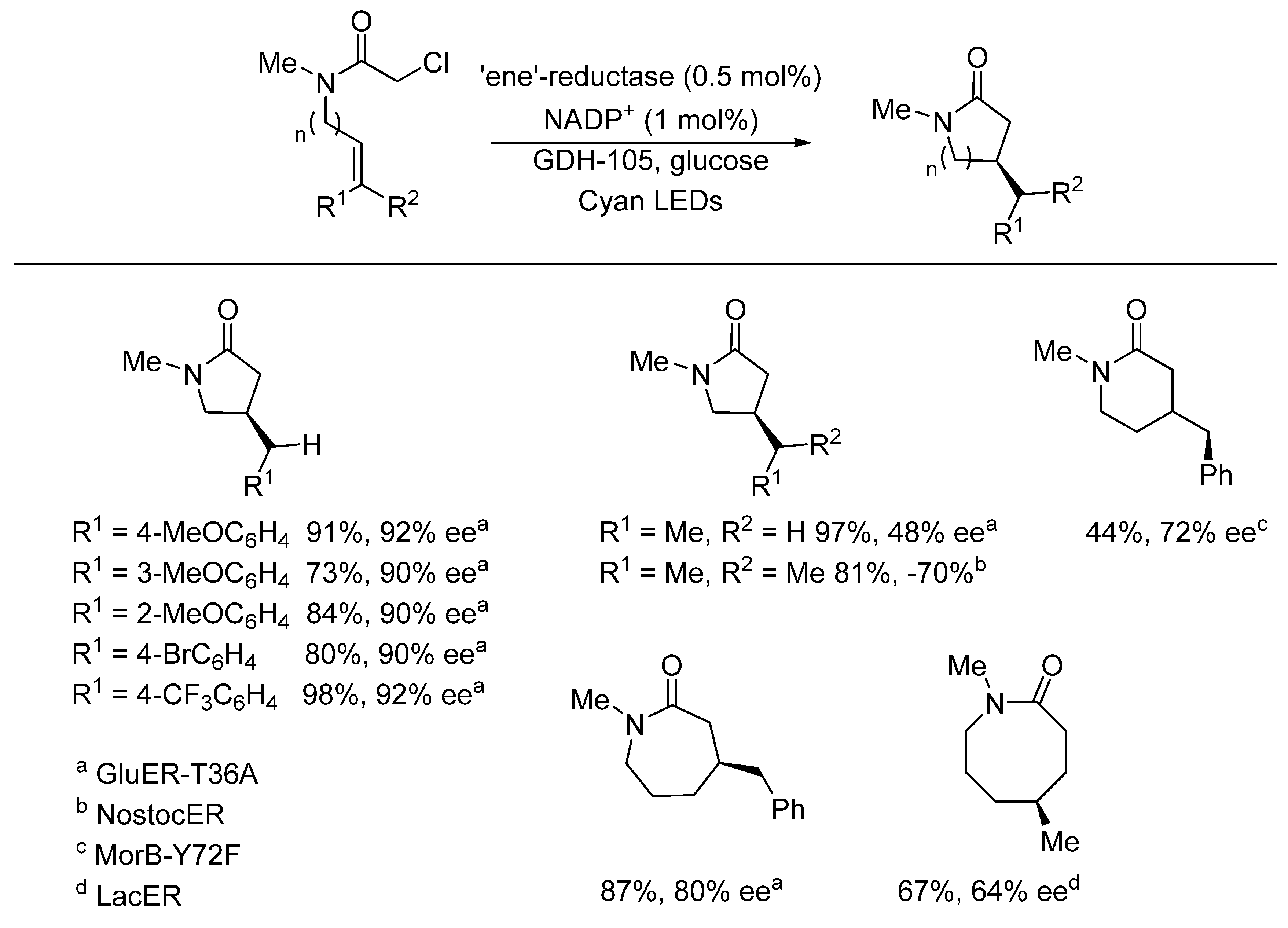{kind=link}
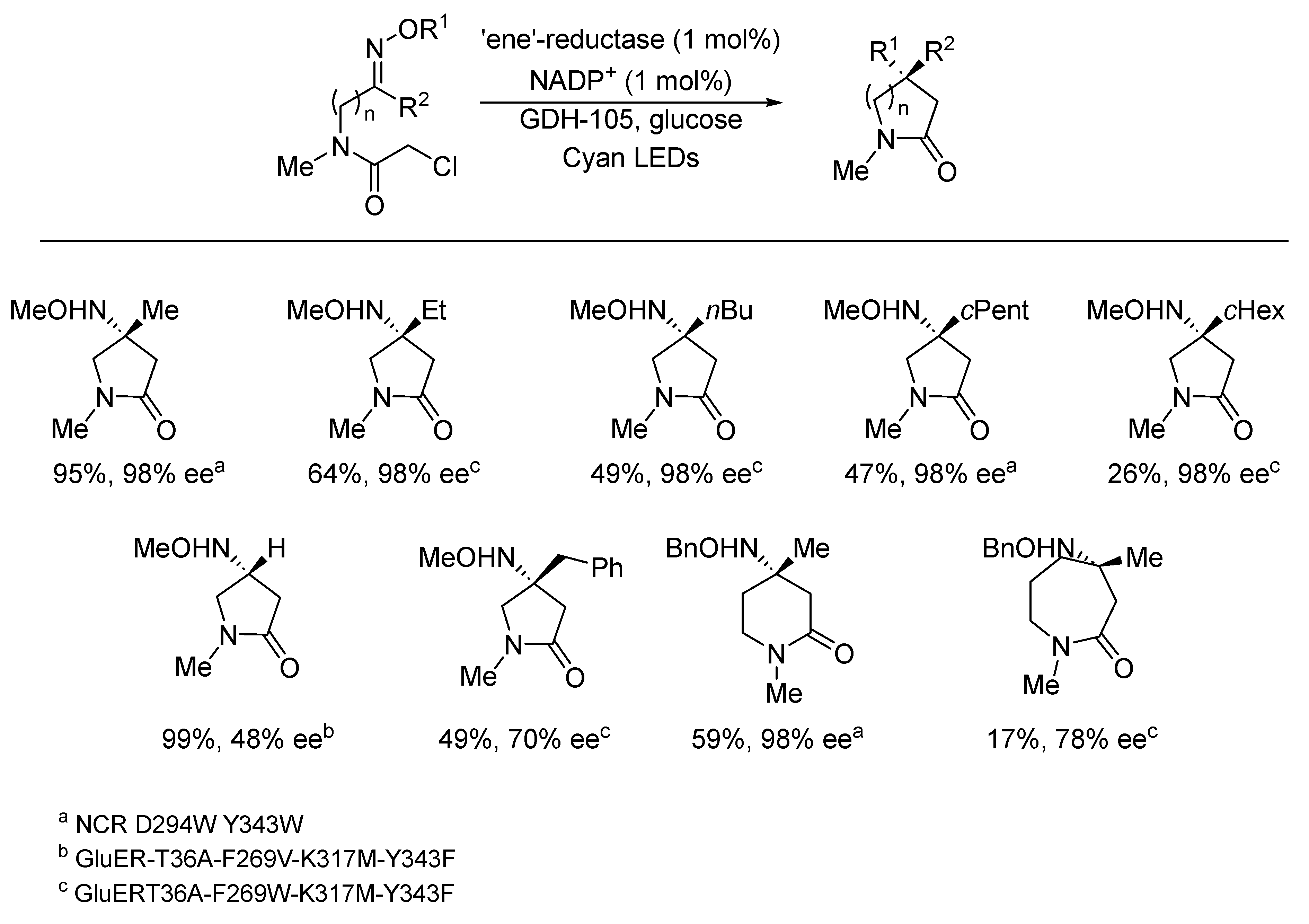{kind=link}
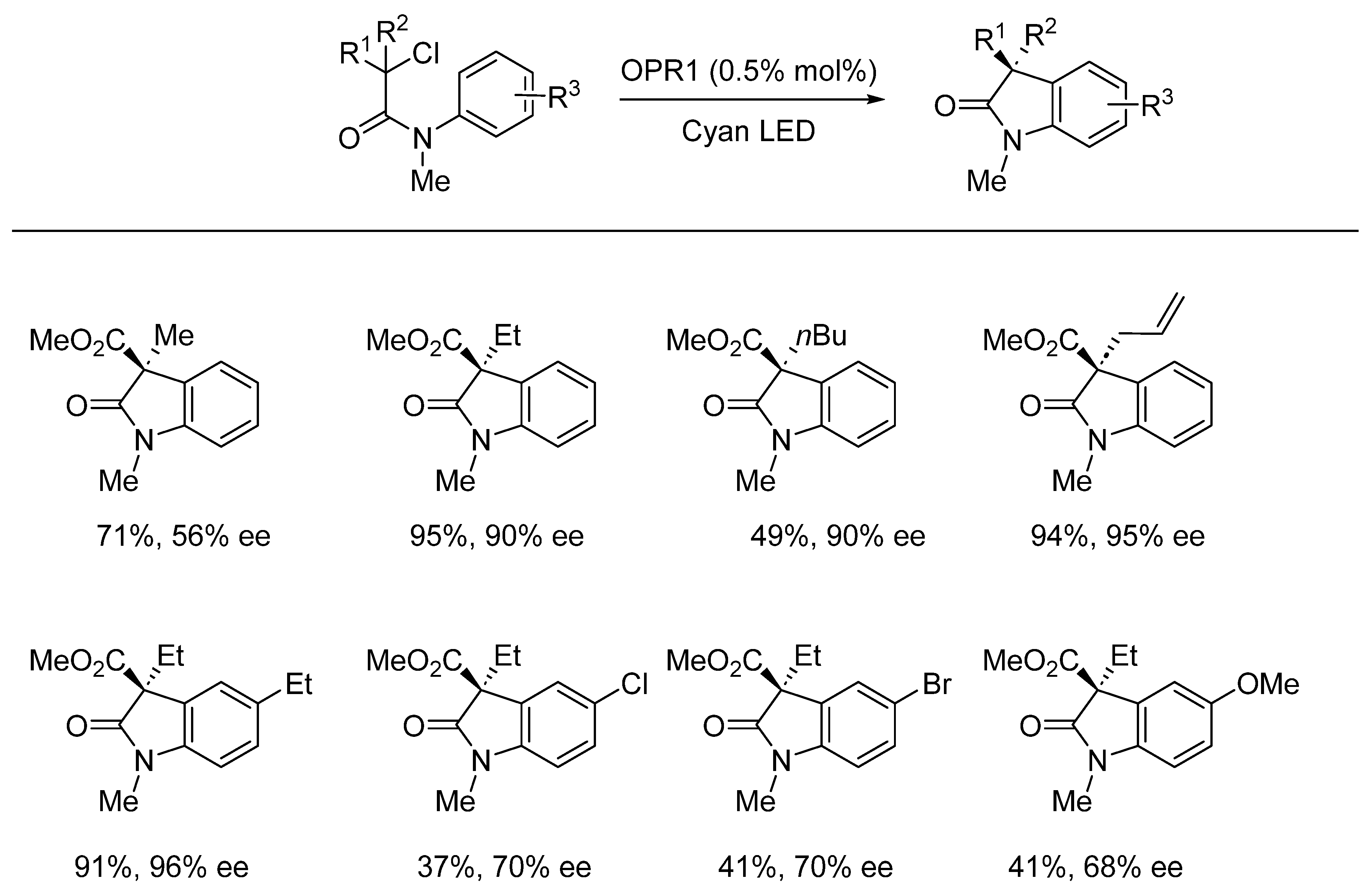{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
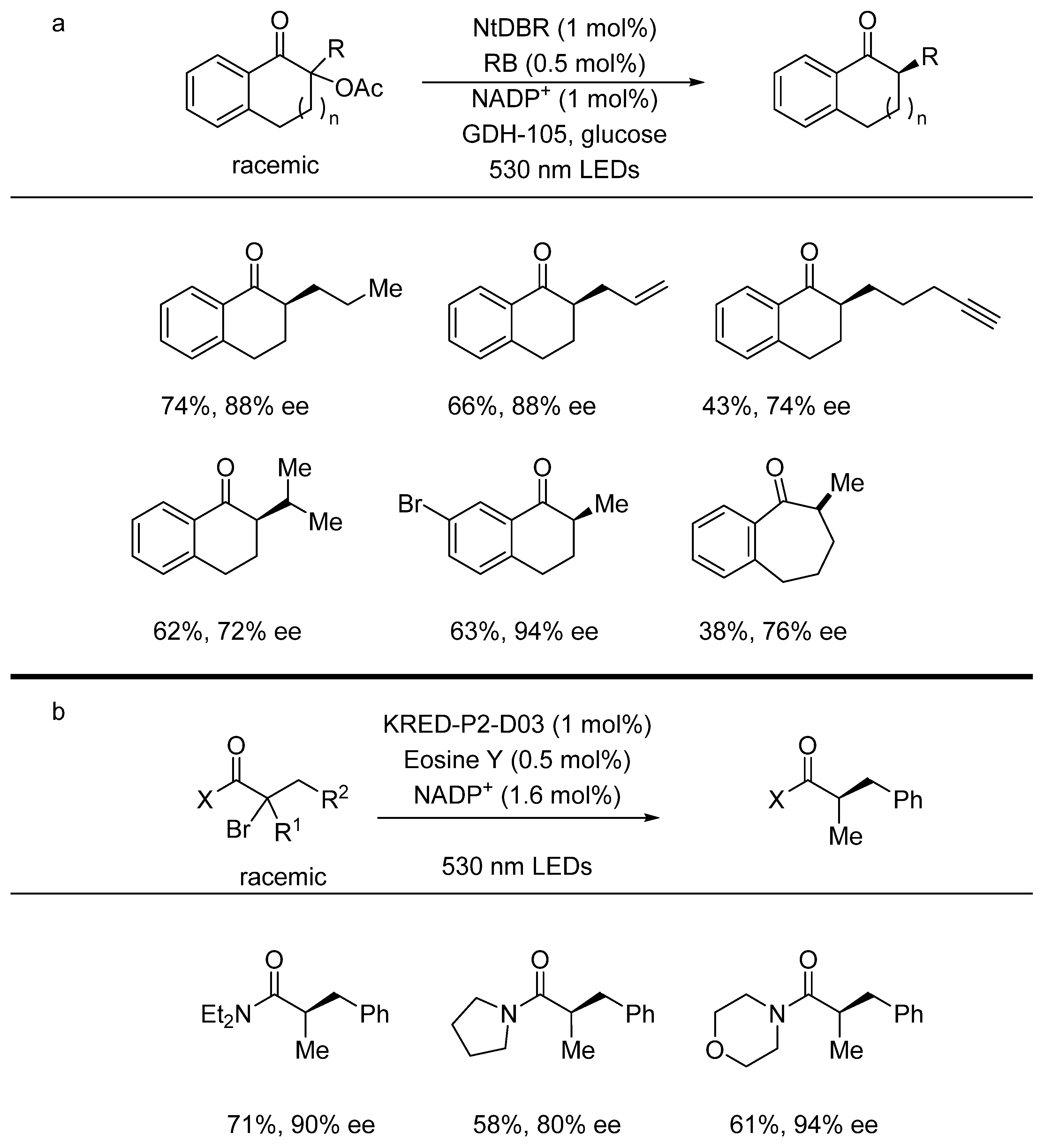{kind=link}
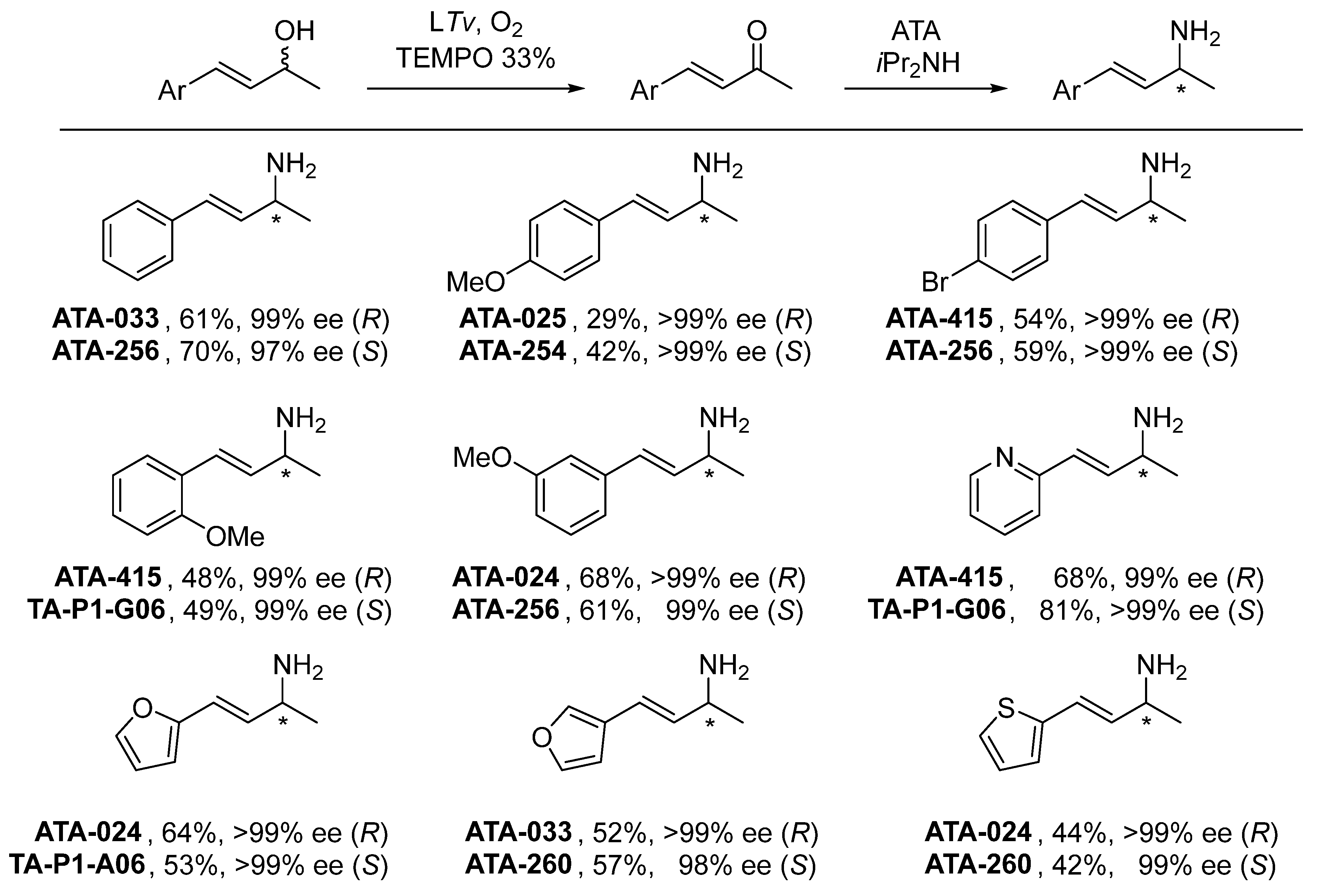{kind=link}
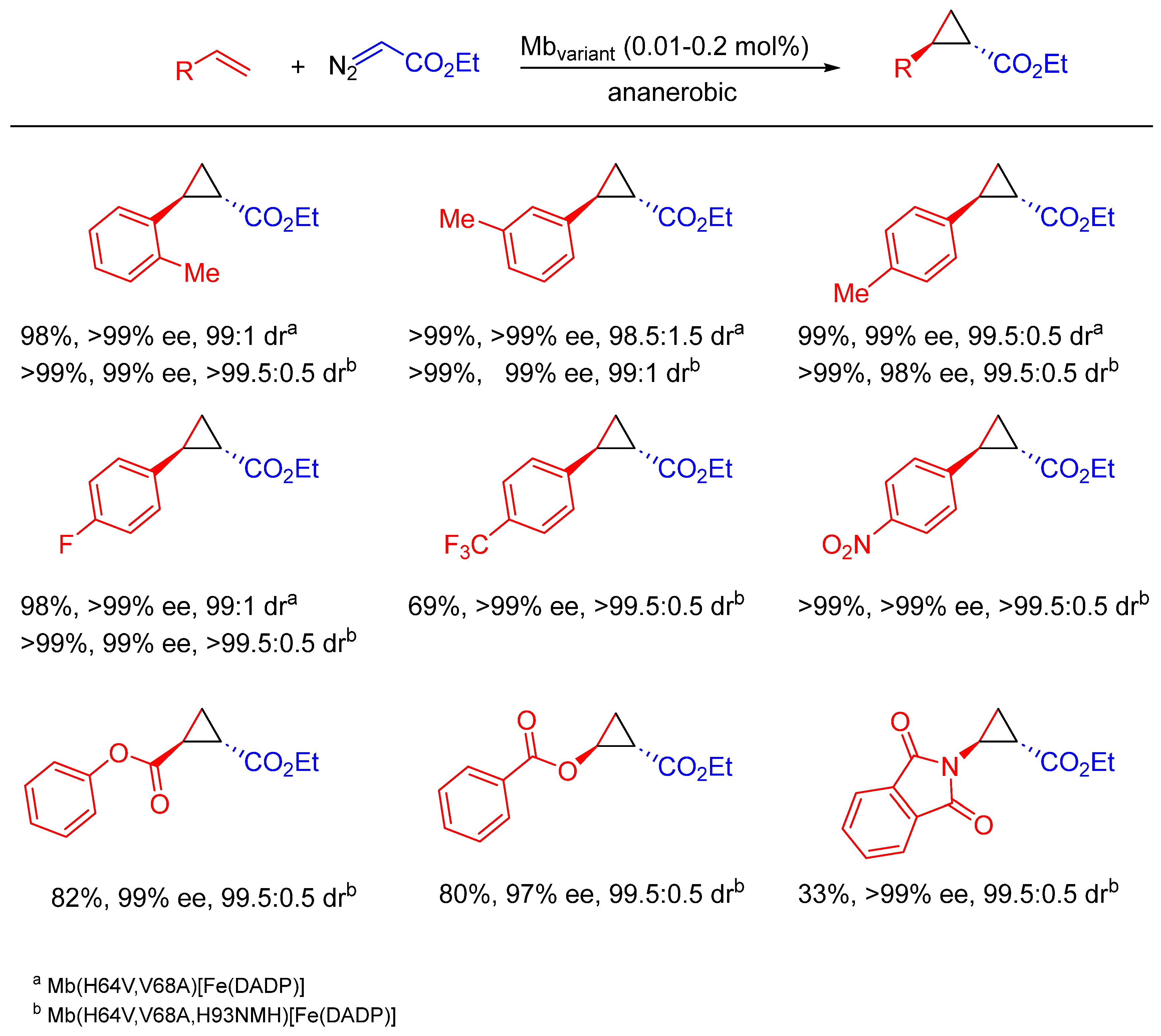{kind=link}
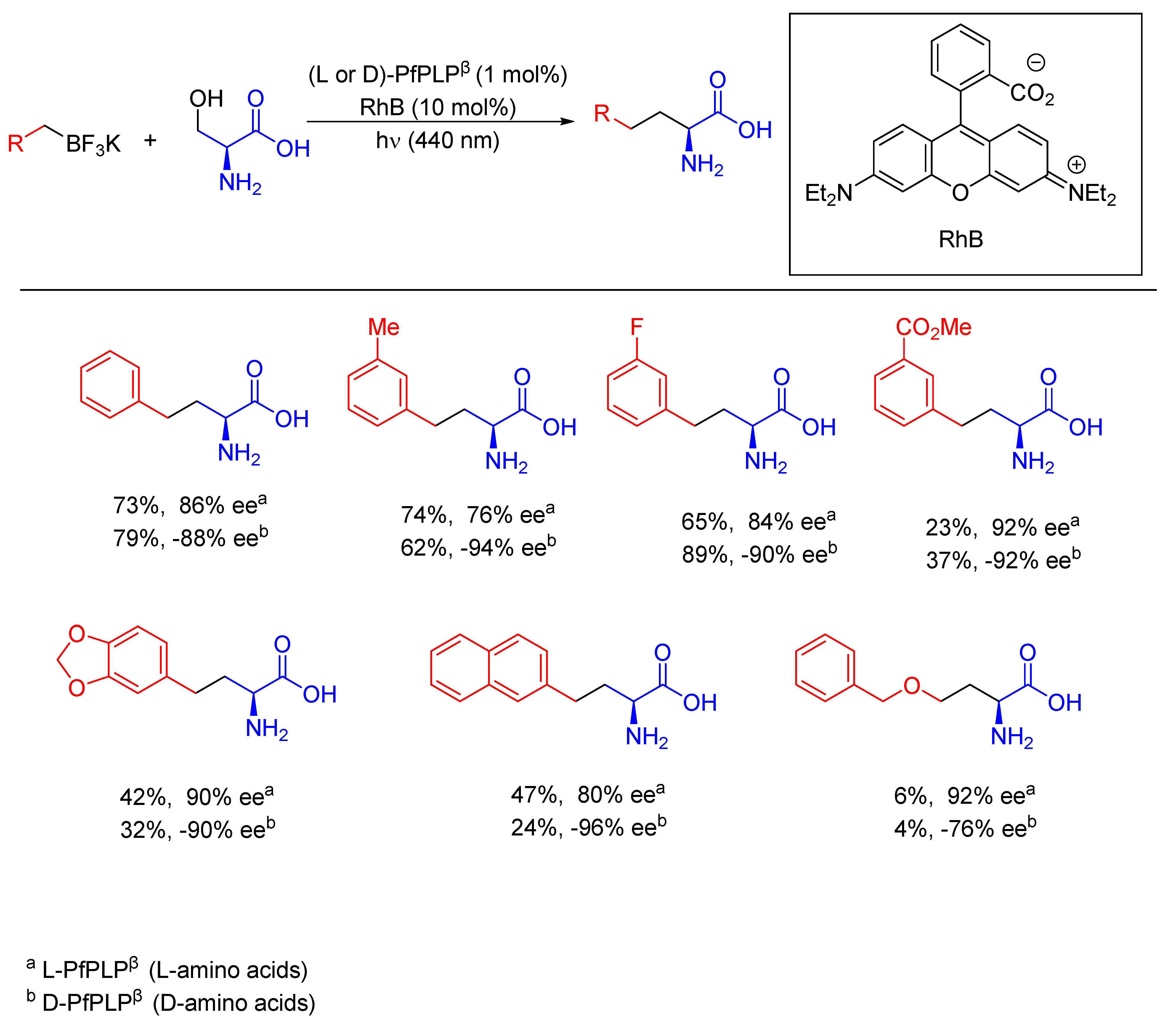{kind=link}
1. Introduction
- Transition metal catalysis—the catalyst not only creates chiral environment by coordination to a substrate, but also plays important role in the radical generation process;
- Visible light photoredox radical reactions combined with transition metal catalysis;
- Visible light photoredox radical reactions combined with organocatalysis including dual transition metal/organocatalytic processes.
2. Transition Metal Catalysis
2.1. Copper-Catalyzed Enantioselective Radical Reactions
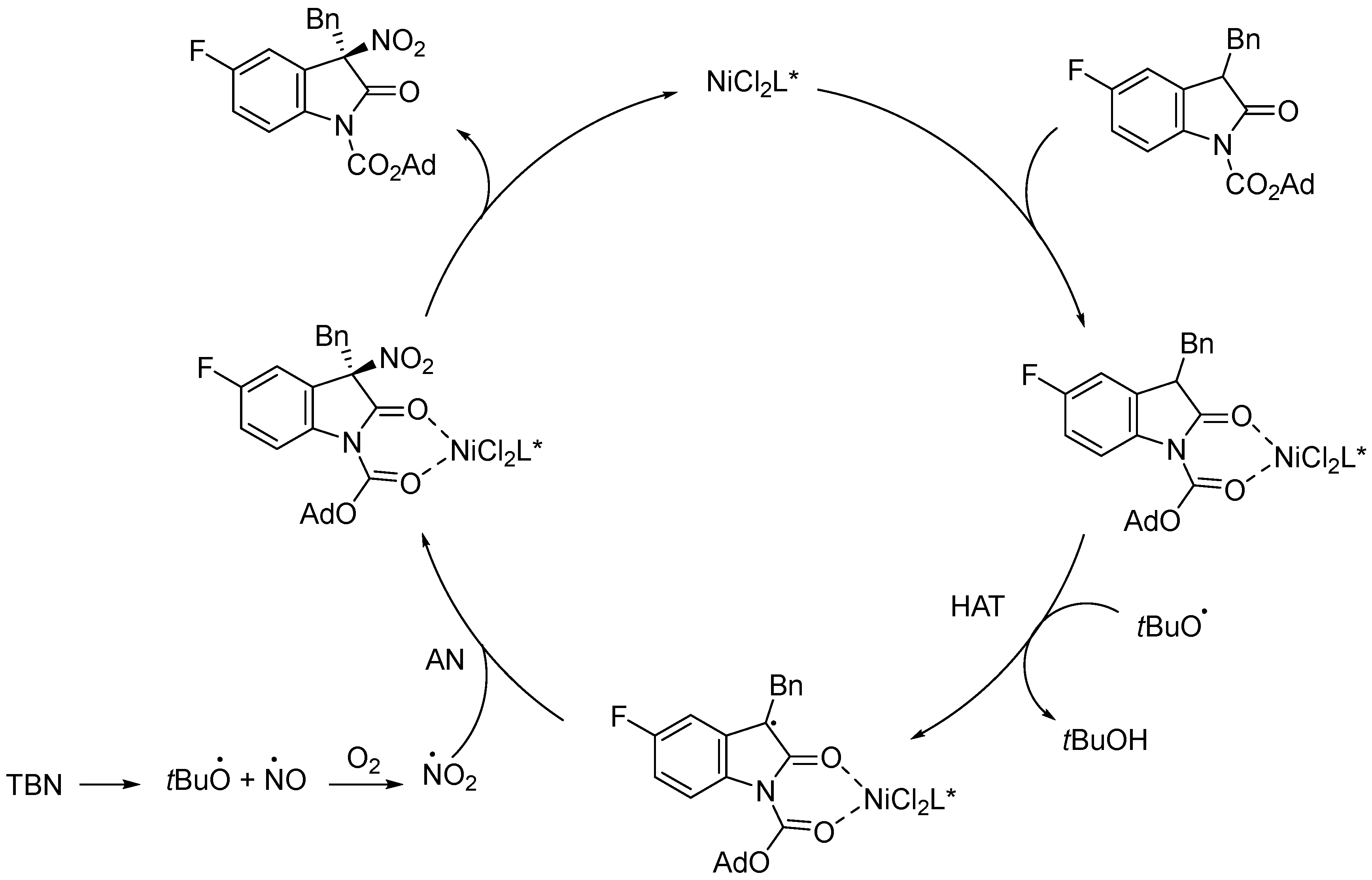
2.2. Nickel-Catalyzed Enantioselective Radical Reactions
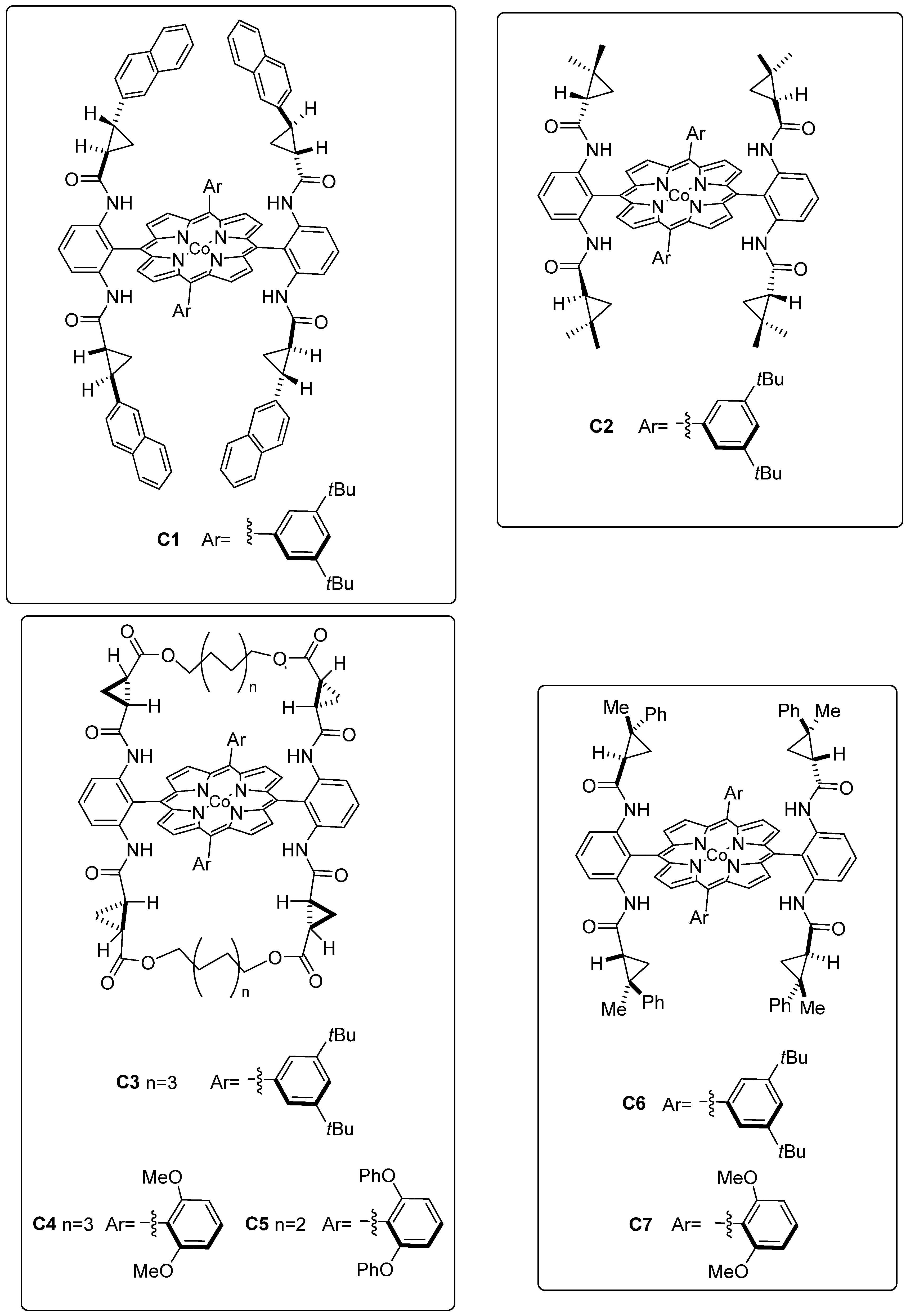
2.3. Cobalt-Catalyzed Enantioselective Radical Reactions
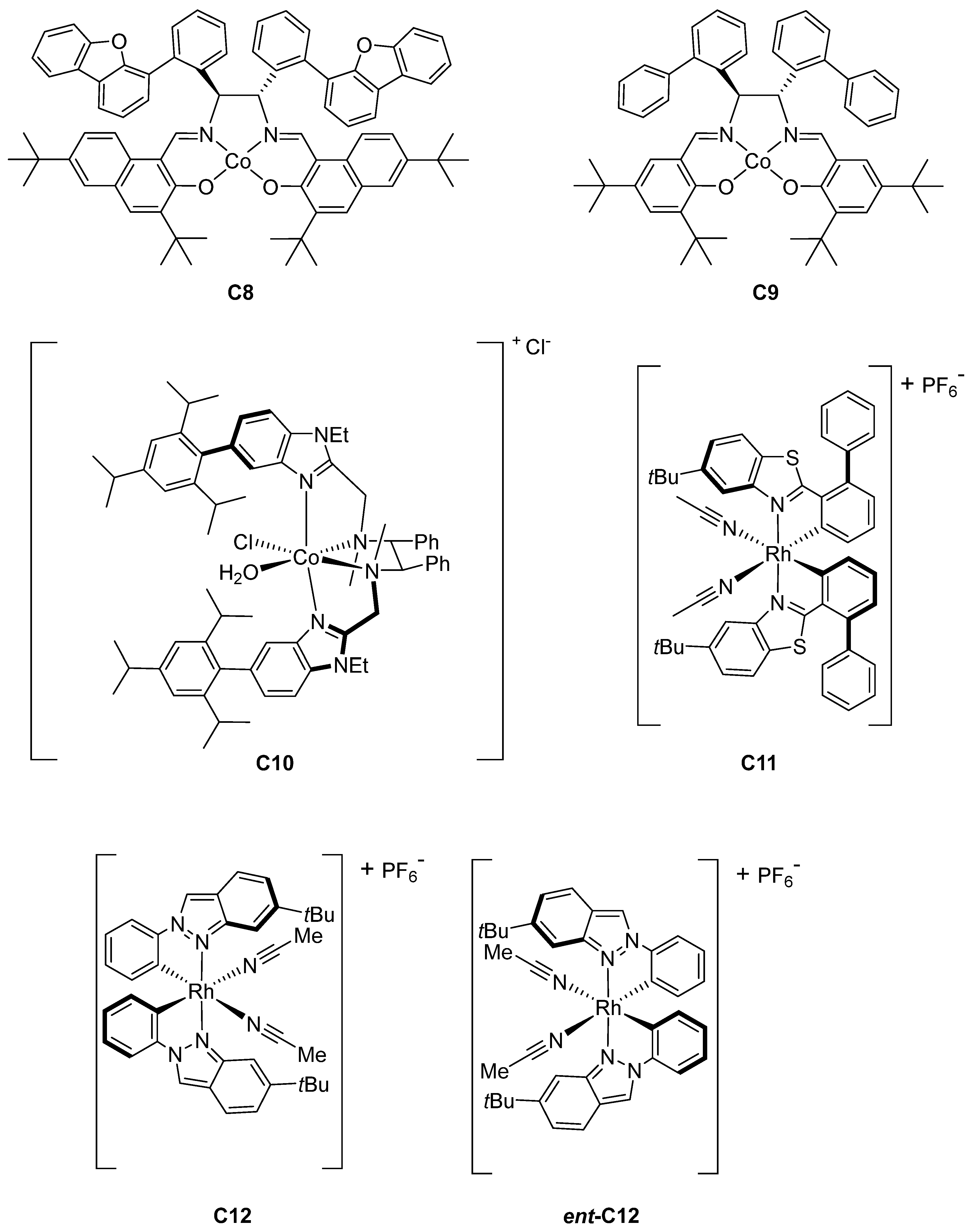
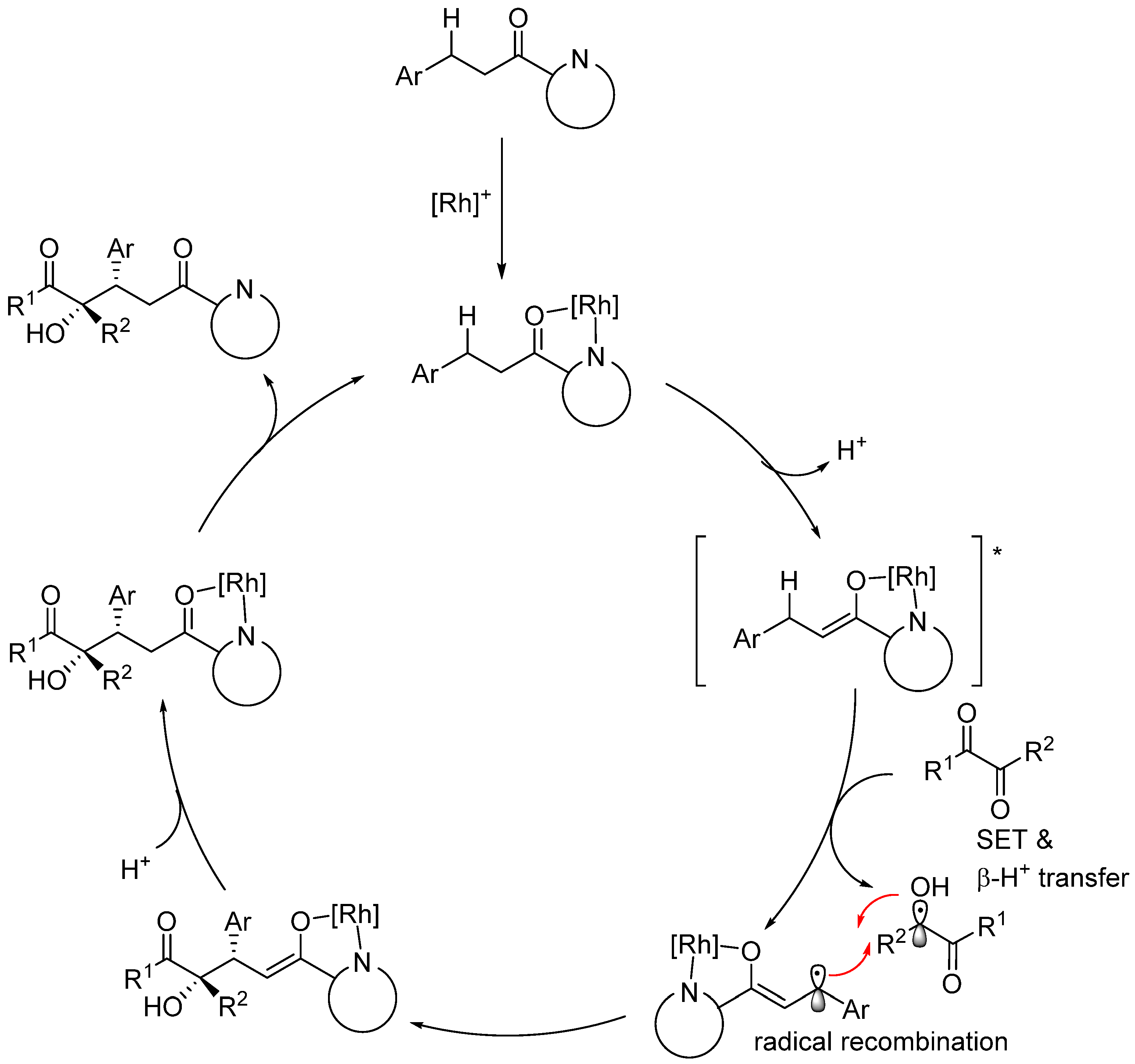
2.4. Rhodium-Catalyzed Enantioselective Radical Reactions
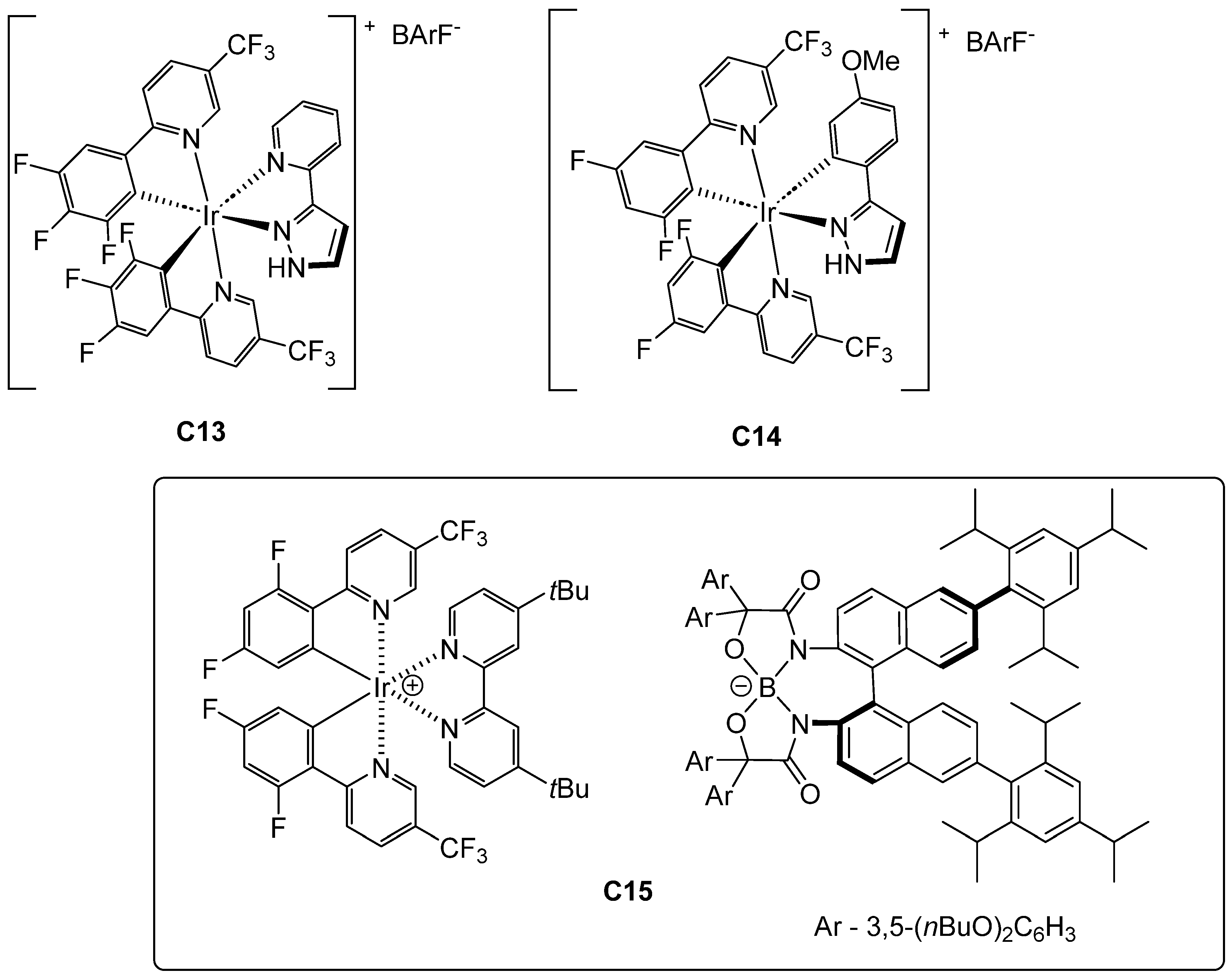
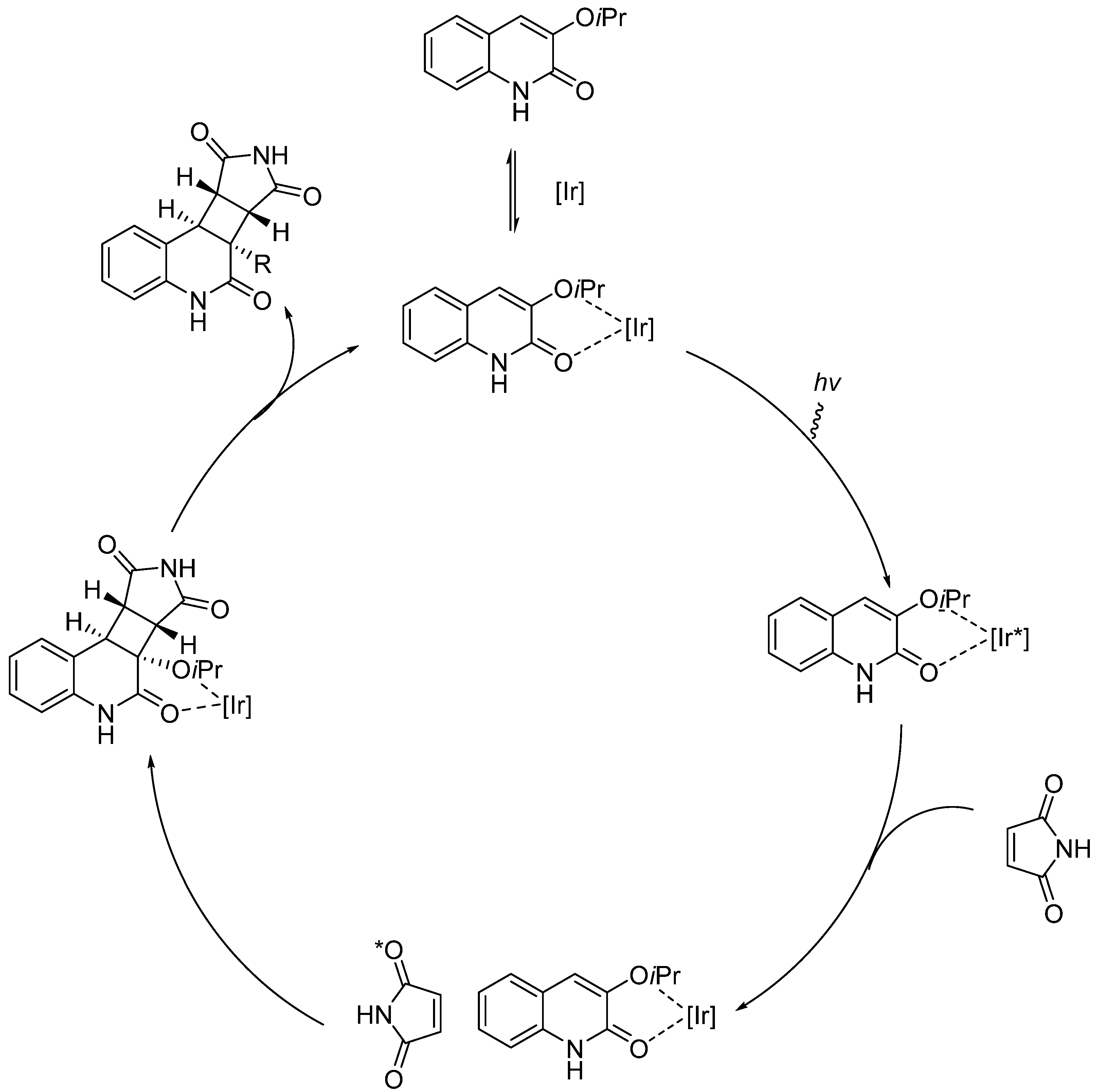
2.5. Iridium-Catalyzed Enantioselective Radical Reactions
2.6. Iron-Catalyzed Enantioselective Radical Reactions
2.7. Catalysis with Complexes of Other Metals
3. Dual Catalysis with Transition Metals and Organocatalysts
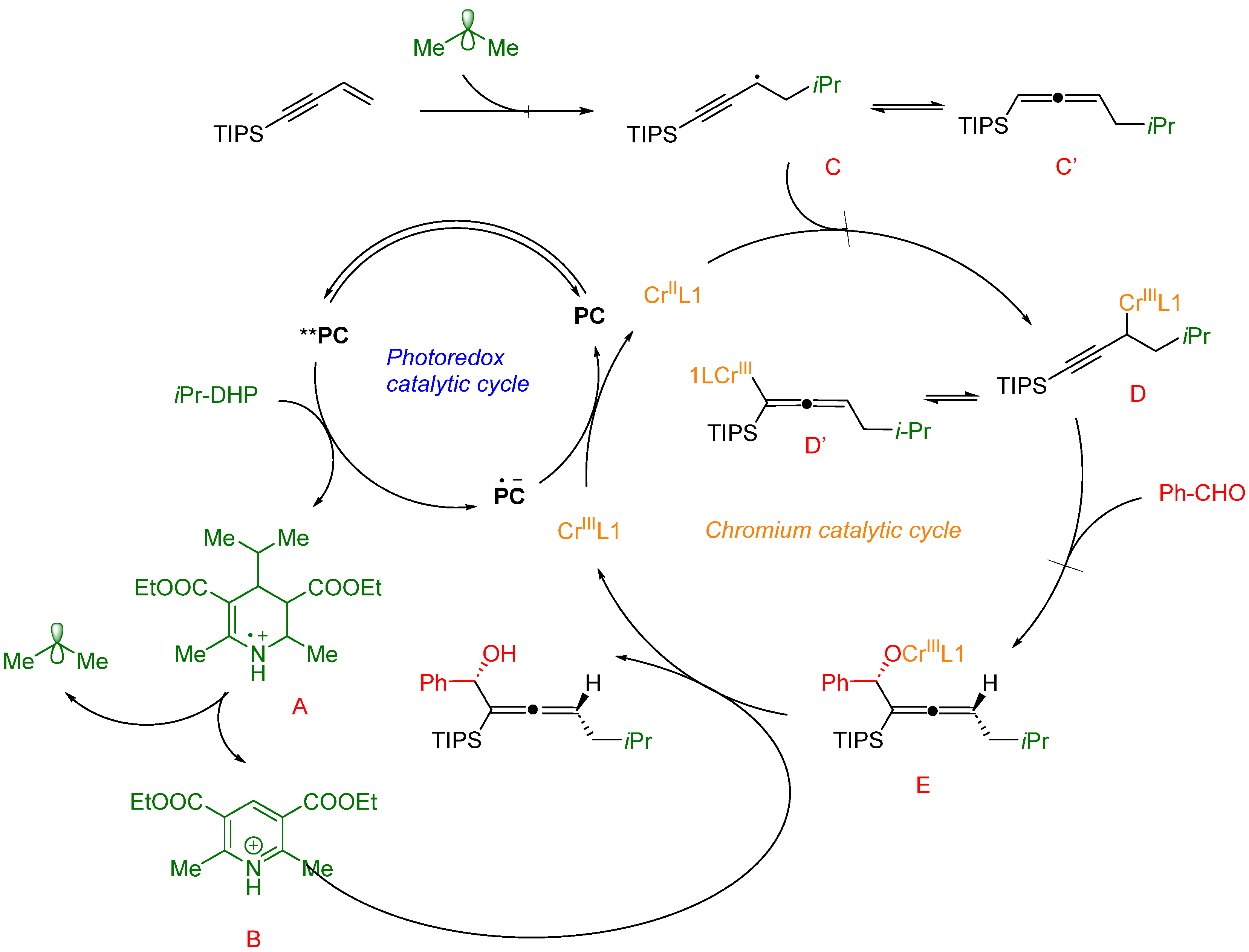
4. Organocatalysis
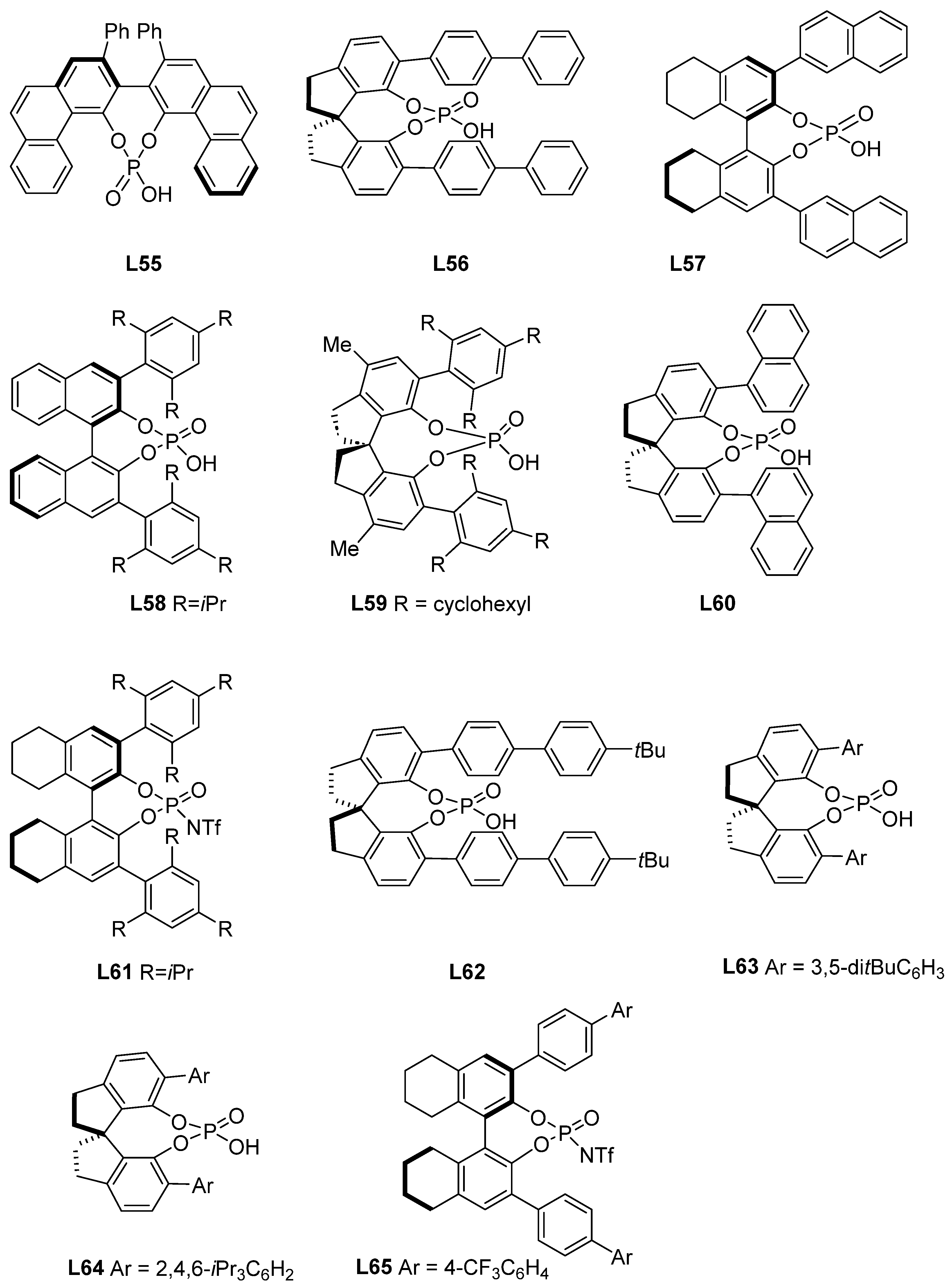
4.1. Chiral Phosphoric Acids as Catalysts
4.2. Double H-Bonding Catalysts
5. Enzyme-Catalyzed Reactions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sibi, M.P.; Ji, J.; Wu, J.H.; Gürtler, S.; Porter, N.A. Chiral Lewis acid catalysis in radical reactions: Enantioselective conjugate radical additions. J. Am. Chem. Soc. 1996, 118, 9200–9201. [Google Scholar] [CrossRef]
- Sibi, M.P.; Porter, N.A. Enantioselective free radical reactions. Acc. Chem. Res. 1999, 32, 163–171. [Google Scholar] [CrossRef]
- Li, Z.-L.; Fang, G.-C.; Gu, Q.-S.; Liu, X.-Y. Recent advances in copper-catalyzed radical involved asymmetric 1,2-difunctionalization of alkenes. Chem. Soc. Rev. 2020, 49, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Larionov, V.A.; Stoletova, N.V.; Maleev, V.I. Advances in Asymmetric Amino Acid Synthesis Enabled by Radical Chemistry. Adv. Synth. Catal. 2020, 362, 4325–4367. [Google Scholar] [CrossRef]
- Garrido-Castro, A.F.; Maestro, M.C.; Alemán, J. Asymmetric induction in photocatalysis—Discovering a new side to light-driven chemistry. Tetrahedron Lett. 2018, 59, 1286–1294. [Google Scholar] [CrossRef]
- Yao, W.; Bazan-Bergamino, E.A.; Ngai, M. Asymmetric Photocatalysis Enabled by Chiral Organocatalysts. ChemCatChem 2022, 14, e202101292. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Meggers, E. Asymmetric Photocatalysis with Bis-cyclometalated Rhodium Complexes. Acc. Chem. Res. 2019, 52, 833–847. [Google Scholar] [CrossRef]
- Tao, Z.-L.; Denmark, S.E. Catalytic, Enantioselective Diamination of Alkenes. Synthesis 2021, 53, 3951–3962. [Google Scholar] [CrossRef]
- Wang, F.; Chen, P.; Liu, G. Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res. 2018, 51, 2036–2046. [Google Scholar] [CrossRef]
- Proctor, R.S.J.; Colgan, A.C.; Phipps, R.J. Exploiting attractive non-covalent interactions for the enantioselective catalysis of reactions involving radical intermediates. Nat. Chem. 2020, 12, 990–1004. [Google Scholar] [CrossRef]
- Chen, D.-F.; Gong, L.-Z. Organo/Transition-Metal Combined Catalysis Rejuvenates Both in Asymmetric Synthesis. J. Am. Chem. Soc. 2022, 144, 2415–2437. [Google Scholar] [CrossRef] [PubMed]
- Vega-Peñaloza, A.; Paria, S.; Bonchio, M.; Dell’amico, L.; Companyó, X. Profiling the Privileges of Pyrrolidine-Based Catalysts in Asymmetric Synthesis: From Polar to Light-Driven Radical Chemistry. ACS Catal. 2019, 9, 6058–6072. [Google Scholar] [CrossRef]
- Wang, K.; Kong, W. Recent Advances in Transition Metal-Catalyzed Asymmetric Radical Reactions. J. Chem. 2018, 36, 247–256. [Google Scholar] [CrossRef]
- Nielsen, C.D.-T.; Linfoot, J.D.; Williams, A.F.; Spivey, A.C. Recent progress in asymmetric synergistic catalysis—The judicious combination of selected chiral aminocatalysts with achiral metal catalysts. Org. Biomol. Chem. 2022, 20, 2764–2778. [Google Scholar] [CrossRef]
- Genzink, M.J.; Kidd, J.B.; Swords, W.B.; Yoon, T.P. Chiral Photocatalyst Structures in Asymmetric Photochemical Synthesis. Chem. Rev. 2022, 122, 1654–1716. [Google Scholar] [CrossRef]
- Mondal, S.; Dumur, F.; Gigmes, D.; Sibi, M.P.; Bertrand, M.P.; Nechab, M. Enantioselective Radical Reactions Using Chiral Cata-lysts. Chem. Rev. 2022, 122, 5842–5976. [Google Scholar] [CrossRef]
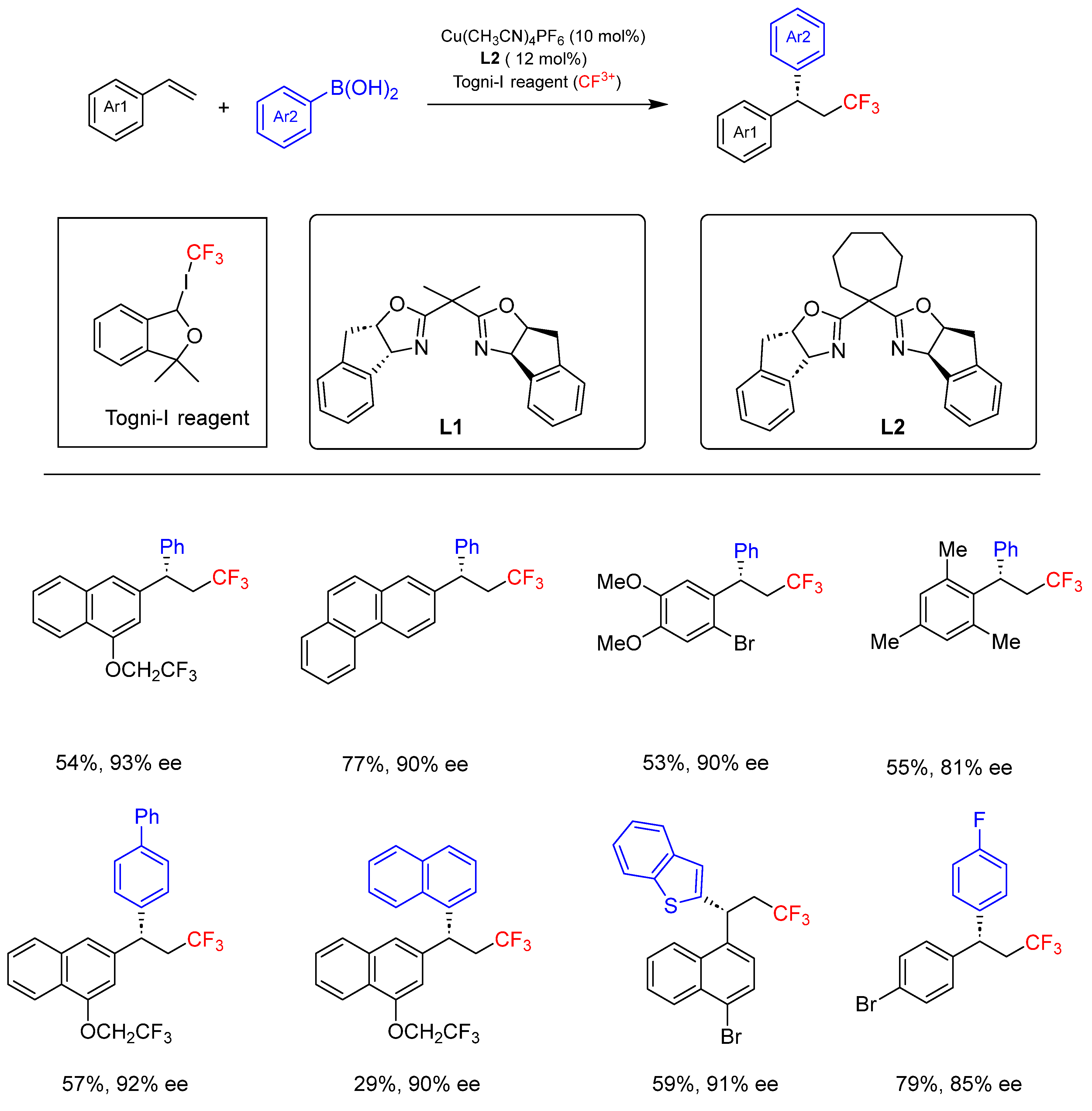
- Wu, L.; Wang, F.; Wan, X.; Wang, D.; Chen, P.; Liu, G. Asymmetric Cu-Catalyzed Intermolecular Trifluoromethylarylation of Styrenes: Enantioselective Arylation of Benzylic Radicals. J. Am. Chem. Soc. 2017, 139, 2904–2907. [Google Scholar] [CrossRef]
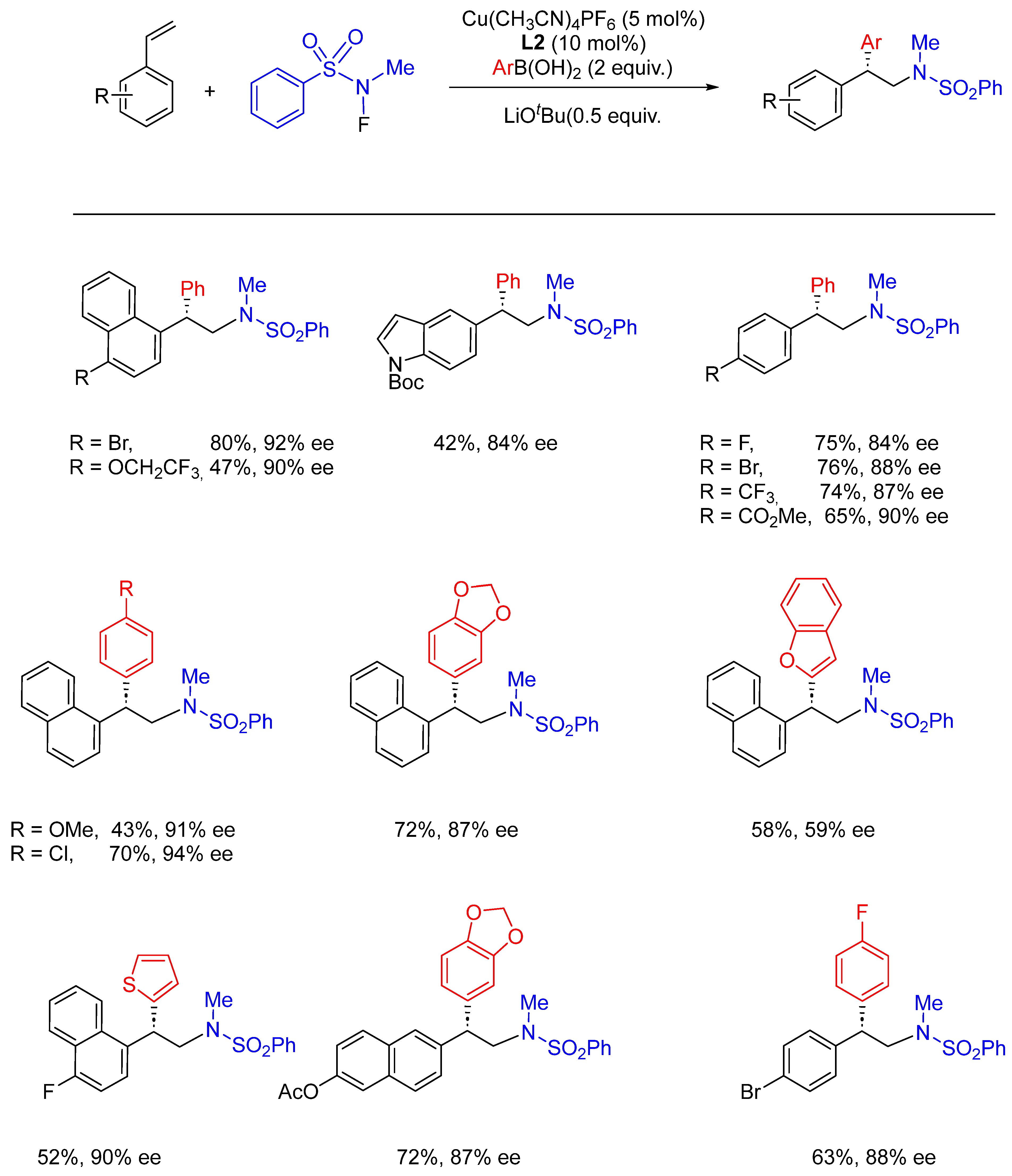
- Wang, D.; Wu, L.; Wang, F.; Wan, X.; Chen, P.; Lin, Z.; Liu, G. Asymmetric Copper-Catalyzed Intermolecular Aminoarylation of Styrenes: Efficient Access to Optical 2,2-Diarylethylamines. J. Am. Chem. Soc. 2017, 139, 6811–6814. [Google Scholar] [CrossRef]
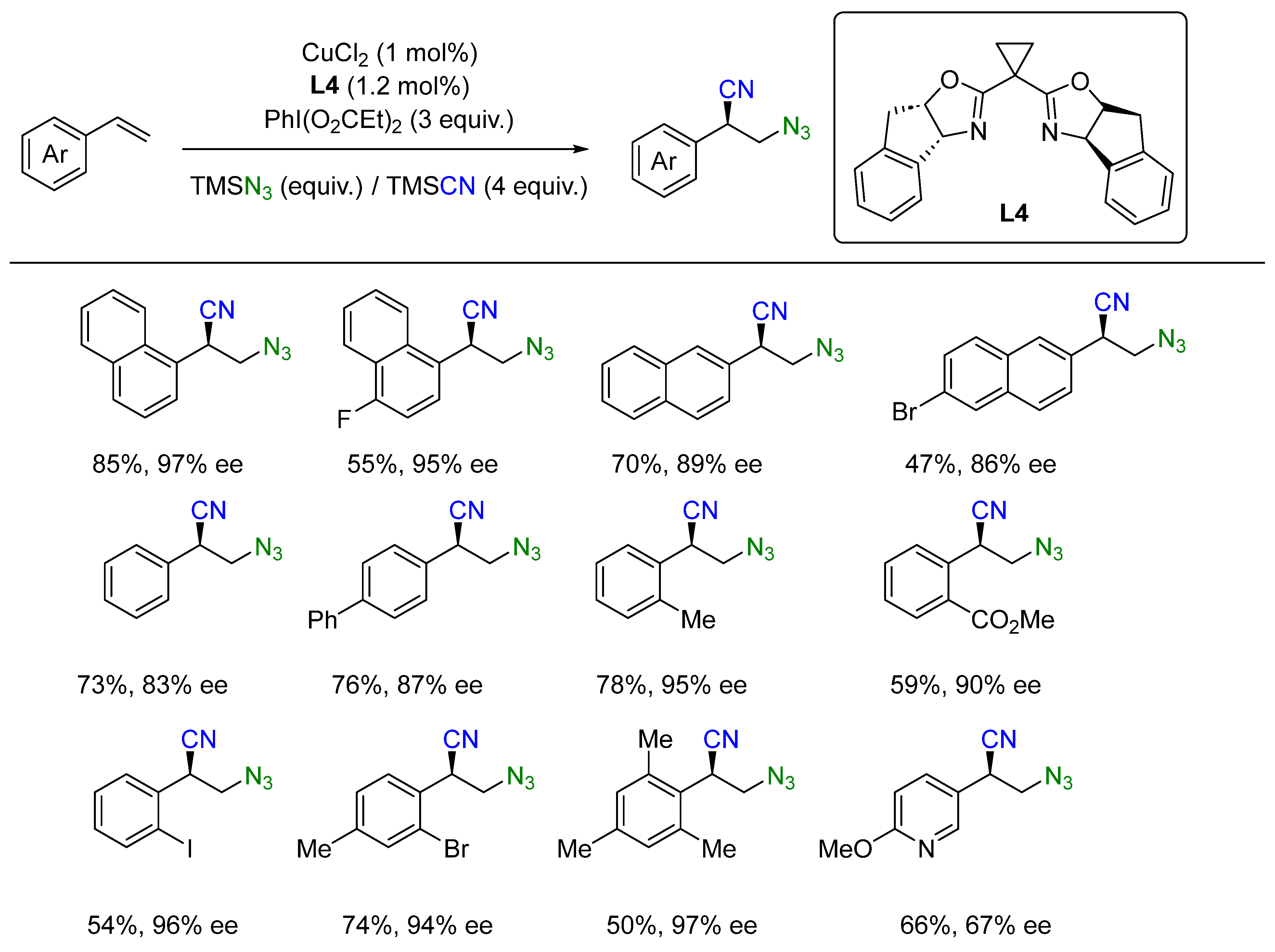
- Wang, D.; Wang, F.; Chen, P.; Lin, Z.; Liu, G. Enantioselective Copper-Catalyzed Intermolecular Amino- and Azidocyanation of Alkenes in a Radical Process. Angew. Chem. Int. Ed. 2017, 56, 2054–2058. [Google Scholar] [CrossRef]
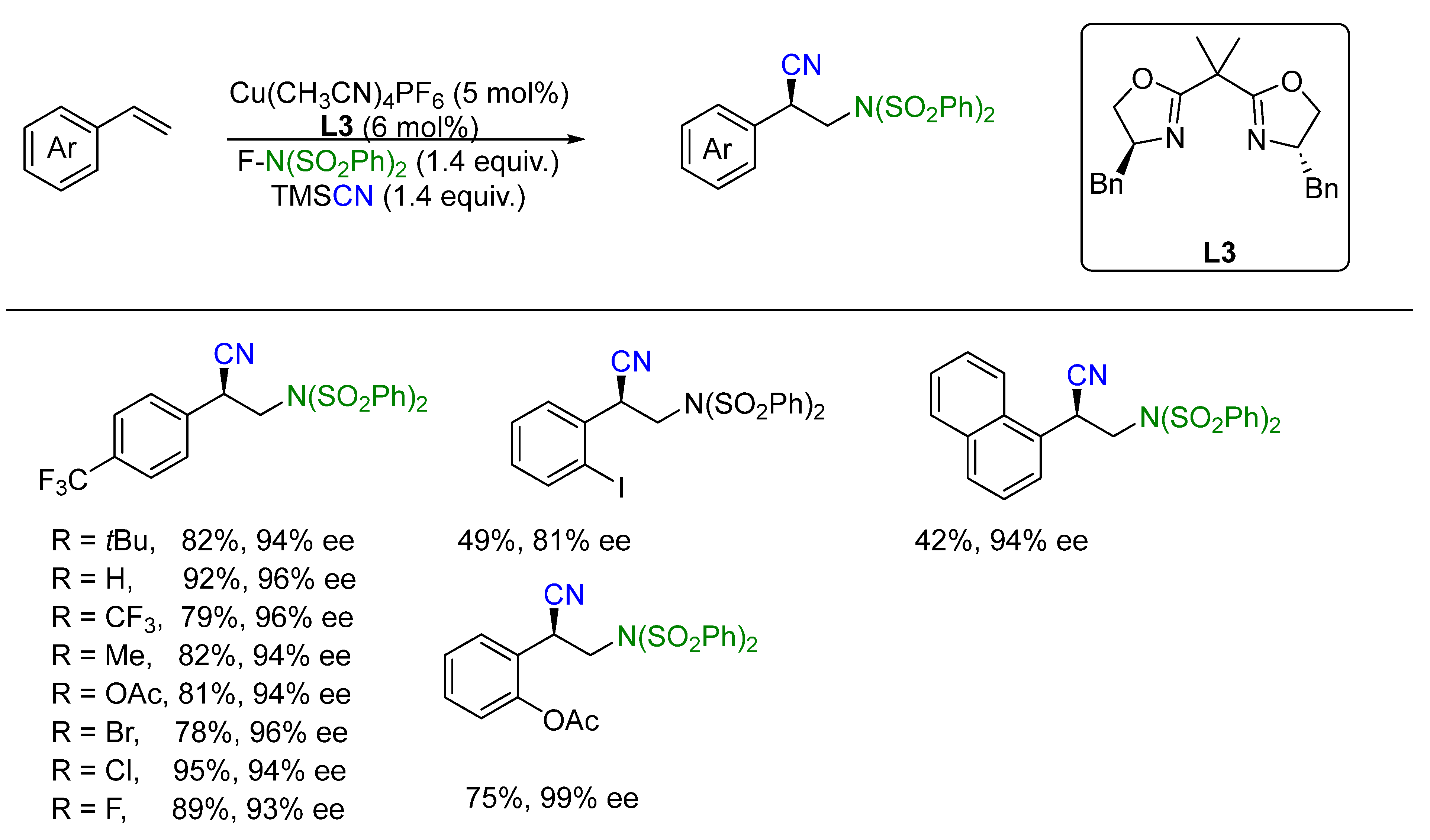
- Zhang, W.; Wang, F.; McCann, S.D.; Wang, D.; Chen, P.; Stahl, S.S.; Liu, G. Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 2016, 353, 1014–1018. [Google Scholar] [CrossRef]
- Wang, F.; Wang, D.; Wan, X.; Wu, L.; Chen, P.; Liu, G. Enantioselective Copper-Catalyzed Intermolecular Cyanotrifluoromethylation of Alkenes via Radical Process. J. Am. Chem. Soc. 2016, 138, 15547–15550. [Google Scholar] [CrossRef] [PubMed]
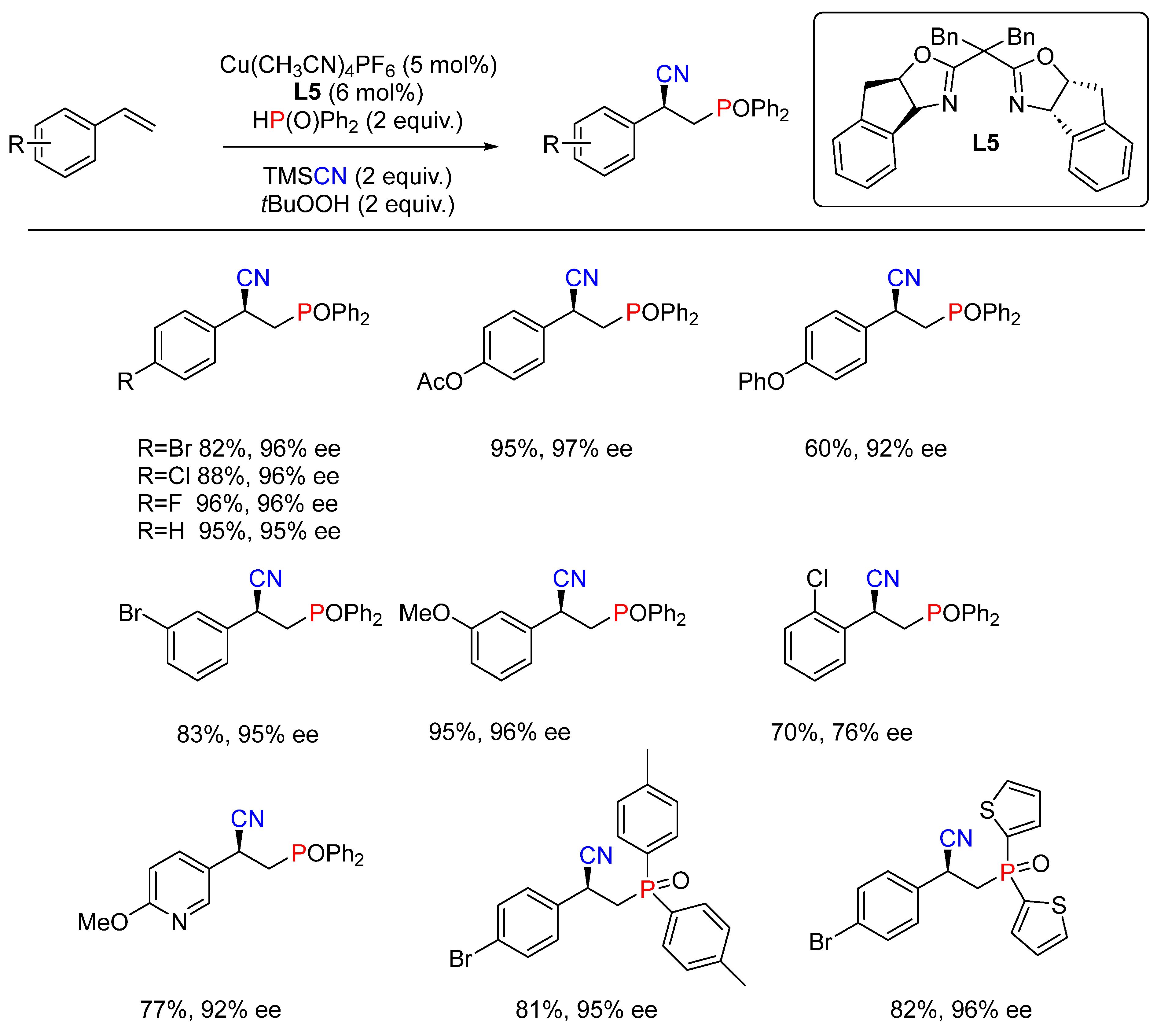
- Zhang, G.; Fu, L.; Chen, P.; Zou, J.; Liu, G. Proton-Coupled Electron Transfer Enables Tandem Radical Relay for Asymmetric Copper-Catalyzed Phosphinoylcyanation of Styrenes. Org. Lett. 2019, 21, 5015–5020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhou, S.; Fu, L.; Chen, P.; Li, Y.; Zou, J.; Liu, G. Asymmetric Coupling of Carbon-Centered Radicals Adjacent to Nitrogen: Copper-Catalyzed Cyanation and Etherification of Enamides. Angew. Chem. Int. Ed. 2020, 59, 20439–20444. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhang, G.; Fu, L.; Chen, P.; Li, Y.; Liu, G. Copper-Catalyzed Asymmetric Cyanation of Alkenes via Carbonyl-Assisted Coupling of Alkyl-Substituted Carbon-Centered Radicals. Org. Lett. 2020, 22, 6299–6303. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Z.; Wu, D.; Wang, F.; Chen, P.; Lin, Z.; Liu, G. Anionic Bisoxazoline Ligands Enable Copper-Catalyzed Asymmetric Radical Azidation of Acrylamides. Angew. Chem. Int. Ed. 2021, 60, 6997–7001. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, G. Asymmetric Alkyl and Aryl/Azolation of Alkenes via a Single Cu(I) Complex. ACS Catal. 2021, 11, 5108–5118. [Google Scholar] [CrossRef]
- Wang, P.-Z.; Gao, Y.; Chen, J.; Huan, X.-D.; Xiao, W.-J.; Chen, J.-R. Asymmetric Three-Component Olefin Dicarbofunctionalization Enabled by Photoredox and Copper Dual Catalysis. Nat. Commun. 2021, 12, 1815. [Google Scholar] [CrossRef]
- Wang, D.; Zhu, N.; Chen, P.; Lin, Z.; Liu, G. Enantioselective Decarboxylative Cyanation Employing Cooperative Photoredox Catalysis and Copper Catalysis. J. Am. Chem. Soc. 2017, 139, 15632–15635. [Google Scholar] [CrossRef]
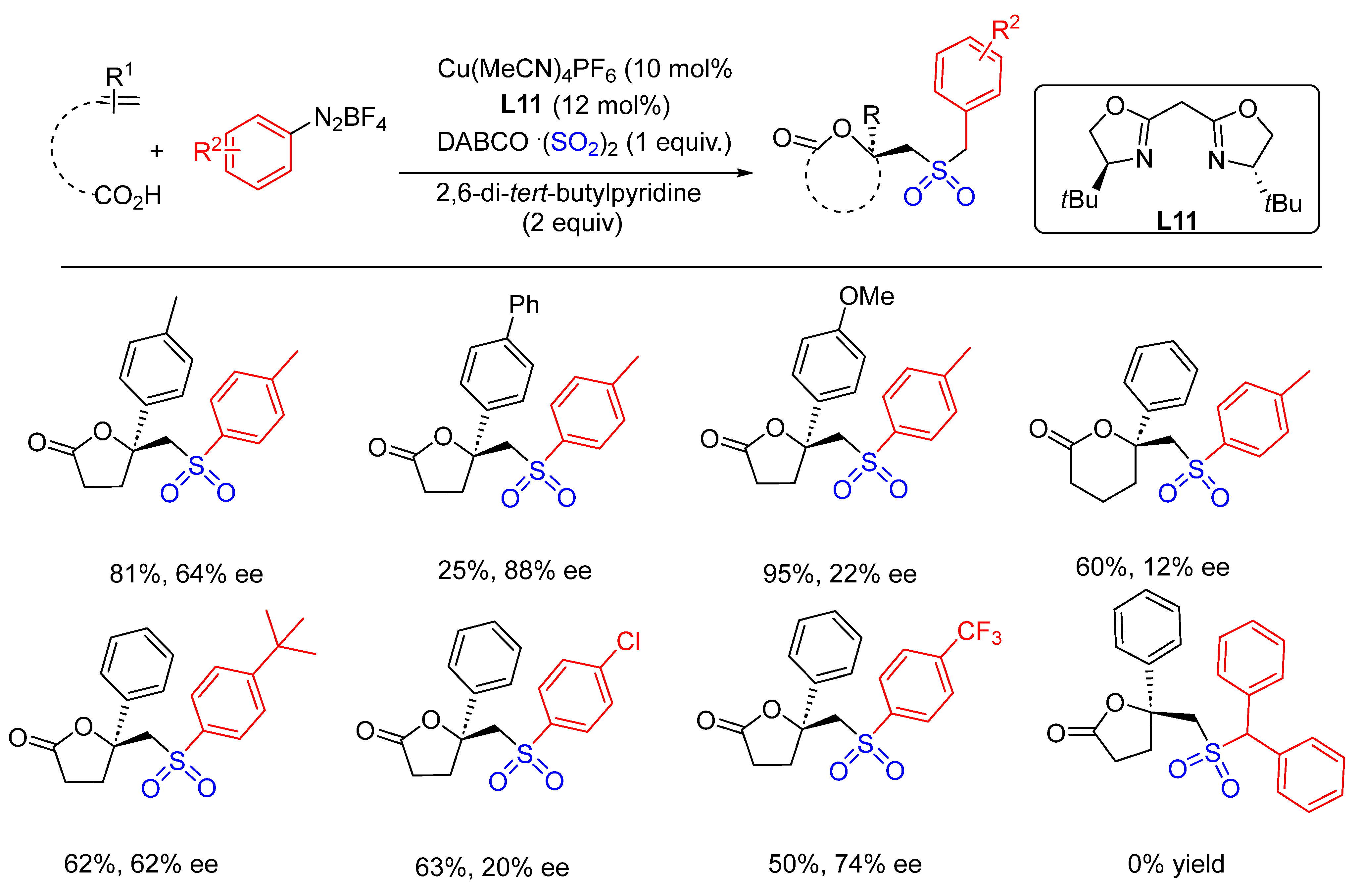
- Wang, Y.; Deng, L.; Zhou, J.; Wang, X.; Mei, H.; Han, J.; Pan, Y. Synthesis of Chiral Sulfonyl Lactones via Copper-Catalyzed Asymmetric Radical Reaction of DABCO⋅(SO2). Adv. Synth. Catal. 2018, 360, 1060–1065. [Google Scholar] [CrossRef]
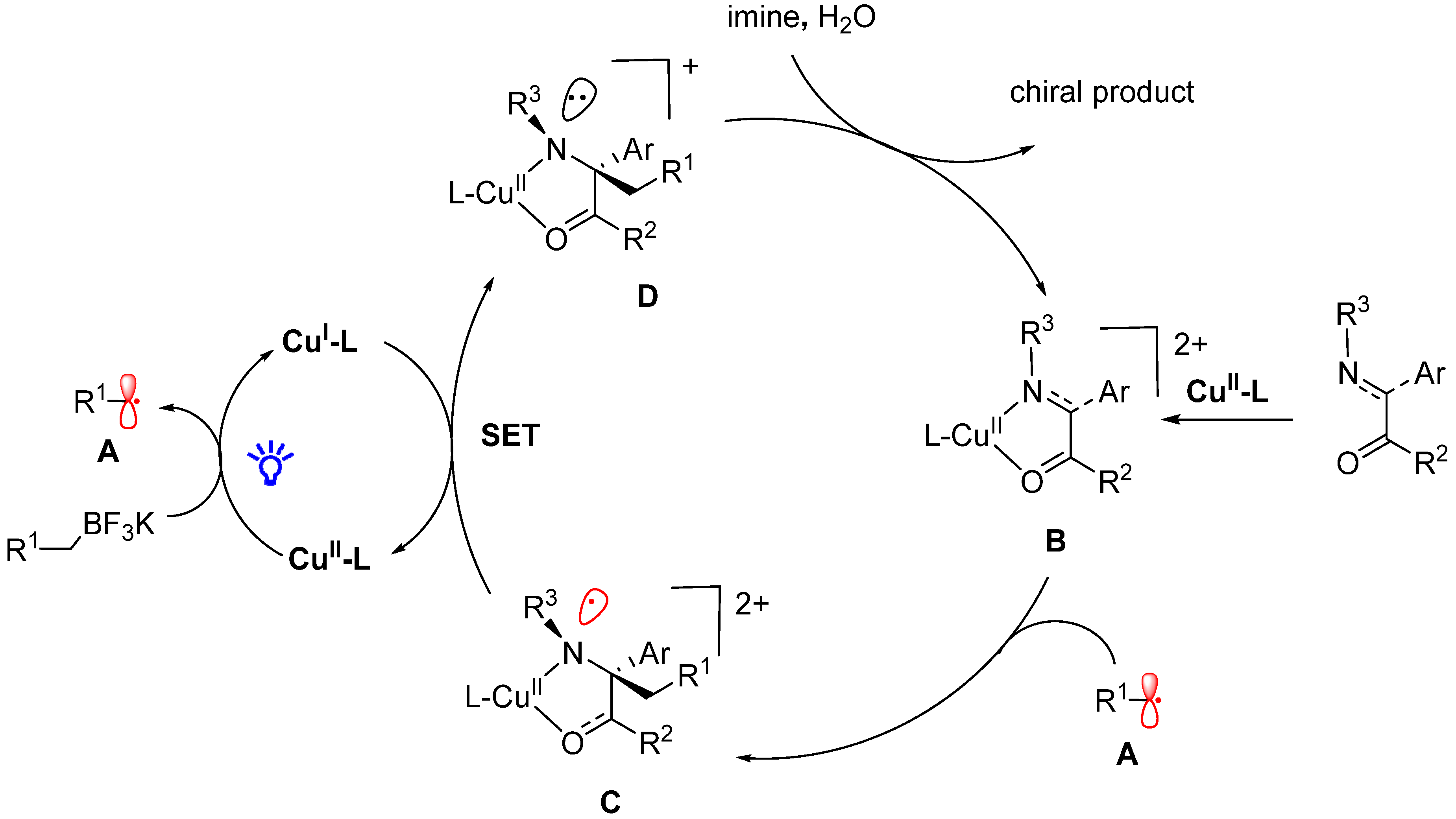
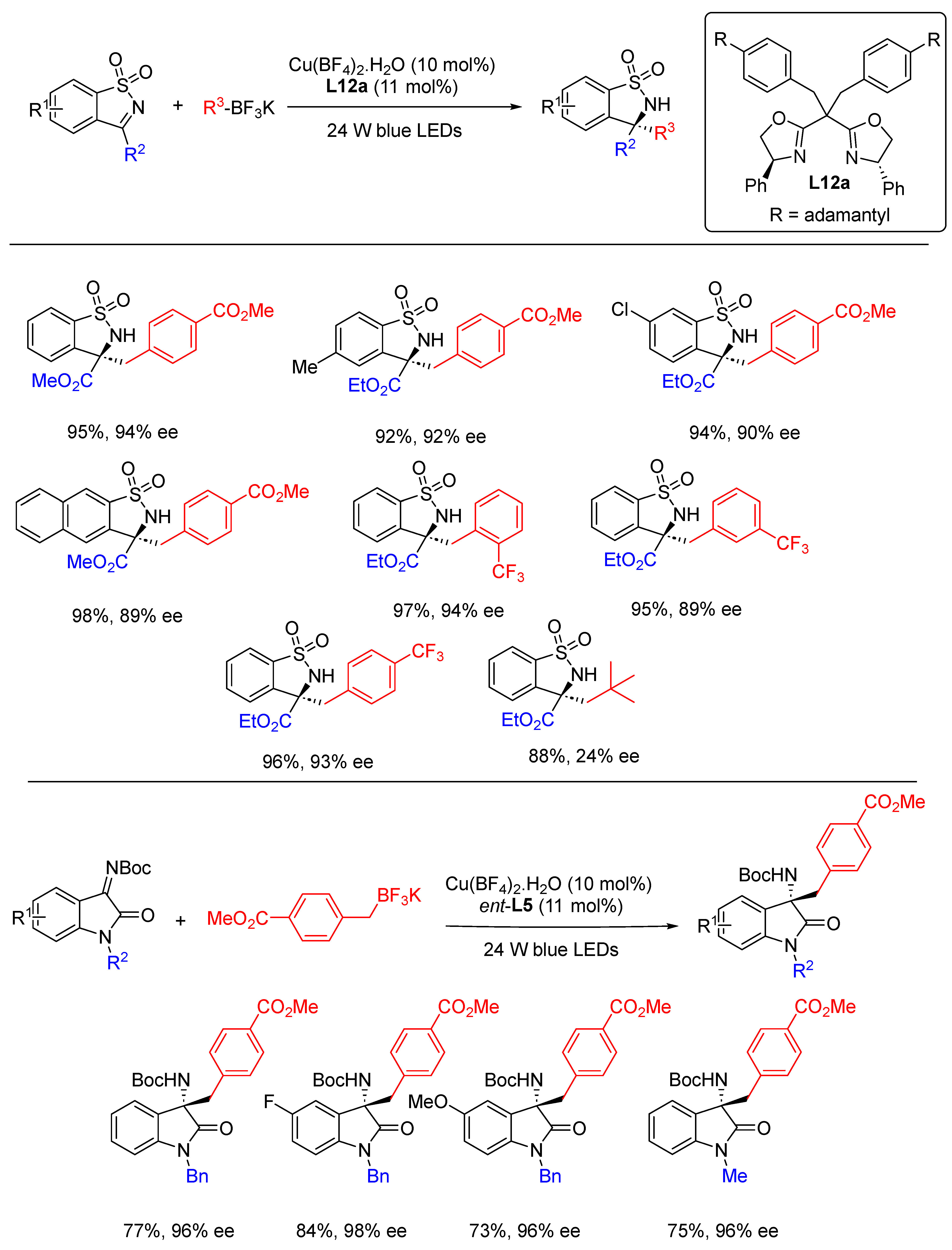
- Li, Y.; Zhou, K.; Wen, Z.; Cao, S.; Shen, X.; Lei, M.; Gong, L. Copper(II)-Catalyzed Asymmetric Photoredox Reactions: Enanti-oselective Alkylation of Imines Driven by Visible Light. J. Am. Chem. Soc. 2018, 140, 15850–15858. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, L.; Chen, P.; Liu, G. Enantioselective Arylation of Benzylic C–H Bonds by Copper-Catalyzed Radical Relay. Angew. Chem. Int. Ed. 2019, 58, 6425–6429. [Google Scholar] [CrossRef]
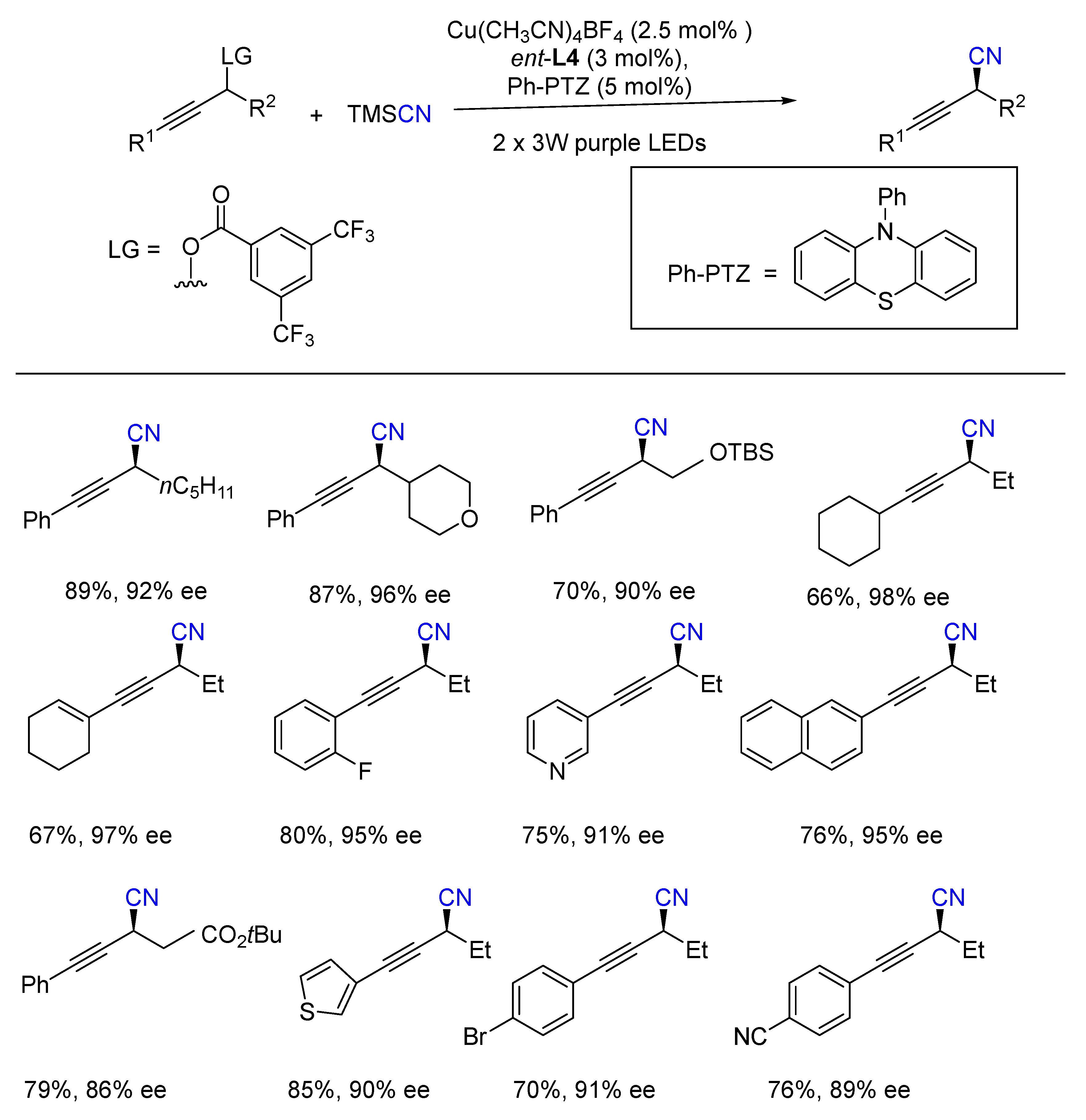
- Lu, F.D.; Liu, D.; Zhu, L.; Lu, L.Q.; Yang, Q.; Zhou, Q.Q.; Wei, Y.; Lan, Y.; Xiao, W.J. Asymmetric Propargylic Radical Cya-nation Enabled by Dual Organophotoredox and Copper Catalysis. J. Am. Chem. Soc. 2019, 141, 6167–6172. [Google Scholar] [CrossRef]
- Yang, S.; Wang, L.; Zhang, H.; Liu, C.; Zhang, L.; Wang, X.; Zhang, G.; Li, Y.; Zhang, Q. Copper-Catalyzed Asymmetric Ami-nocyanation of Arylcyclopropanes for Synthesis of γ-Amino Nitriles. ACS Catal. 2019, 9, 716–721. [Google Scholar] [CrossRef]
- Wdowik, T.; Galster, S.L.; Carmo, R.L.L.; Chemler, S.R. Enantioselective, Aerobic Copper-Catalyzed Intramolecular Carbo-amination and Carboetherification of Unactivated Alkenes. ACS Catal. 2020, 10, 8535–8541. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Jian, W.; Huang, M.; Li, D.; Li, Y.; Zhang, X.; Bao, H. Asymmetric Radical Carboesterification of Dienes. Nat. Commun. 2021, 12, 6670. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Luo, X.; Ge, Y.; Wang, H.; Liu, J.; Wang, Y.; Luan, X. Catalytic Asymmetric [4+1] Spiroannulation of α-Bromo-Beta-Naphthols with Azoalkenes by an Electrophilic Dearomatization-SRN1-Debromination Approach. CCS Chem. 2021, 4, 1054–1064. [Google Scholar] [CrossRef]
- Zheng, M.; Gao, K.; Qin, H.; Li, G.; Lu, H. Metal-to-Ligand Ratio-Dependent Chemodivergent Asymmetric Synthesis. Angew. Chem. Int. Ed. 2021, 60, 22892–22899. [Google Scholar] [CrossRef]
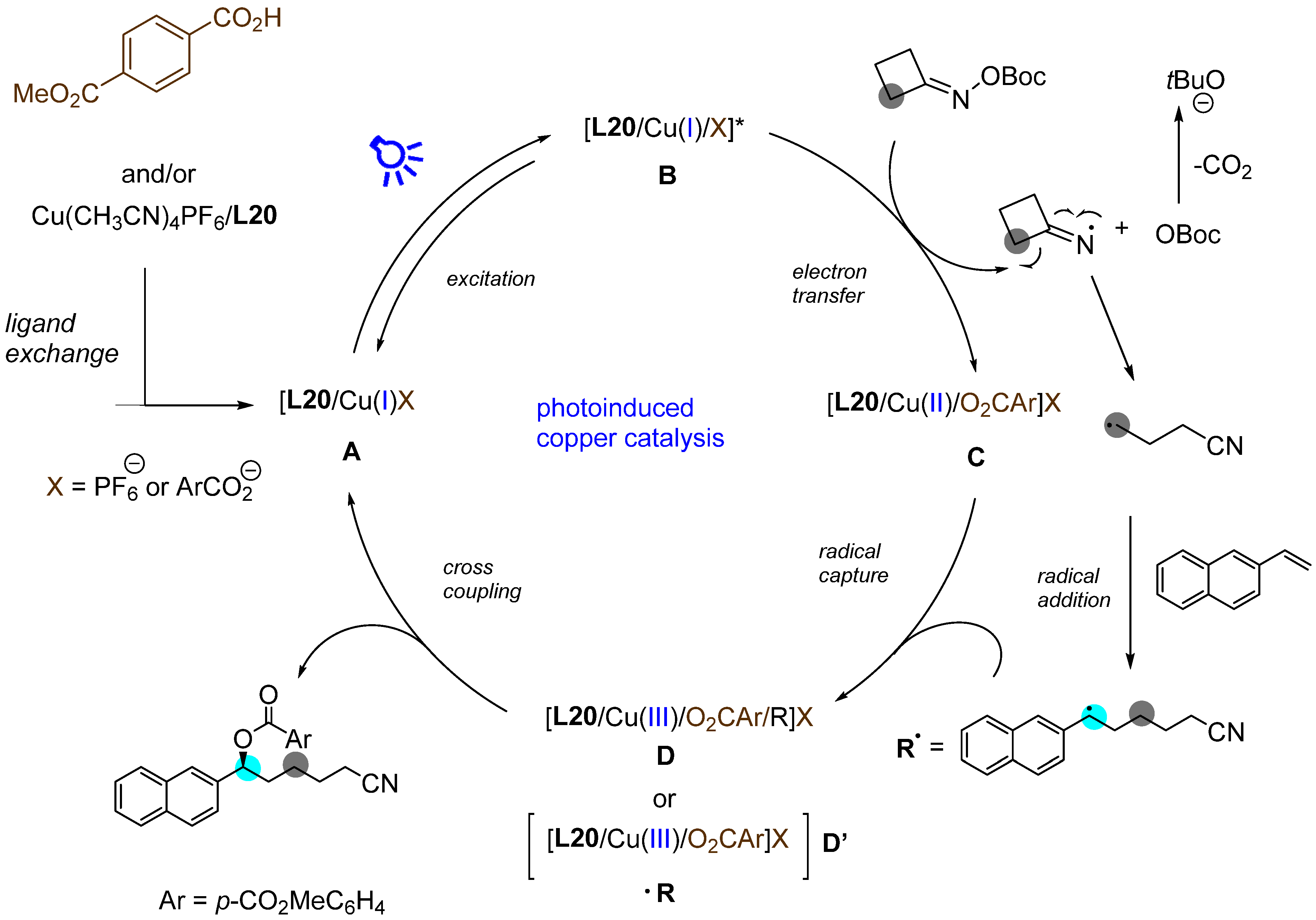
- Chen, J.; Liang, Y.J.; Wang, P.Z.; Li, G.Q.; Zhang, B.; Qian, H.; Huan, X.D.; Guan, W.; Xiao, W.J.; Chen, J.R. Photoinduced Copper-Catalyzed Asymmetric C-O Cross-Coupling. J. Am. Chem. Soc. 2021, 143, 13382–13392. [Google Scholar] [CrossRef]
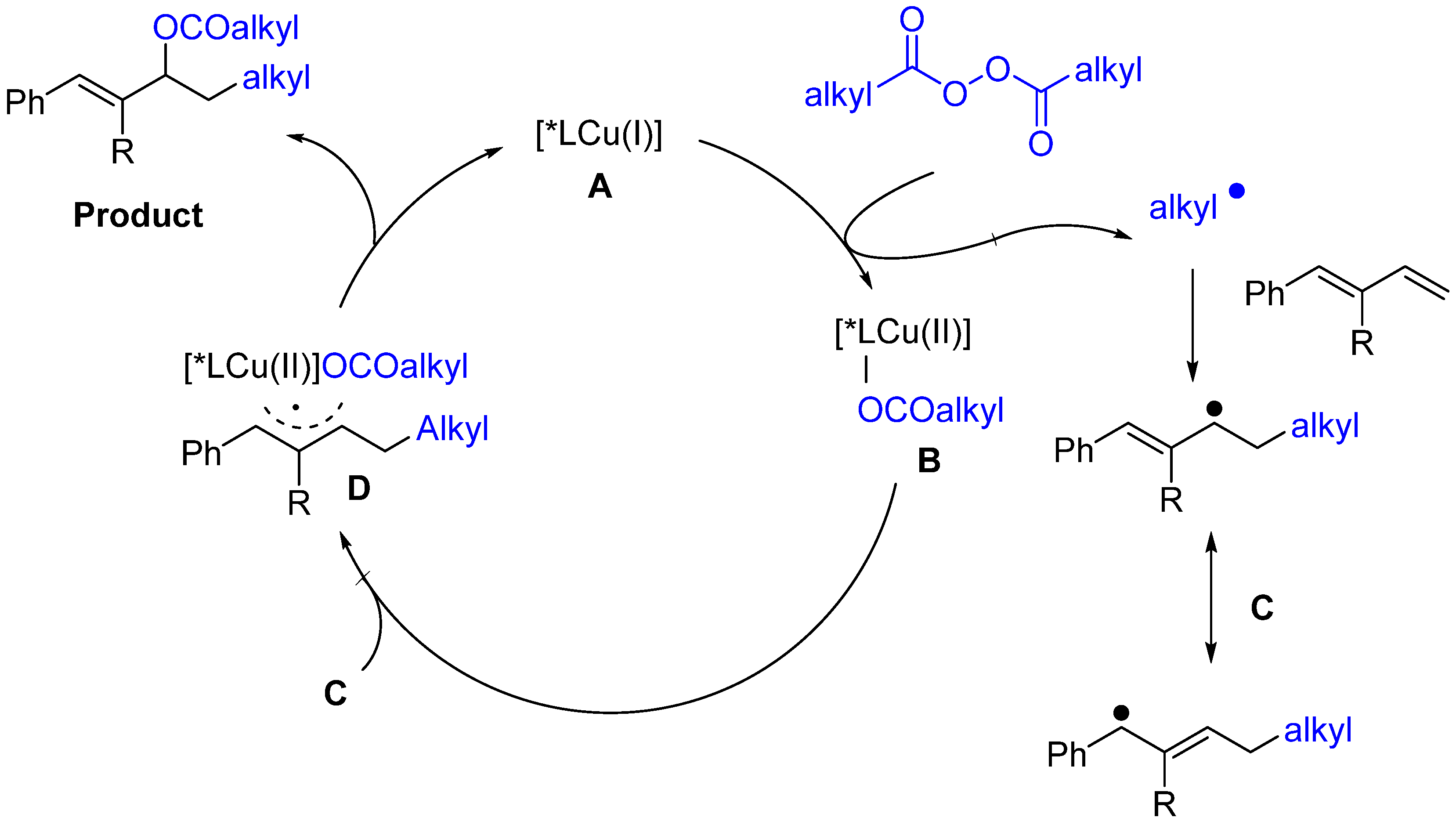
- Wang, P.Z.; Wu, X.; Cheng, Y.; Jiang, M.; Xiao, W.J.; Chen, J.R. Photoinduced Copper-Catalyzed Asymmetric Three-Component Coupling of 1,3-Dienes: An Alternative to Kharasch–Sosnovsky Reaction. Angewe. Chem. Int. Ed. 2021, 60, 22956–22962. [Google Scholar] [CrossRef]
- Wang, P.Z.; Liang, Y.J.; Wu, X.; Guan, W.; Xiao, W.J.; Chen, J.R. Copper-Catalyzed Three-Component Photo-ATRA-Type Reaction for Asymmetric Intermolecular C-O Coupling. ACS Catal. 2022, 12, 10925–10937. [Google Scholar] [CrossRef]
- Dai, L.; Zhu, Q.; Zeng, J.; Liu, Y.; Zhong, G.; Han, X.; Zeng, X. Asymmetric Synthesis of Chiral Imidazolidines by Merging Copper and Visible Light-Induced Photoredox Catalysis. Org. Chem. Front. 2022, 9, 2994–2999. [Google Scholar] [CrossRef]
- Wang, B.C.; Fan, T.; Xiong, F.Y.; Chen, P.; Fang, K.X.; Tan, Y.; Lu, L.Q.; Xiao, W.J. De Novo Construction of Chiral Ami-noindolines by Cu-Catalyzed Asymmetric Cyclization and Subsequent Discovery of an Unexpected Sulfonyl Migration. J. Am. Chem. Soc. 2022, 144, 19932–19941. [Google Scholar] [CrossRef]
- Sakurai, S.; Matsumoto, A.; Kano, T.; Maruoka, K. Cu-Catalyzed Enantioselective Alkylarylation of Vinylarenes Enabled by Chiral Binaphthyl–BOX Hybrid Ligands. J. Am. Chem. Soc. 2020, 142, 19017–19022. [Google Scholar] [CrossRef]
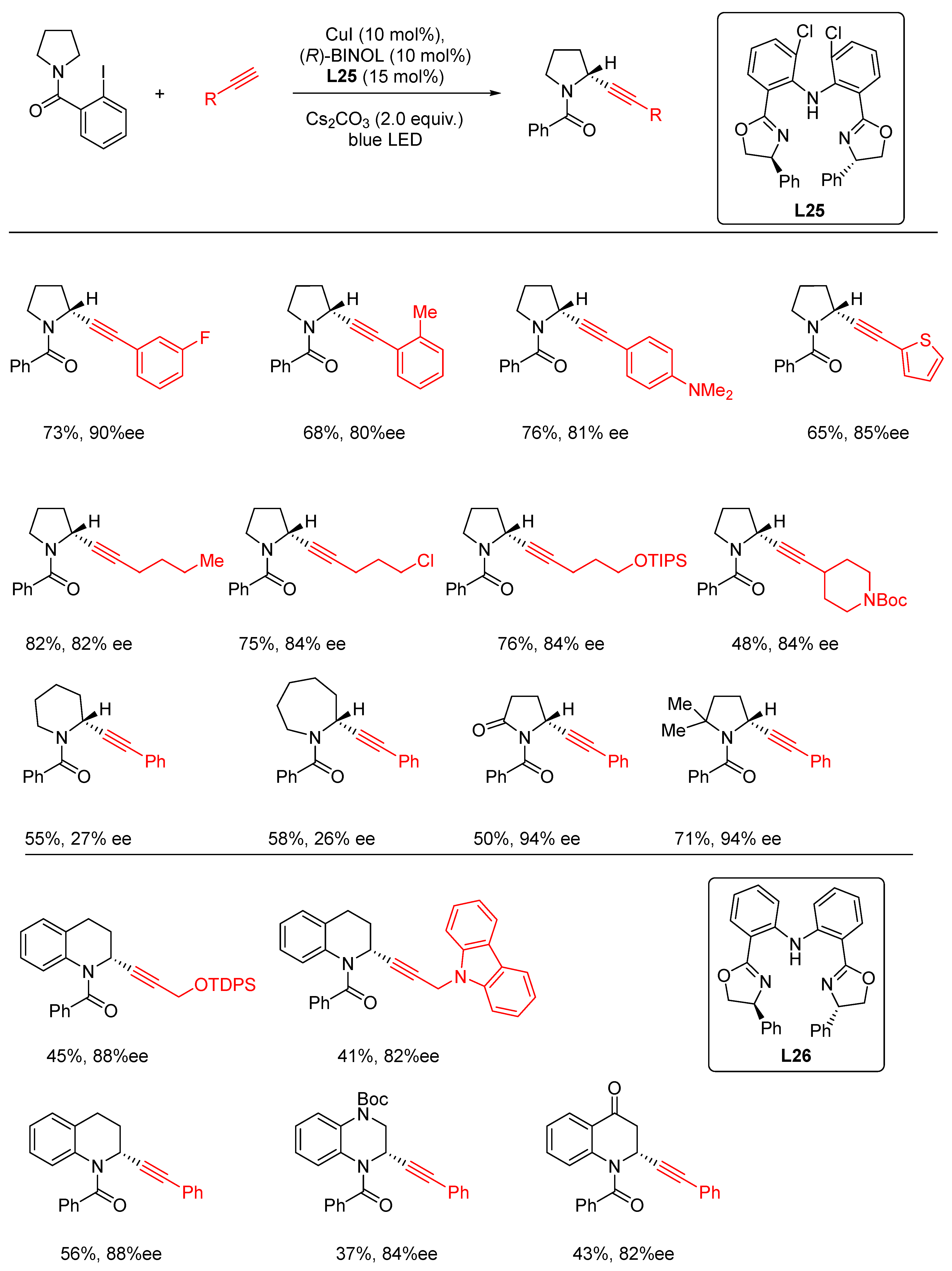
- Guo, R.; Xiao, H.; Li, S.; Luo, Y.; Bai, J.; Zhang, M.; Guo, Y.; Qi, X.; Zhang, G. Photoinduced Copper-Catalyzed Asymmetric C(sp3)–H Alkynylation of Cyclic Amines by Intramolecular 1,5-Hydrogen Atom Transfer. Angew. Chem. Int. Ed. 2022, 61, e202208232. [Google Scholar] [CrossRef]
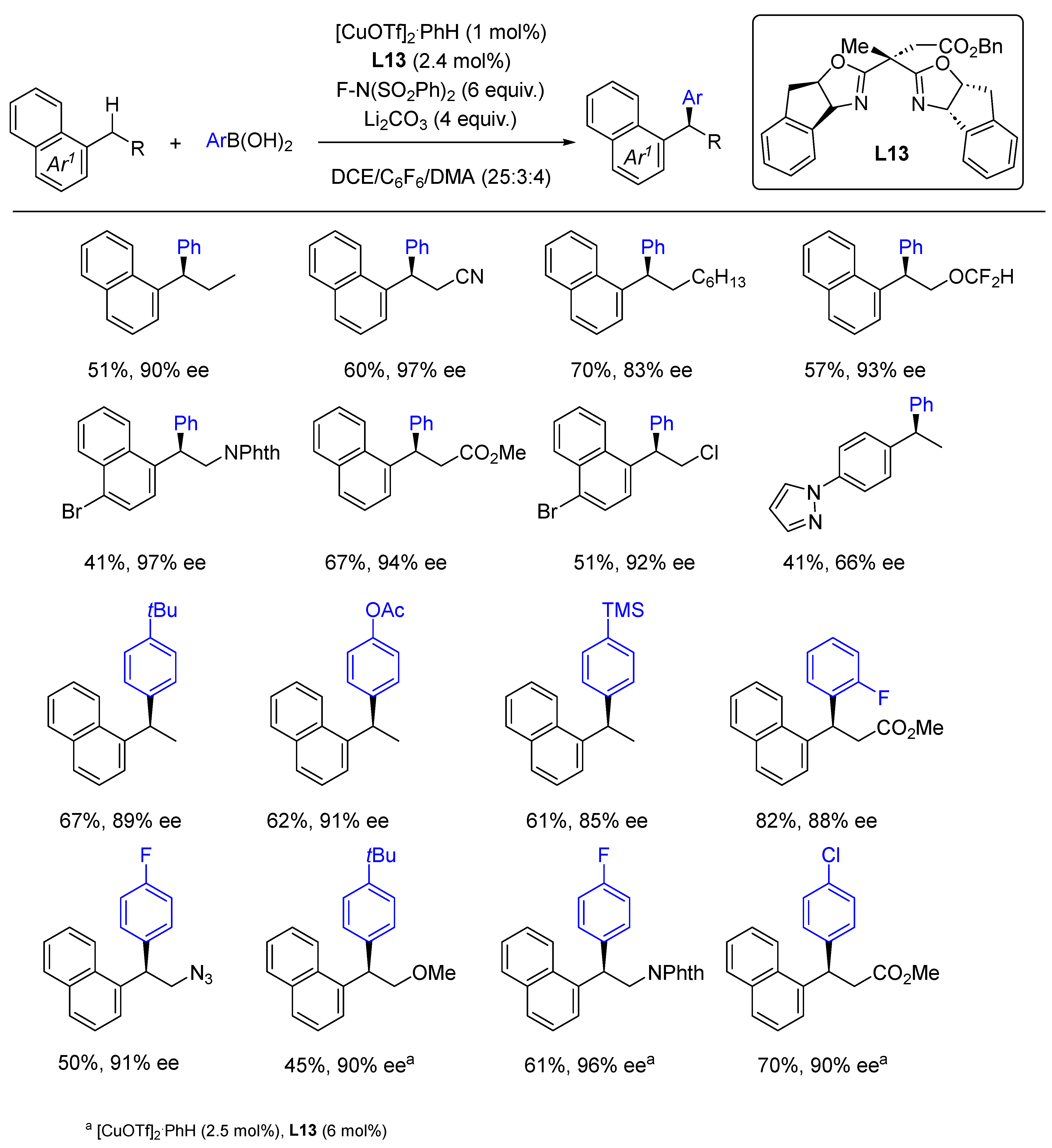
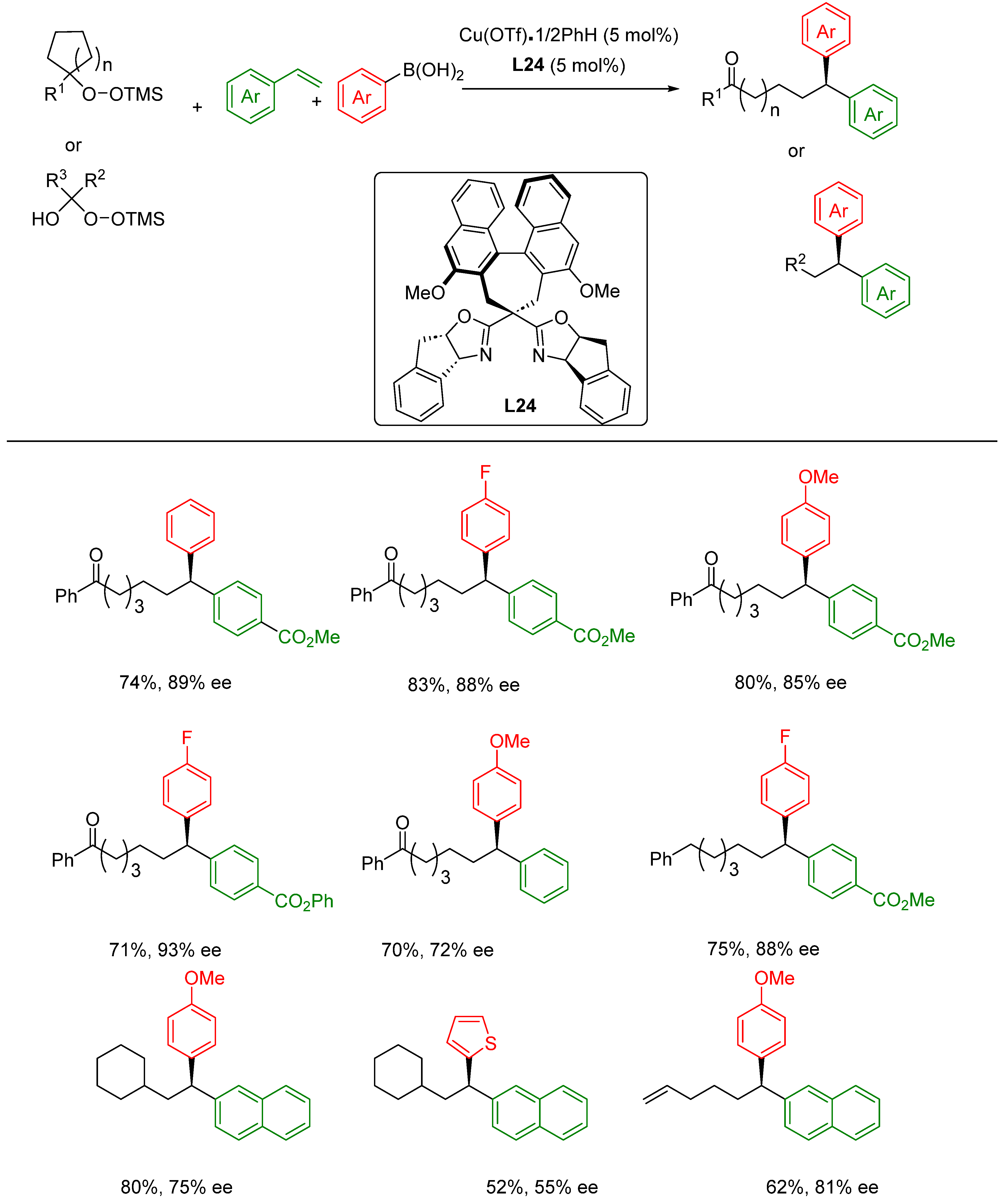
- Liu, L.; Guo, K.X.; Tian, Y.; Yang, C.J.; Gu, Q.S.; Li, Z.L.; Ye, L.; Liu, X.Y. Copper-Catalyzed Intermolecular Enantioselective Radical Oxidative C(sp3)–H/C(sp)–H Cross-Coupling with Rationally Designed Oxazoline-Derived N,N,P(O)-Ligands. Angew. Chem. Int. Ed. 2021, 60, 26710–26717. [Google Scholar] [CrossRef]
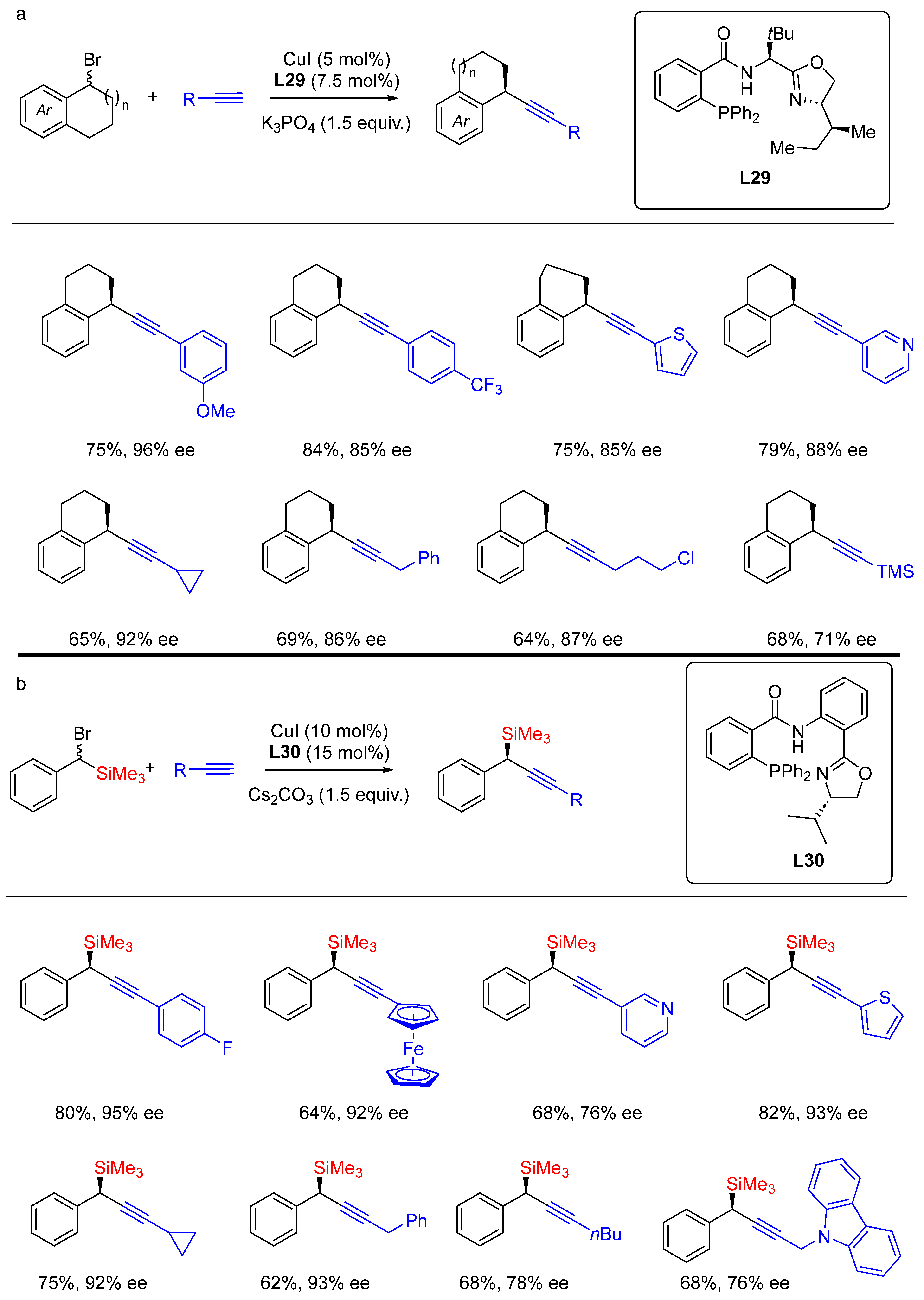
- Guo, R.; Sang, J.; Xiao, H.; Li, J.; Zhang, G. Development of Novel Phosphino-Oxazoline Ligands and Their Application in Asymmetric Alkynlylation of Benzylic Halides. Chin. J. Chem. 2022, 40, 1337–1345. [Google Scholar] [CrossRef]
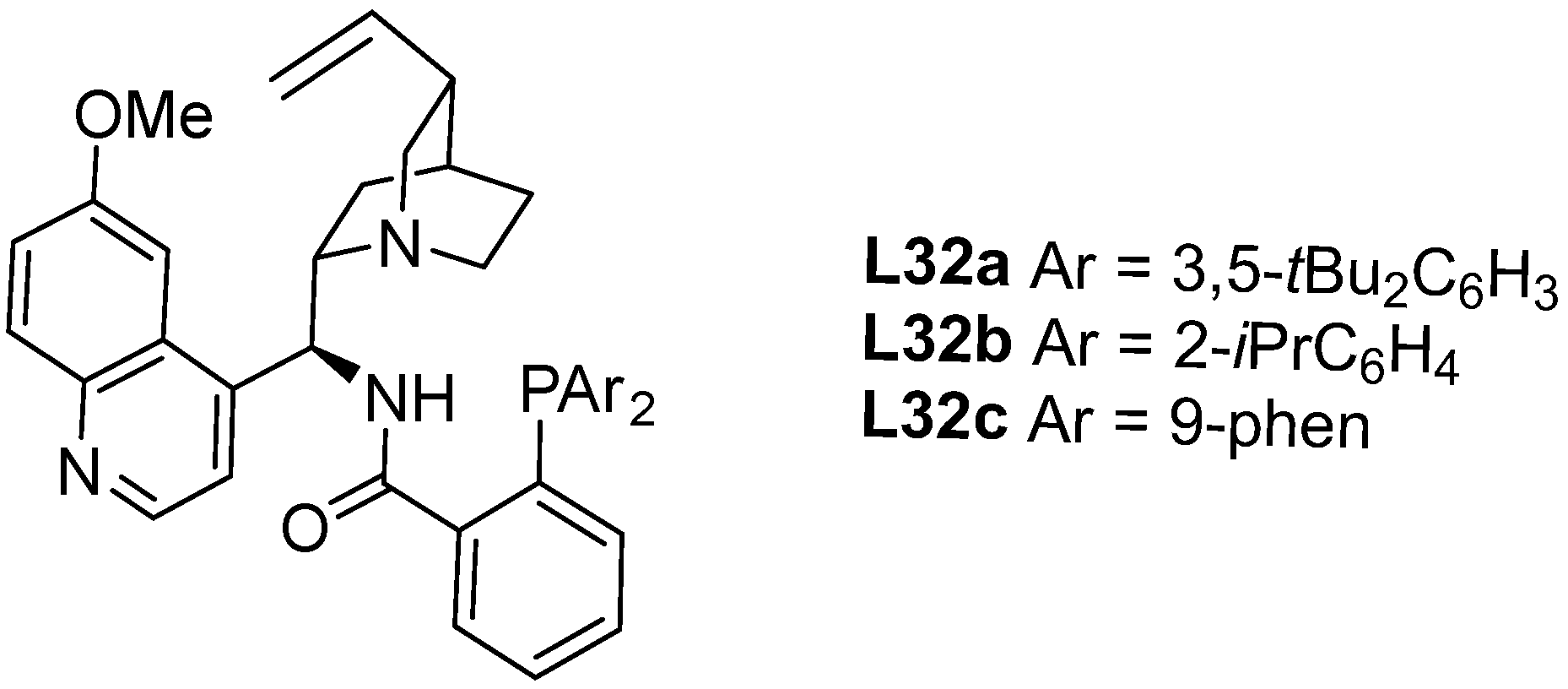
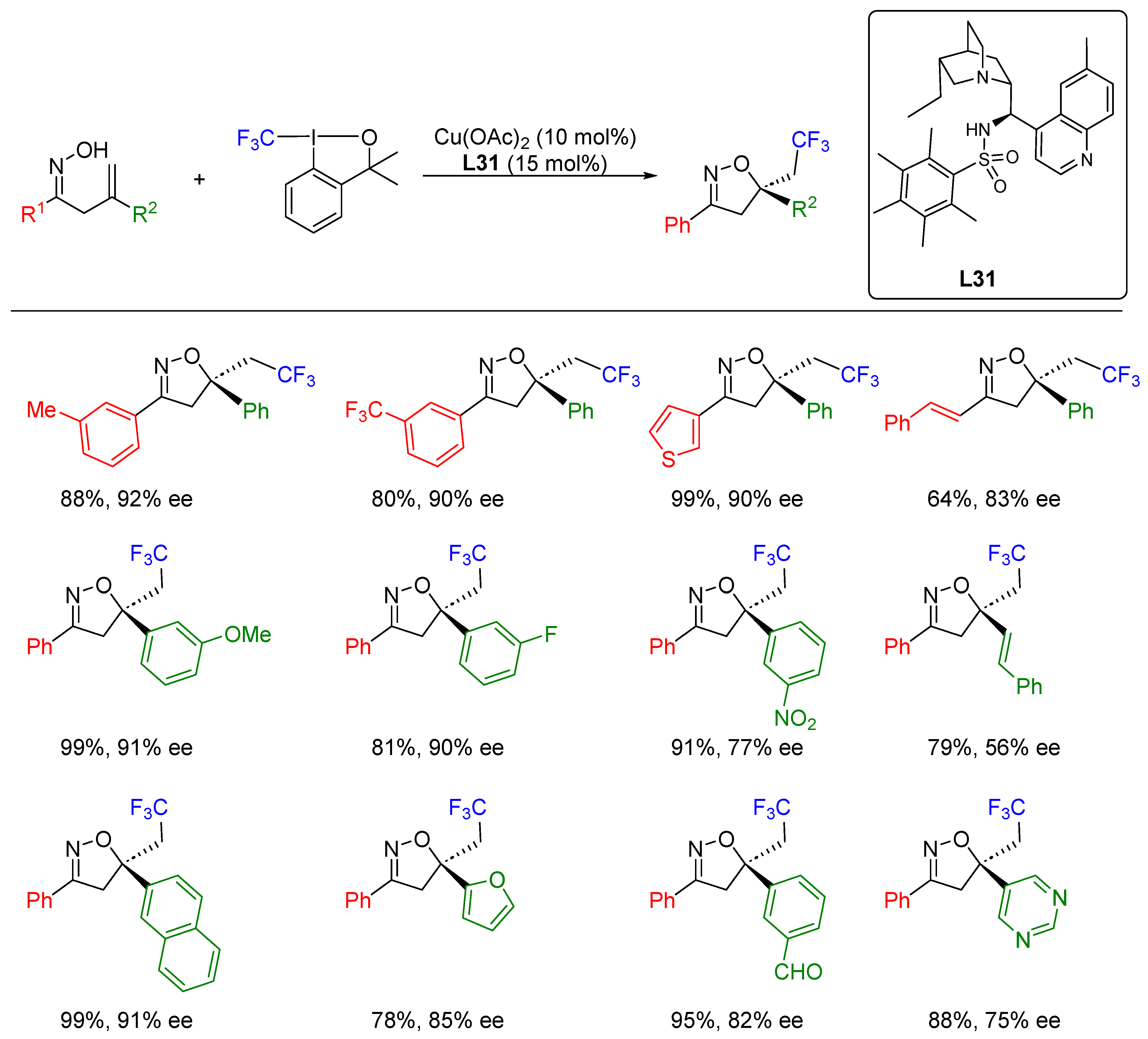
- Li, X.-T.; Gu, Q.-S.; Dong, X.-Y.; Meng, X.; Liu, X.-Y. A Copper Catalyst with a Cinchona-Alkaloid-Based Sulfonamide Ligand for Asymmetric Radical Oxytrifluoromethylation of Alkenyl Oximes. Angew. Chem. Int. Ed. 2018, 57, 7668–7672. [Google Scholar] [CrossRef] [PubMed]
- Sladojevic, F.; Trabocch, A.; Guarna, A.; Dixon, D.J. A New Family of Cinchona-Derived Amino Phosphine Precatalysts: Appli-cation to the Highly Enantio- and Diastereoselective Silver-Catalyzed Isocyanoacetate Aldol Reaction. J. Am. Chem. Soc. 2011, 133, 1710–1713. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-Y.; Zhang, Y.-F.; Ma, C.-L.; Gu, Q.-S.; Wang, F.-L.; Li, Z.-L.; Jiang, S.-P.; Liu, X.-Y. A general asymmetric copper-catalysed Sonogashira C(sp3)–C(sp) coupling. Nat. Chem. 2019, 11, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
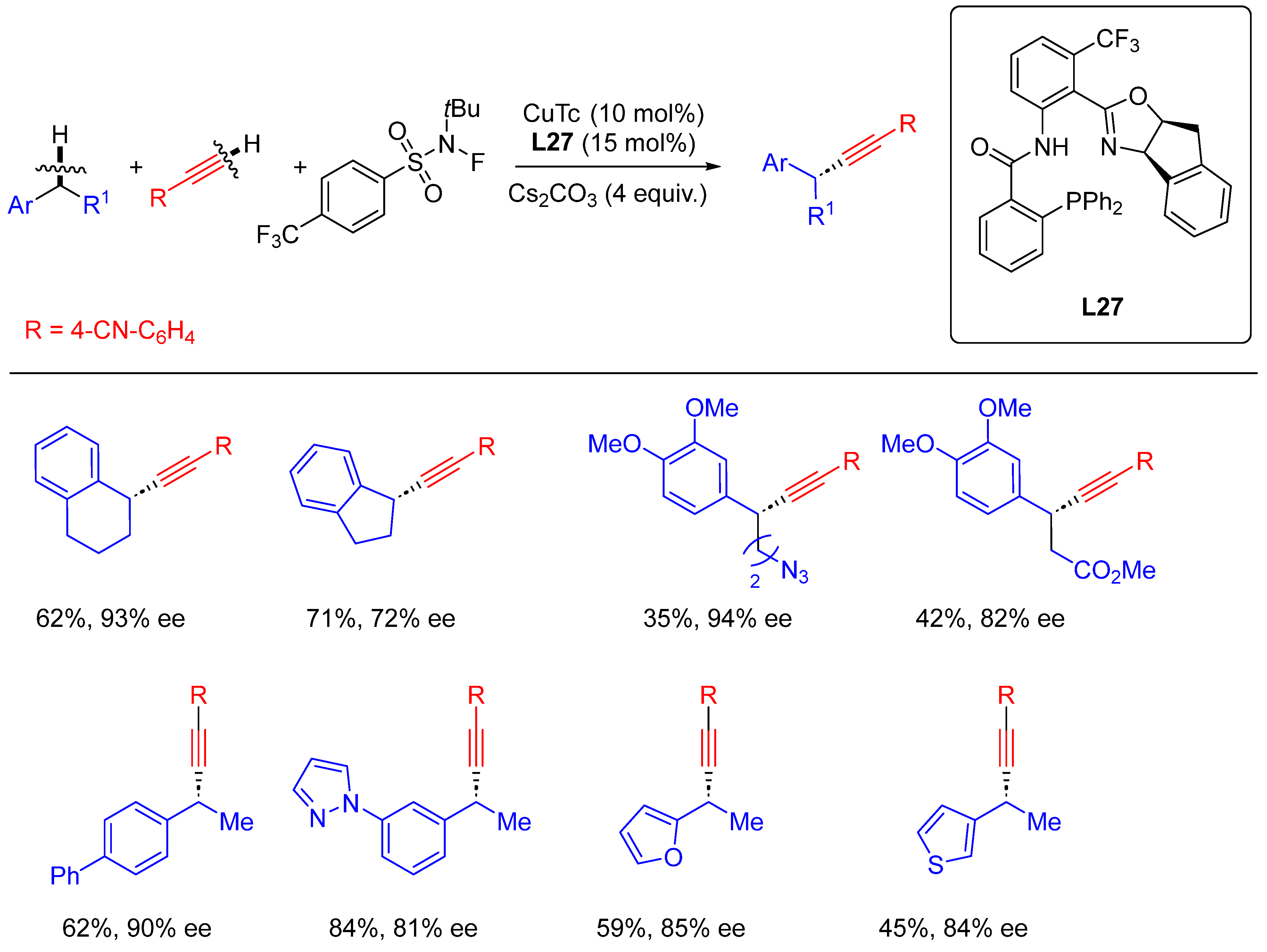
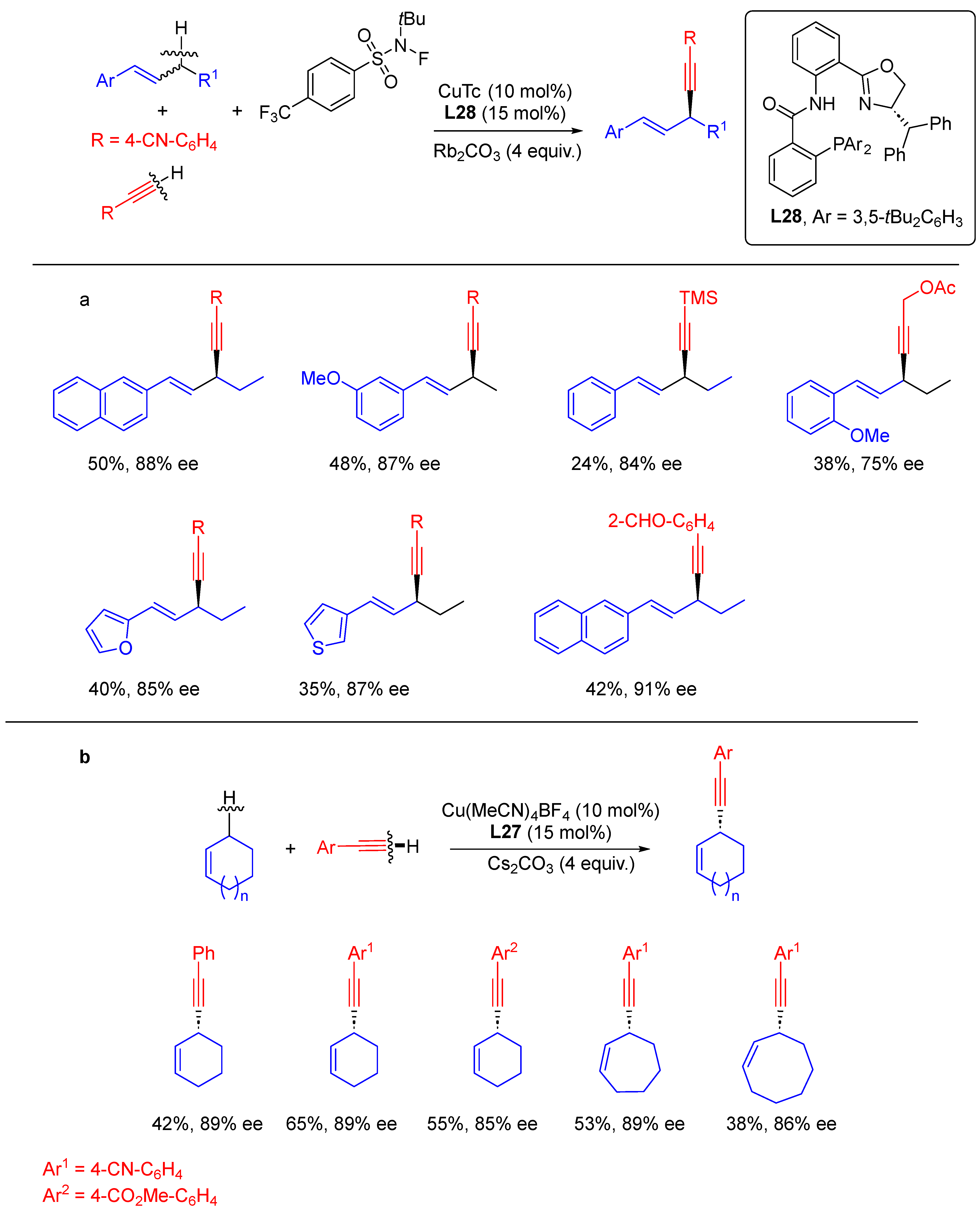
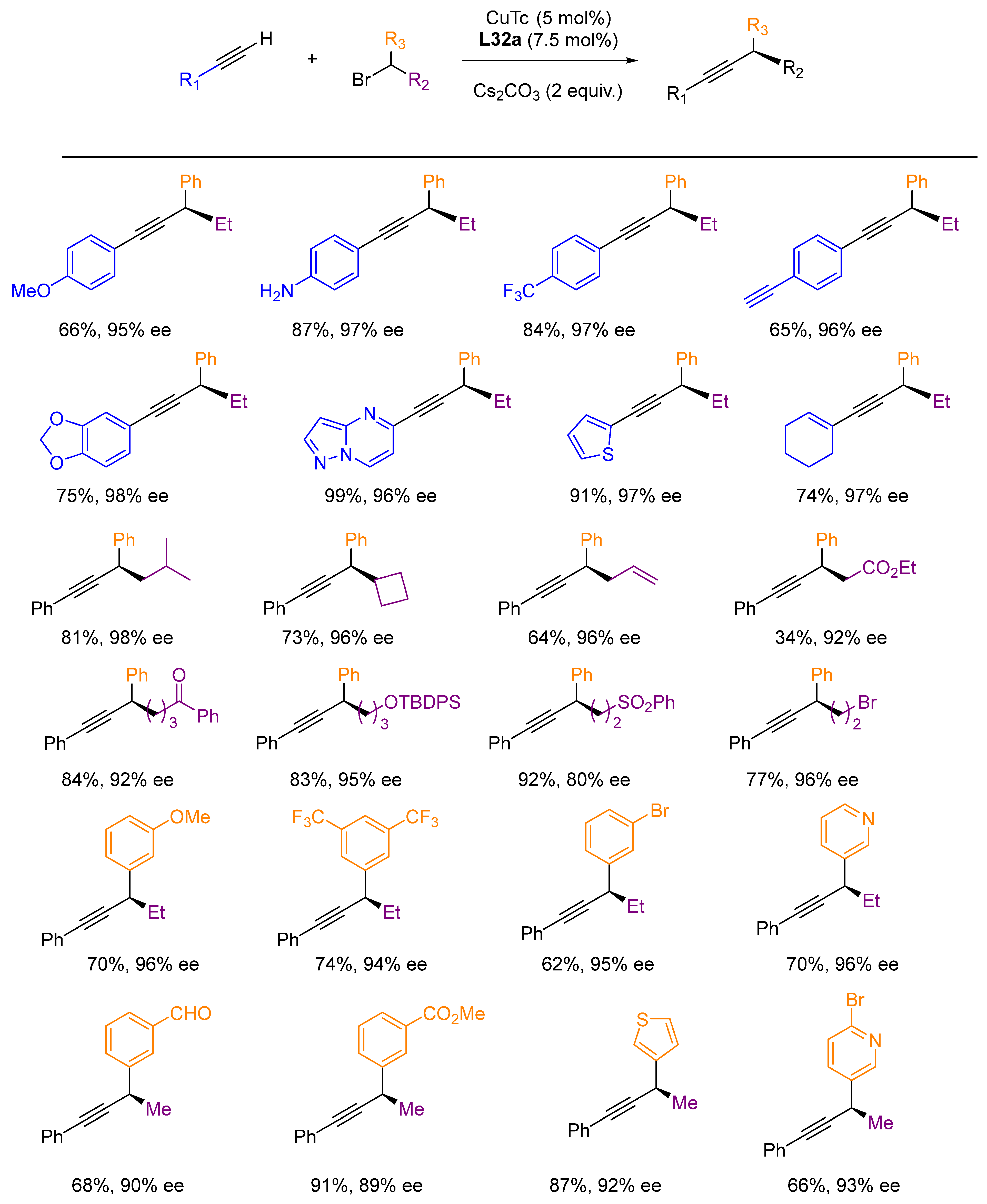
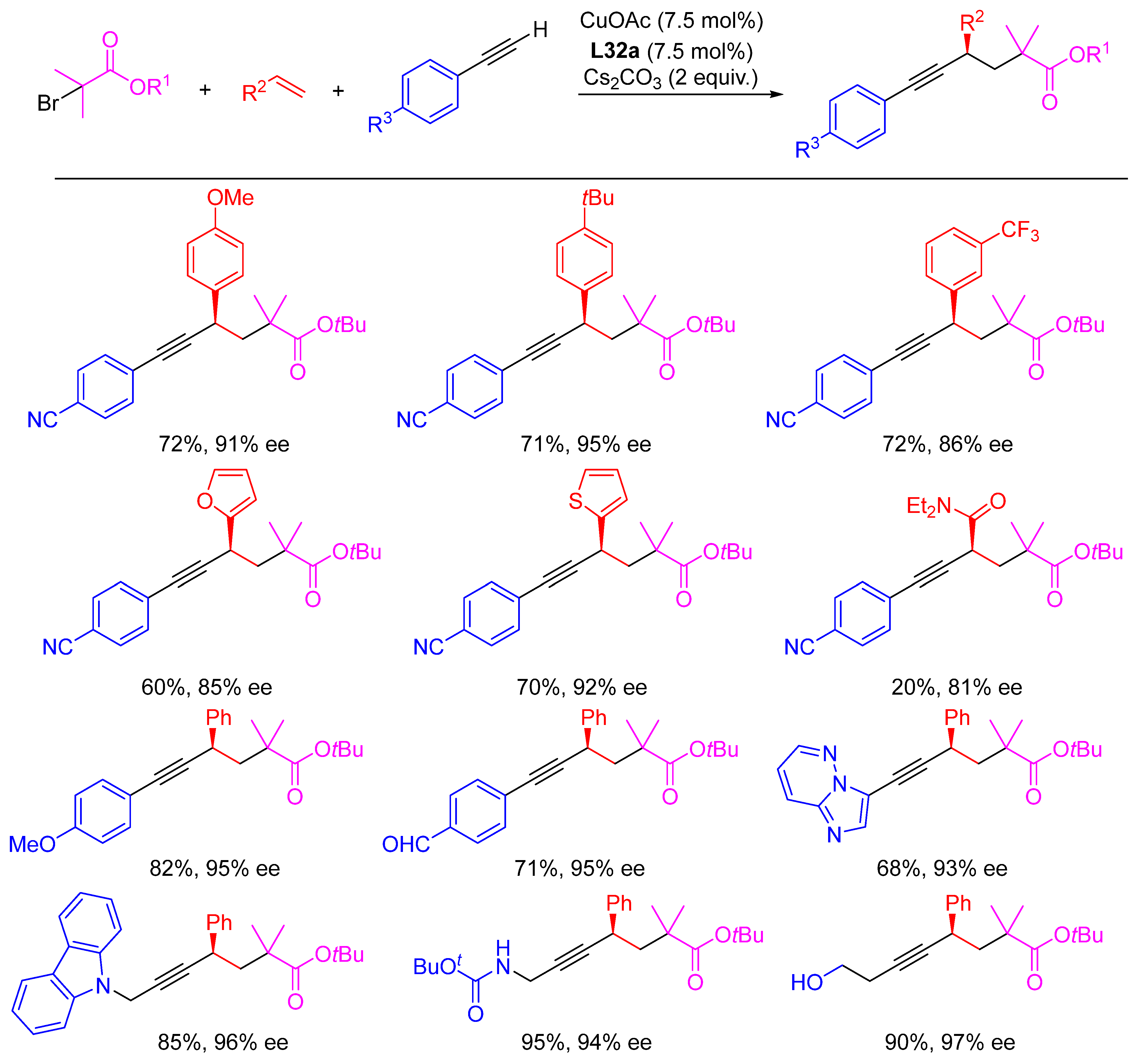
- Dong, X.-Y.; Cheng, J.-T.; Zhang, Y.-F.; Li, Z.-L.; Zhan, T.-Y.; Chen, J.-J.; Wang, F.-L.; Yang, N.-Y.; Ye, L.; Gu, Q.-S.; et al. Copper-Catalyzed Asymmetric Radical 1,2-Carboalkynylation of Alkenes with Alkyl Halides and Terminal Alkynes. J. Am. Chem. Soc. 2020, 142, 9501–9509. [Google Scholar] [CrossRef]
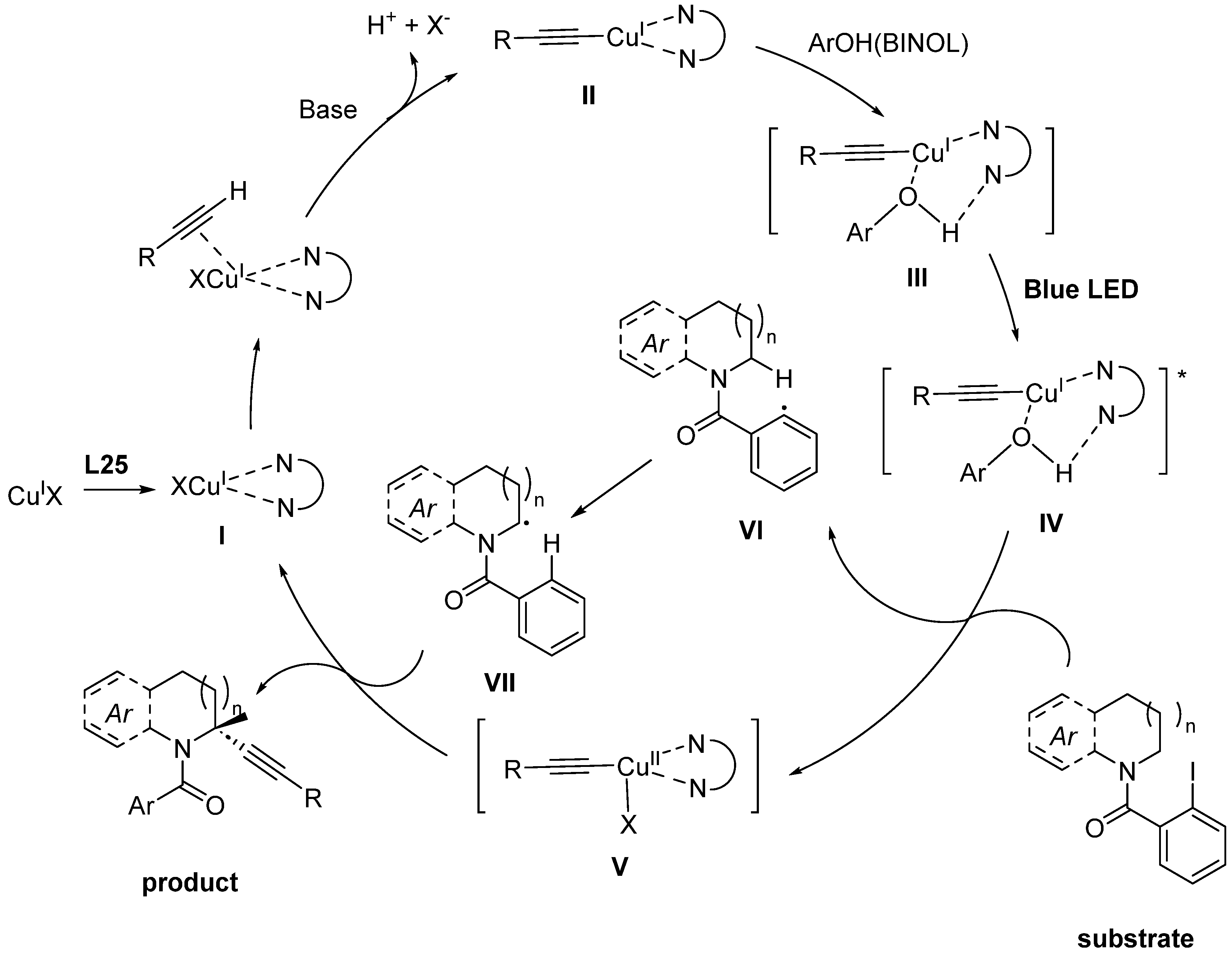
- Xia, H.D.; Li, Z.L.; Gu, Q.S.; Dong, X.Y.; Fang, J.H.; Du, X.Y.; Wang, L.L.; Liu, X.Y. Photoinduced Copper-Catalyzed Asymmetric Decarboxylative Alkynylation with Terminal Alkynes. Angew. Chem. Int. Ed. 2020, 59, 16926–16932. [Google Scholar] [CrossRef] [PubMed]
- Su, X.L.; Ye, L.; Chen, J.J.; Liu, X.D.; Jiang, S.P.; Wang, F.L.; Liu, L.; Yang, C.J.; Chang, X.Y.; Li, Z.L.; et al. Copper-Catalyzed Enantioconvergent Cross-Coupling of Racemic Alkyl Bromides with Azole C(sp2)–H Bonds. Angew. Chem. Int. Ed. 2021, 60, 380–384. [Google Scholar] [CrossRef]
- Dong, X.Y.; Zhan, T.Y.; Jiang, S.P.; Liu, X.D.; Ye, L.; Li, Z.L.; Gu, Q.S.; Liu, X.Y. Copper-Catalyzed Asymmetric Coupling of Allenyl Radicals with Terminal Alkynes to Access Tetrasubstituted Allenes. Angew. Chem. Int. Ed. 2021, 60, 2160–2164. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Masuda, Y.; Iwai, T.; Imaeda, K.; Takeuchi, H.; Ueno, K.; Gao, M.; Hasegawa, J.Y.; Sawamura, M. Photoinduced Cop-per-Catalyzed Asymmetric Acylation of Allylic Phosphates with Acylsilanes. J. Am. Chem. Soc. 2022, 144, 2218–2224. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Li, Y.; Wen, Z.; Cao, S.; Hou, X.; Gong, L. A chiral nickel DBFOX complex as a bifunctional catalyst for visible-light-promoted asymmetric photoredox reactions. Chem. Sci. 2018, 9, 4562–4568. [Google Scholar] [CrossRef]
- Gandolfo, E.; Tang, X.; Roy, R.R.; Melchiorre, P. Photochemical Asymmetric Nickel-Catalyzed Acyl Cross-Coupling. Angew. Chem. Int. Ed. 2019, 58, 16854–16858. [Google Scholar] [CrossRef]
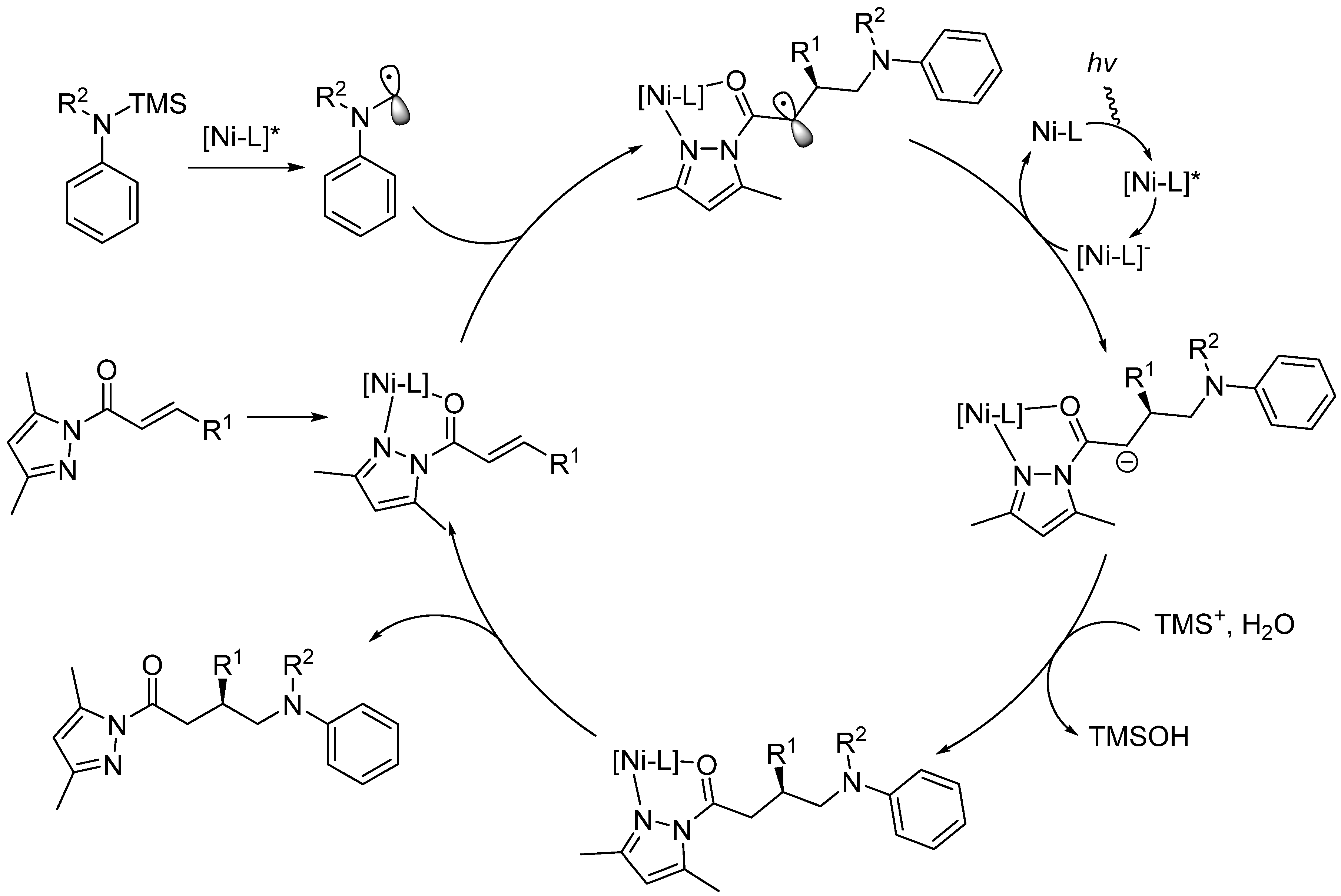
- Huan, L.; Shu, X.; Zu, W.; Zhong, D.; Huo, H. Asymmetric benzylic C(sp3)–H acylation via dual nickel and photoredox catalysis. Nat. Commun. 2021, 12, 3536. [Google Scholar] [CrossRef]
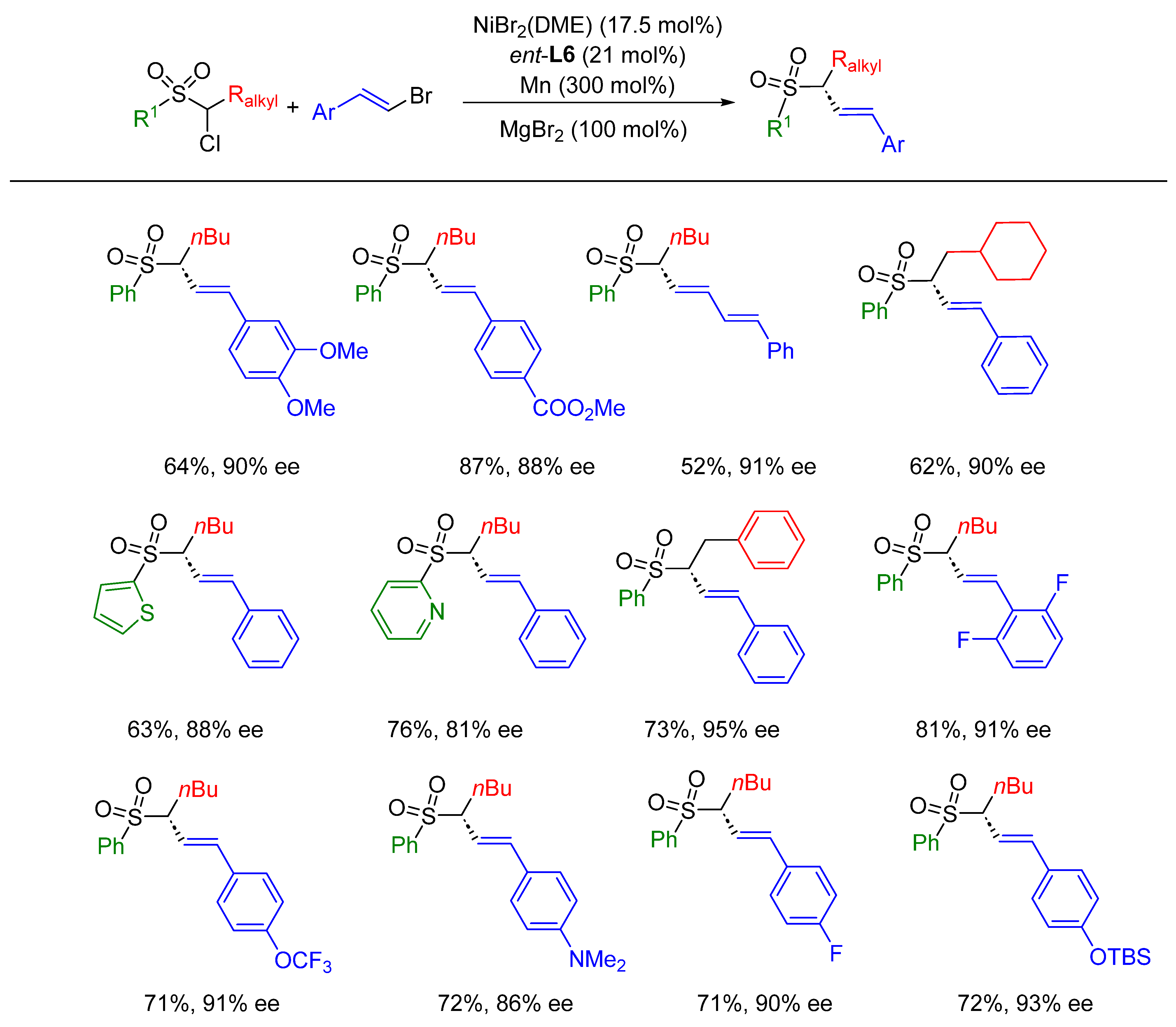
- Geng, J.; Sun, D.; Song, Y.; Tong, W.; Wu, F. Ni-Catalyzed Asymmetric Reductive Alkenylation of α-Chlorosulfones with Vinyl Bromides. Org. Lett. 2022, 24, 1807–1811. [Google Scholar] [CrossRef]
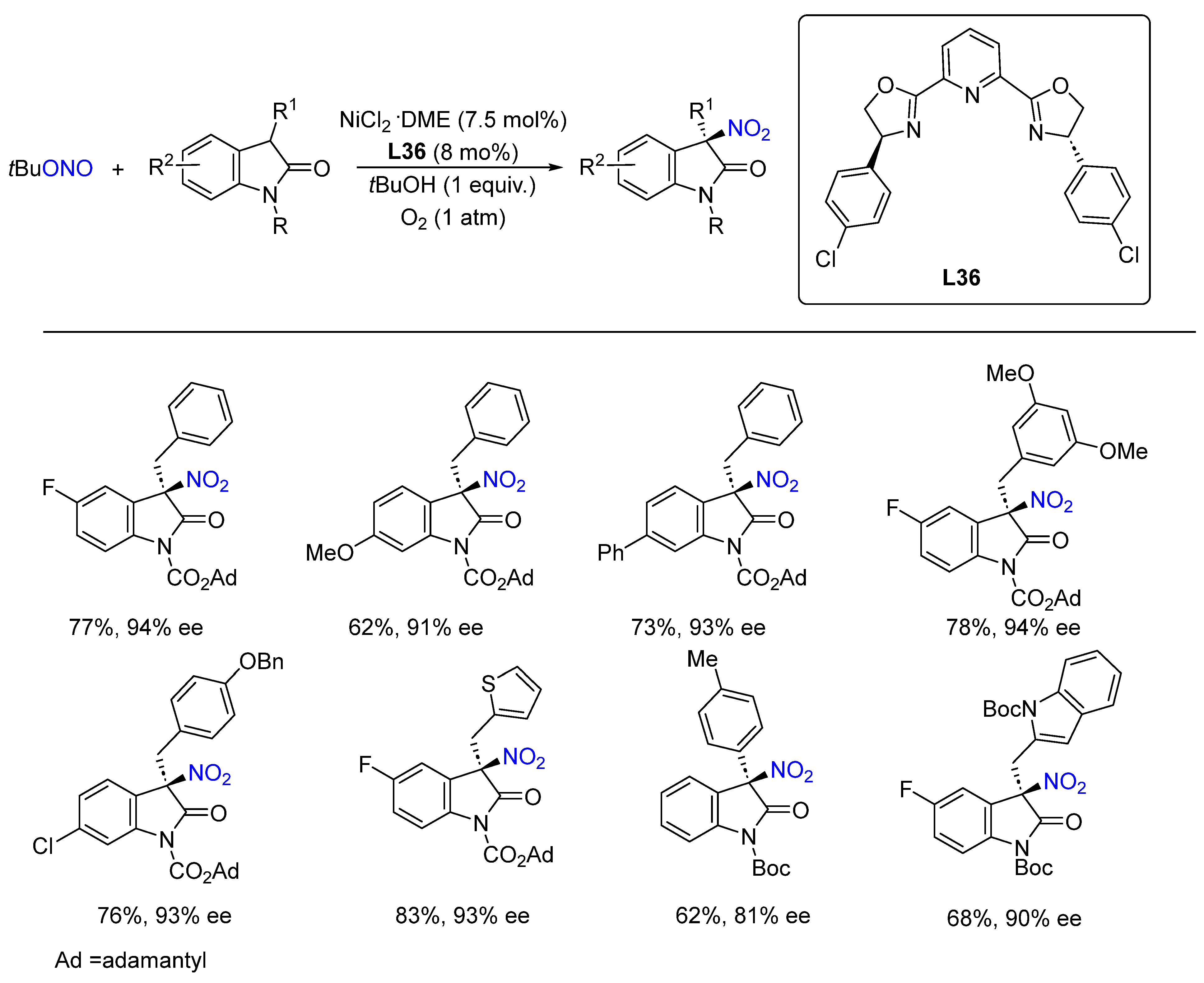
- Lv, M.; Li, X. Ni(II)-Catalyzed Asymmetric Nitration of Oxindoles: Construction of Cipargamin Analogues. ACS Catal. 2021, 11, 14829–14835. [Google Scholar] [CrossRef]
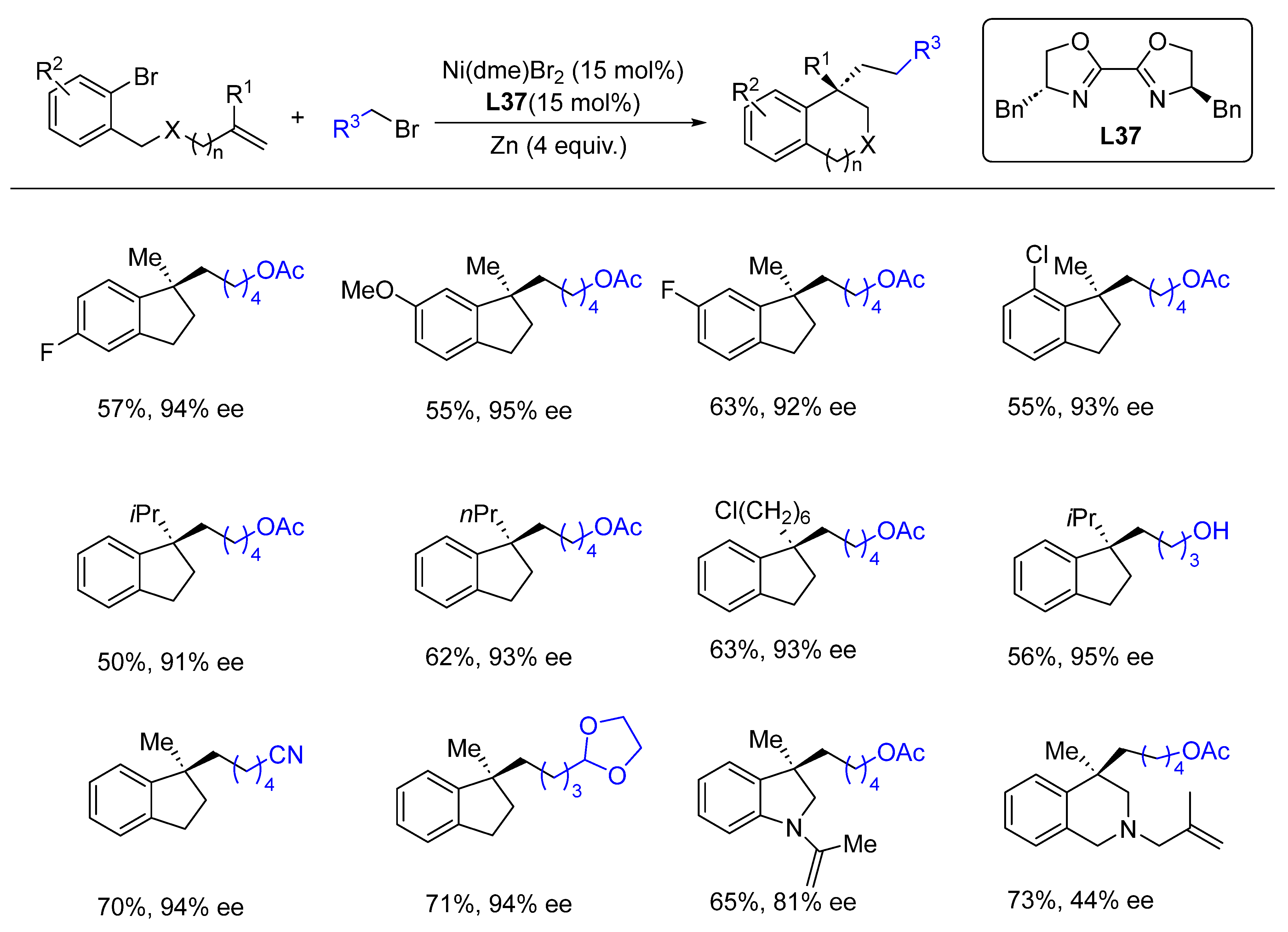
- Jin, Y.; Wang, C. Nickel-Catalyzed Asymmetric Reductive Arylalkylation of Unactivated Alkenes. Angew. Chem. Int. Ed. 2019, 58, 6722–6726. [Google Scholar] [CrossRef]
- Anthony, D.; Lin, Q.; Baudet, J.; Diao, T. Nickel-Catalyzed Asymmetric Reductive Diarylation of Vinylarenes. Angew. Chem. Int. Ed. 2019, 58, 3198–3202. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Shu, W.; García-Domínguez, A.; Merino, E.; Nevado, C. Asymmetric Ni-Catalyzed Radical Relayed Reductive Coupling. J. Am. Chem. Soc. 2020, 142, 13515–13522. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, Z.-P.; Fu, G.C. Quaternary stereocentres via catalytic enantioconvergent nucleophilic substitution reactions of tertiary alkyl halides. Nat. Chem. 2021, 13, 236–242. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, H.; Wang, C. Nickel-Catalyzed Asymmetric Reductive Arylbenzylation of Unactivated Alkenes. Org. Lett. 2020, 22, 2724–2729. [Google Scholar] [CrossRef] [PubMed]
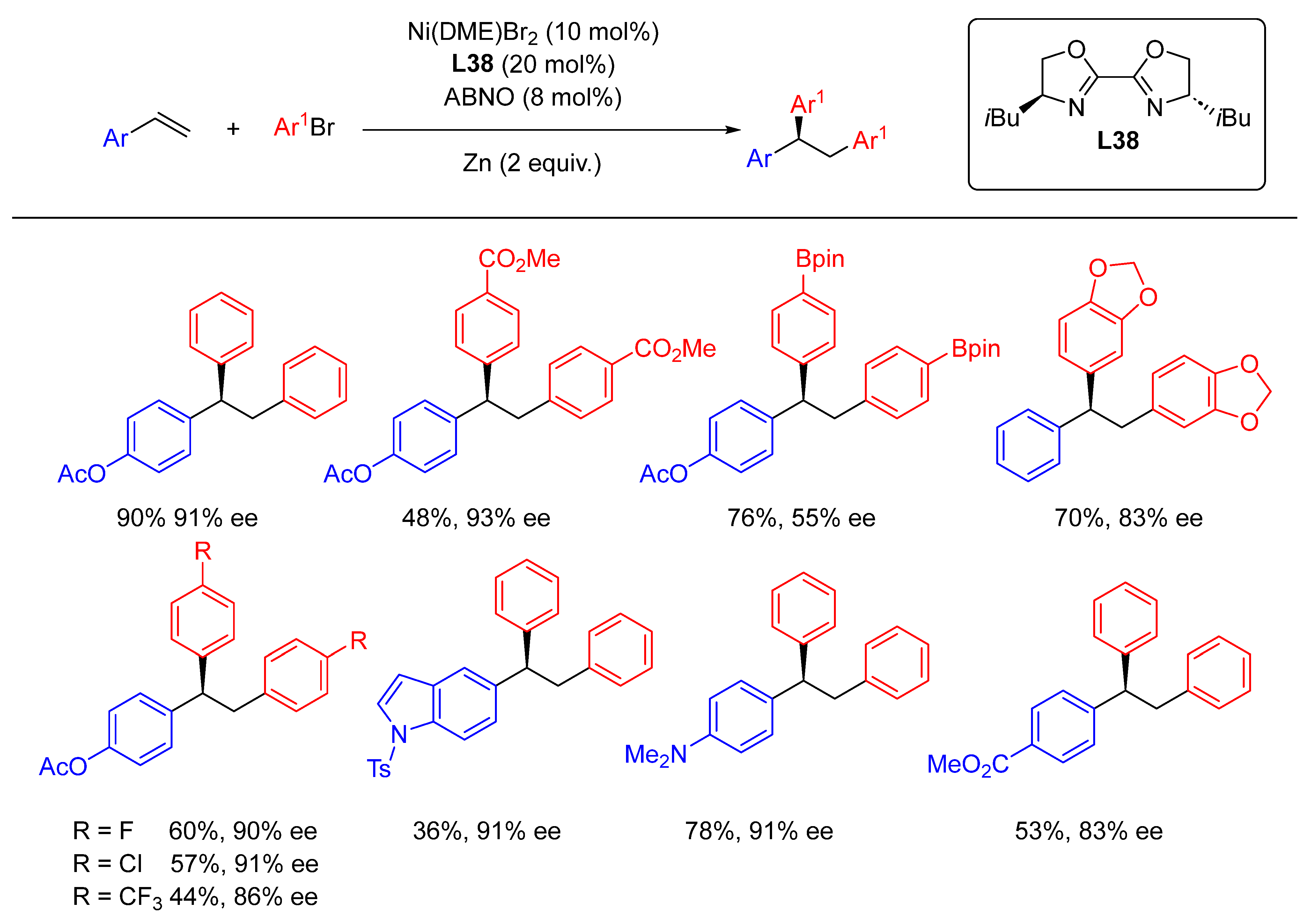
- Zhang, X.; Wu, W.; Cao, W.; Yu, H.; Xu, X.; Liu, X.; Feng, X. Enantioselective Radical-Polar Crossover Reactions of Indanonecarboxamides with Alkenes. Angew. Chem. Int. Ed. 2020, 59, 4846–4850. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wen, X.; Cui, X.; Wojtas, L.; Zhang, X.P. Asymmetric Radical Cyclopropanation of Alkenes with In Situ-Generated Donor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc. 2017, 139, 1049–1052. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-C.C.; Wang, D.-S.; Zhang, C.; Xie, J.; Li, B.; Zhang, X.P. Asymmetric radical cyclopropanation of dehydroaminocarboxylates: Stereoselective synthesis of cyclopropyl α-amino acids. Chem 2021, 7, 1588–1601. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ke, J.; Zhu, Y.; Deb, A.; Xu, Y.; Zhang, X.P. Asymmetric Radical Process for General Synthesis of Chiral Heteroaryl Cyclopropanes. J. Am. Chem. Soc. 2021, 143, 11121–11129. [Google Scholar] [CrossRef]
- Ke, J.; Lee, W.-C.C.; Wang, X.; Wang, Y.; Wen, X.; Zhang, X.P. Metalloradical Activation of In Situ-Generated α-Alkynyldiazomethanes for Asymmetric Radical Cyclopropanation of Alkenes. J. Am. Chem. Soc. 2022, 144, 2368–2378. [Google Scholar] [CrossRef]
- Xie, J.; Xu, P.; Zhu, Y.; Wang, J.; Lee, W.-C.C.; Zhang, X.P. New Catalytic Radical Process Involving 1,4-Hydrogen Atom Abstraction: Asymmetric Construction of Cyclobutanones. J. Am. Chem. Soc. 2021, 143, 11670–11678. [Google Scholar] [CrossRef]
- Jiang, H.; Lang, K.; Lu, H.; Wojtas, L.; Zhang, X.P. Asymmetric Radical Bicyclization of Allyl Azidoformates via Cobalt(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc. 2017, 139, 9164–9167. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Torker, S.; Wojtas, L.; Zhang, X.P. Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H Amination via Enantiodifferentiative H-Atom Abstraction and Stereoretentive Radical Substitution. J. Am. Chem. Soc. 2019, 141, 12388–12396. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhong, J.; Zhou, Y.; Gao, Z.; Walsh, P.J.; Wang, X.; Ma, S.; Hou, S.; Liu, S.; Wang, M.; et al. Cobalt-Catalyzed Enantioselective Negishi Cross-Coupling of Racemic α-Bromo Esters with Arylzincs. Chem. A Eur. J. 2018, 24, 2059–2064. [Google Scholar] [CrossRef]
- Discolo, C.A.; Touney, E.E.; Pronin, S.V. Catalytic Asymmetric Radical–Polar Crossover Hydroalkoxylation. J. Am. Chem. Soc. 2019, 141, 17527–17532. [Google Scholar] [CrossRef]
- Qin, T.; Lv, G.; Meng, Q.; Zhang, G.; Xiong, T.; Zhang, Q. Cobalt-Catalyzed Radical Hydroamination of Alkenes with N -Fluorobenzenesulfonimides. Angew. Chem. Int. Ed. 2021, 60, 25949–25957. [Google Scholar] [CrossRef]
- Zhang, K.; Lu, L.; Jia, Y.; Wang, Y.; Lu, F.; Pan, F.; Xiao, W. Exploration of a Chiral Cobalt Catalyst for Visible-Light-Induced Enantioselective Radical Conjugate Addition. Angew. Chem. Int. Ed. 2019, 58, 13375–13379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Meggers, E. Steering Asymmetric Lewis Acid Catalysis Exclusively with Octahedral Metal-Centered Chirality. Acc. Chem. Res. 2017, 50, 320–330. [Google Scholar] [CrossRef]
- Ma, J.; Rosales, A.R.; Huang, X.; Harms, K.; Riedel, R.; Wiest, O.; Meggers, E. Visible-Light-Activated Asymmetric β-C–H Functionalization of Acceptor-Substituted Ketones with 1,2-Dicarbonyl Compounds. J. Am. Chem. Soc. 2017, 139, 17245–17248. [Google Scholar] [CrossRef]
- Ma, J.; Lin, J.; Zhao, L.; Harms, K.; Marsch, M.; Xie, X.; Meggers, E. Synthesis of β-Substituted γ-Aminobutyric Acid Derivatives through Enantioselective Photoredox Catalysis. Angew. Chem. Int. Ed. 2018, 57, 11193–11197. [Google Scholar] [CrossRef]
- Chen, S.; Huang, X.; Meggers, E.; Houk, K.N. Origins of Enantioselectivity in Asymmetric Radical Additions to Octahedral Chiral-at-Rhodium Enolates: A Computational Study. J. Am. Chem. Soc. 2017, 139, 17902–17907. [Google Scholar] [CrossRef]
- Tutkowski, B.; Meggers, E.; Wiest, O. Understanding Rate Acceleration and Stereoinduction of an Asymmetric Giese Reaction Mediated by a Chiral Rhodium Catalyst. J. Am. Chem. Soc. 2017, 139, 8062–8065. [Google Scholar] [CrossRef] [PubMed]
- Steinlandt, P.S.; Zuo, W.; Harms, K.; Meggers, E. Bis-Cyclometalated Indazole Chiral-at-Rhodium Catalyst for Asymmetric Photoredox Cyanoalkylations. Chem. A Eur. J. 2019, 25, 15333–15340. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Fang, C.; Liu, P.; Ready, J.M. Rhodium-Catalyzed Enantioselective Radical Addition of CX4 Reagents to Olefins. Angew. Chem. Int. Ed. 2017, 56, 8780–8784. [Google Scholar] [CrossRef] [PubMed]
- Skubi, K.L.; Kidd, J.B.; Jung, H.; Guzei, I.A.; Baik, M.-H.; Yoon, T.P. Enantioselective Excited-State Photoreactions Controlled by a Chiral Hydrogen-Bonding Iridium Sensitizer. J. Am. Chem. Soc. 2017, 139, 17186–17192. [Google Scholar] [CrossRef]
- Zheng, J.; Swords, W.B.; Jung, H.; Skubi, K.L.; Kidd, J.B.; Meyer, G.J.; Baik, M.-H.; Yoon, T.P. Enantioselective Intermolecular Excited-State Photoreactions Using a Chiral Ir Triplet Sensitizer: Separating Association from Energy Transfer in Asymmetric Photocatalysis. J. Am. Chem. Soc. 2019, 141, 13625–13634. [Google Scholar] [CrossRef]
- Uraguchi, D.; Kimura, Y.; Ueoka, F.; Ooi, T. Urea as a Redox-Active Directing Group under Asymmetric Photocatalysis of Iridium-Chiral Borate Ion Pairs. J. Am. Chem. Soc. 2020, 142, 19462–19467. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Faraone, A.; Gandolfo, E.; Mazzarella, D.; Melchiorre, P. Catalytic asymmetric C–C cross-couplings enabled by photoexcitation. Nat. Chem. 2021, 13, 575–580. [Google Scholar] [CrossRef]
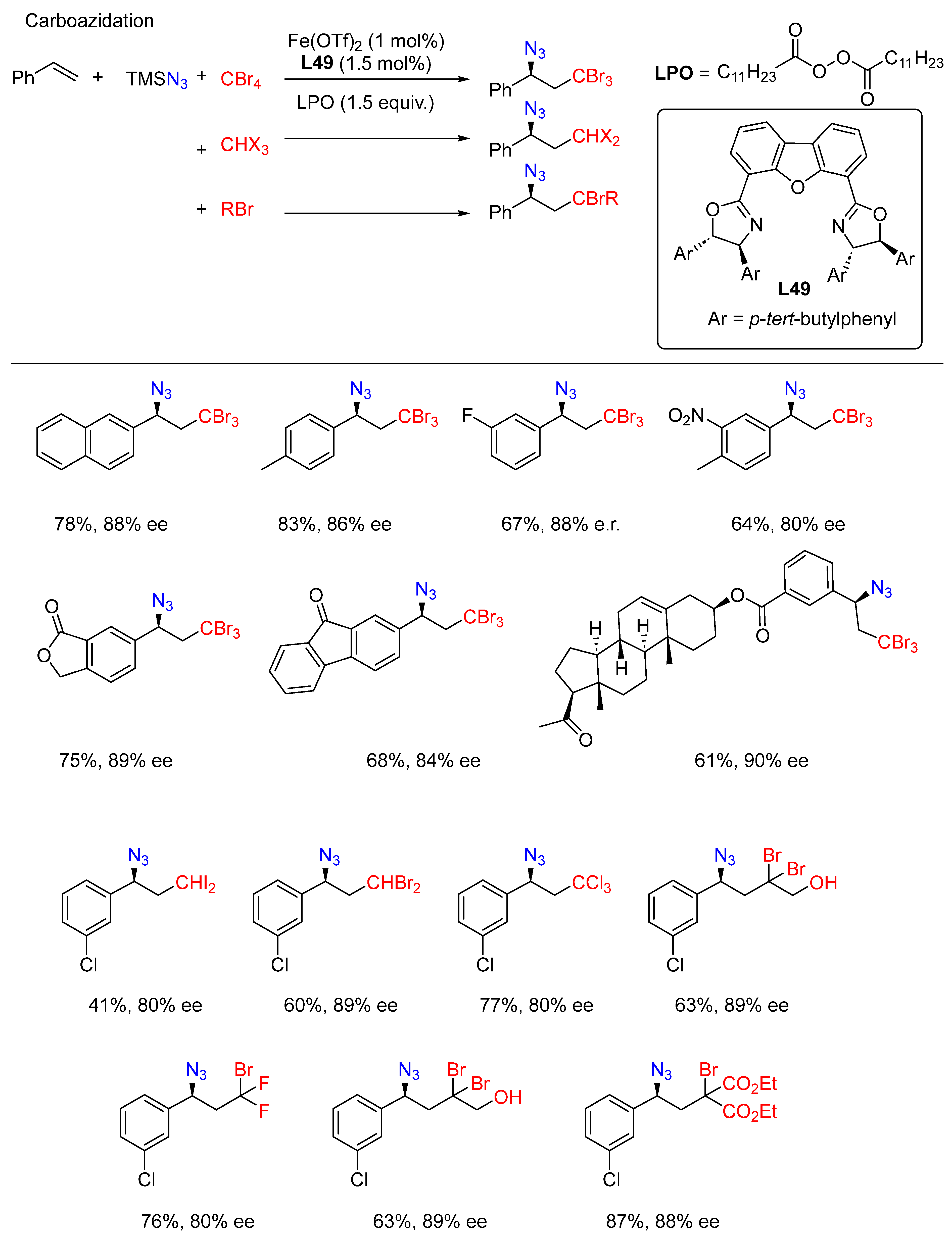
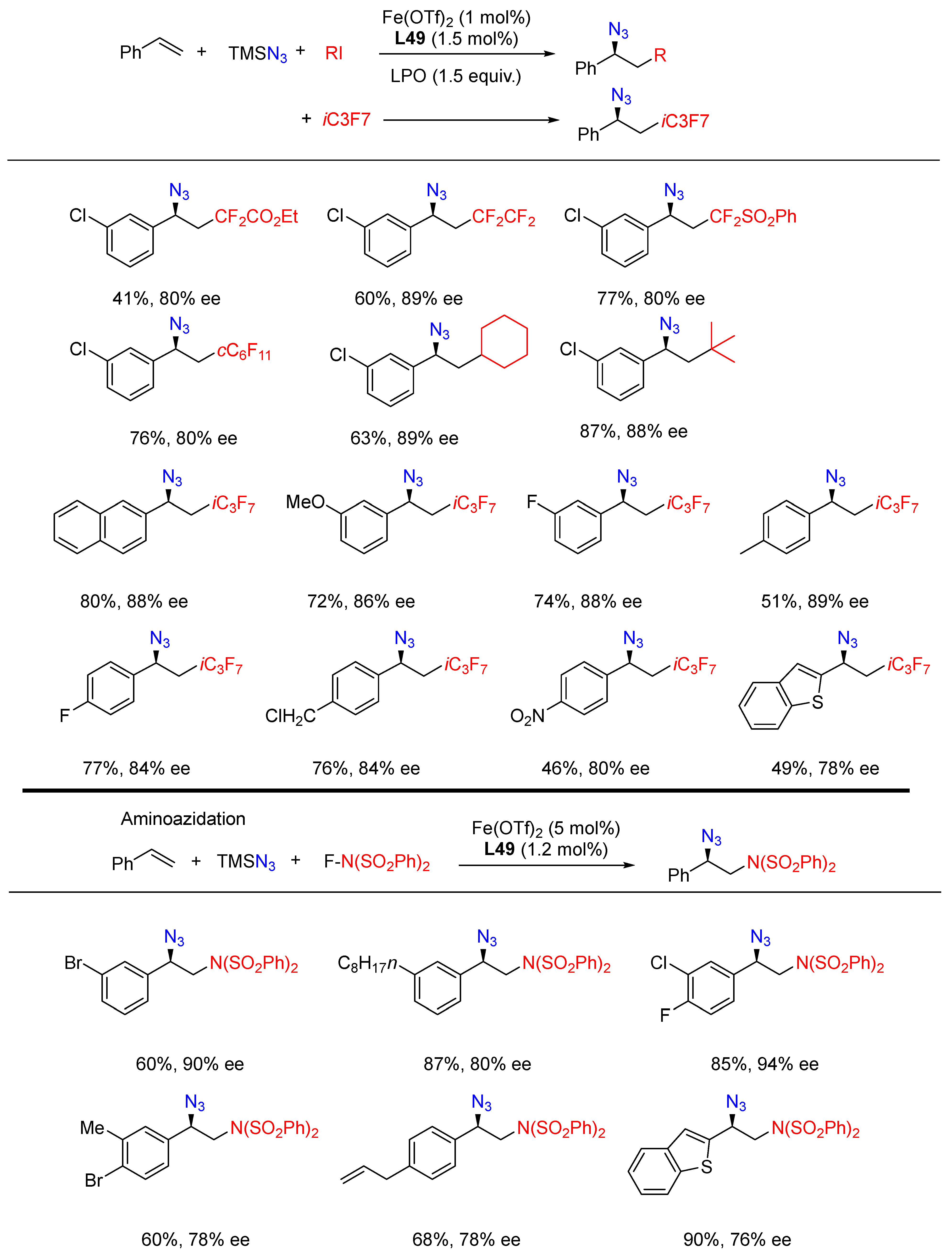
- Ge, L.; Zhou, H.; Chiou, M.-F.; Jiang, H.; Jian, W.; Ye, C.; Li, X.; Zhu, X.; Xiong, H.; Li, Y.; et al. Iron-catalysed asymmetric carboazidation of styrenes. Nat. Catal. 2021, 4, 28–35. [Google Scholar] [CrossRef]
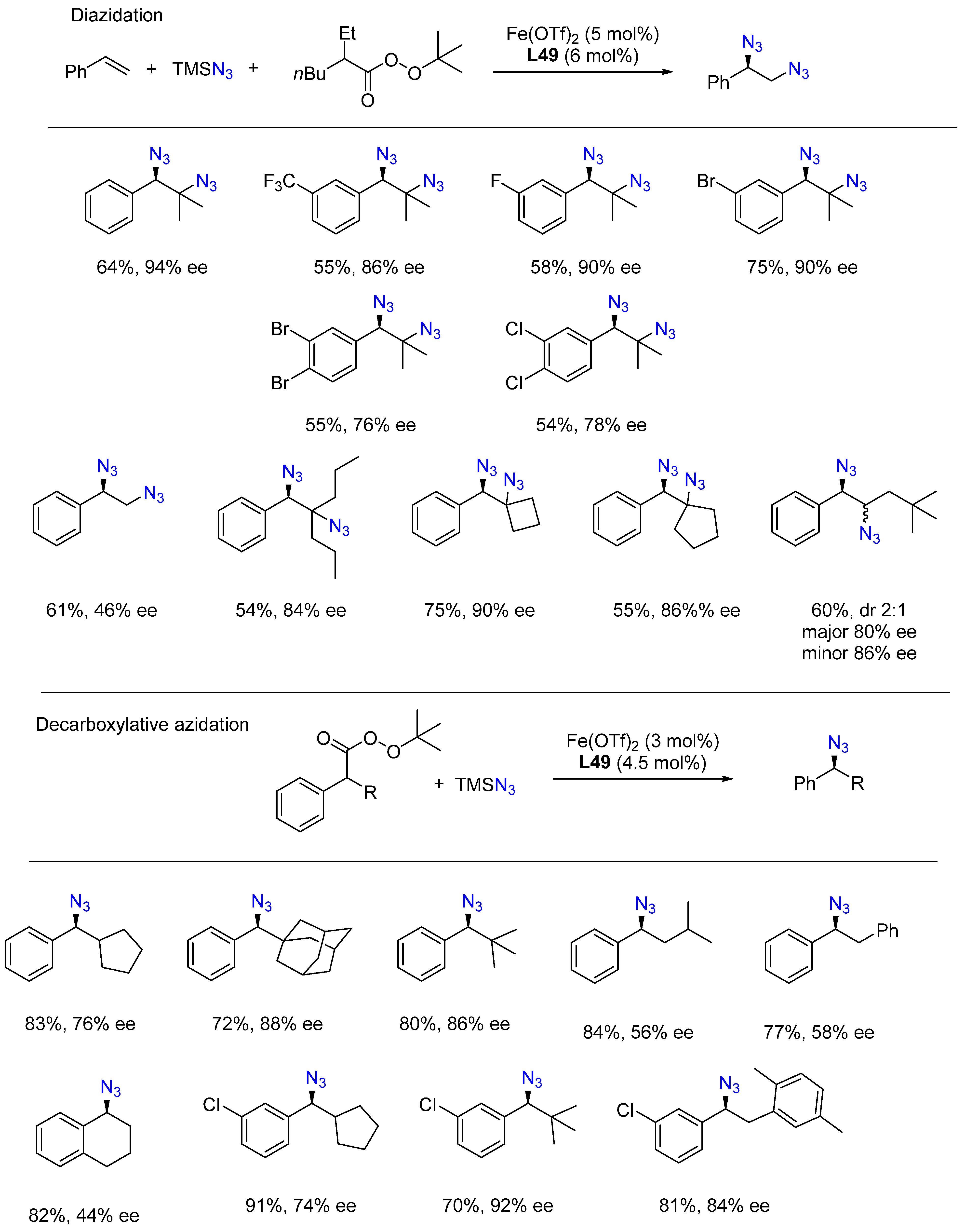
- Lv, D.; Sun, Q.; Zhou, H.; Ge, L.; Qu, Y.; Li, T.; Ma, X.; Li, Y.; Bao, H. Iron-Catalyzed Radical Asymmetric Aminoazidation and Diazidation of Styrenes. Angew. Chem. Int. Ed. 2021, 60, 12455–12460. [Google Scholar] [CrossRef]
- Wang, K.; Li, Y.; Li, X.; Li, D.; Bao, H. Iron-Catalyzed Asymmetric Decarboxylative Azidation. Org. Lett. 2021, 23, 8847–8851. [Google Scholar] [CrossRef]
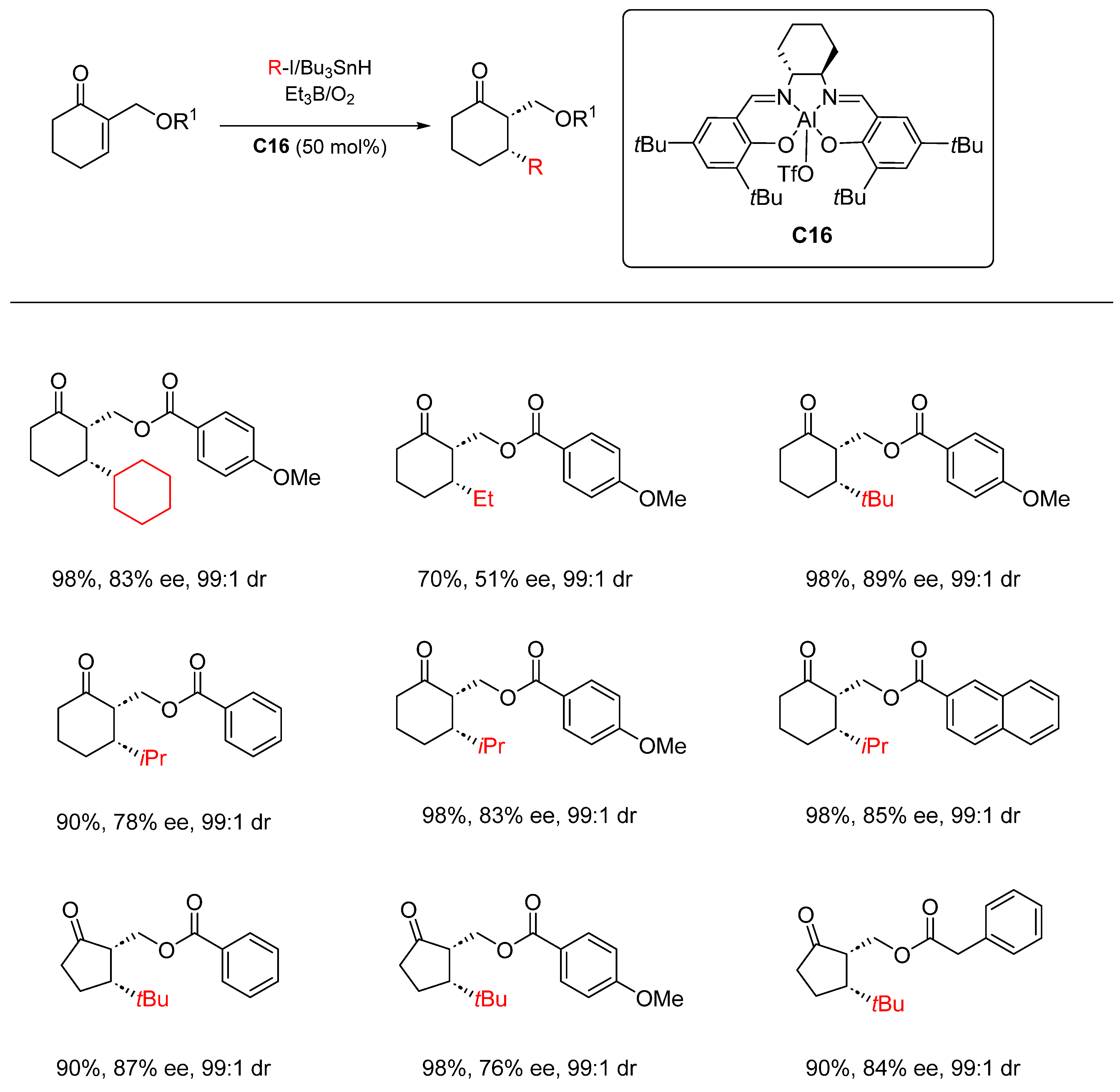
- Nad, S.; Sibi, M.P. Asymmetric Synthesis of 2,3-Disubstituted Cyclic Ketones by Enantioselective Conjugate Radical Additions Helv. Chim. Acta 2019, 102, e1900223. [Google Scholar] [CrossRef]
- Zhang, F.-H.; Guo, X.; Zeng, X.; Wang, Z. Asymmetric 1,4-Functionalization of 1,3-Enynes via Dual Photoredox and Chromium Catalysis. Nat. Commun. 2022, 13, 5036. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhan, T.; Zhou, Y.; Chen, L.; Liu, X.; Feng, X. Visible-Light-Activated Asymmetric Addition of Hydrocarbons to Pyridine-Based Ketones. ACS Catal. 2022, 12, 5136–5144. [Google Scholar] [CrossRef]
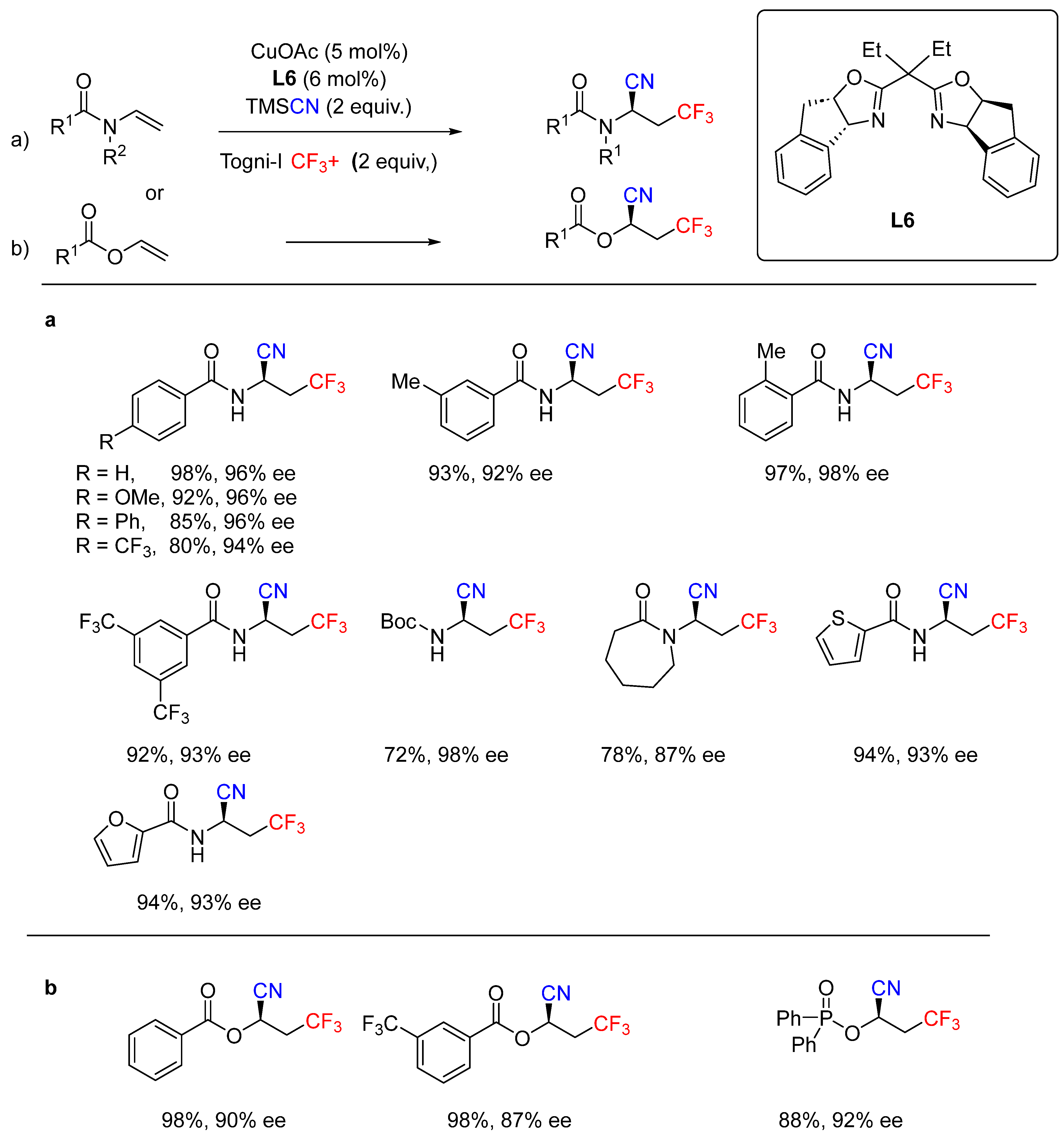
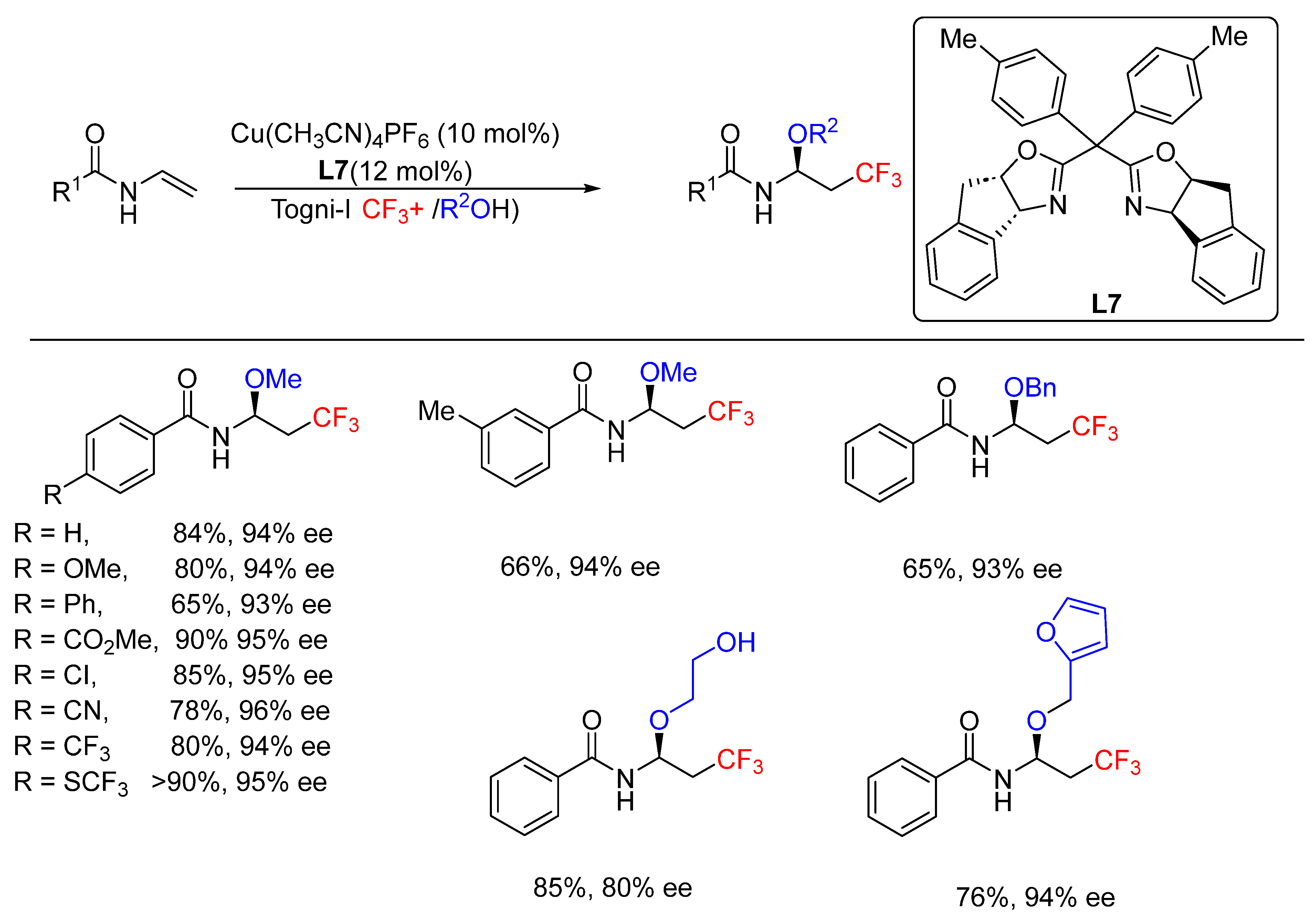
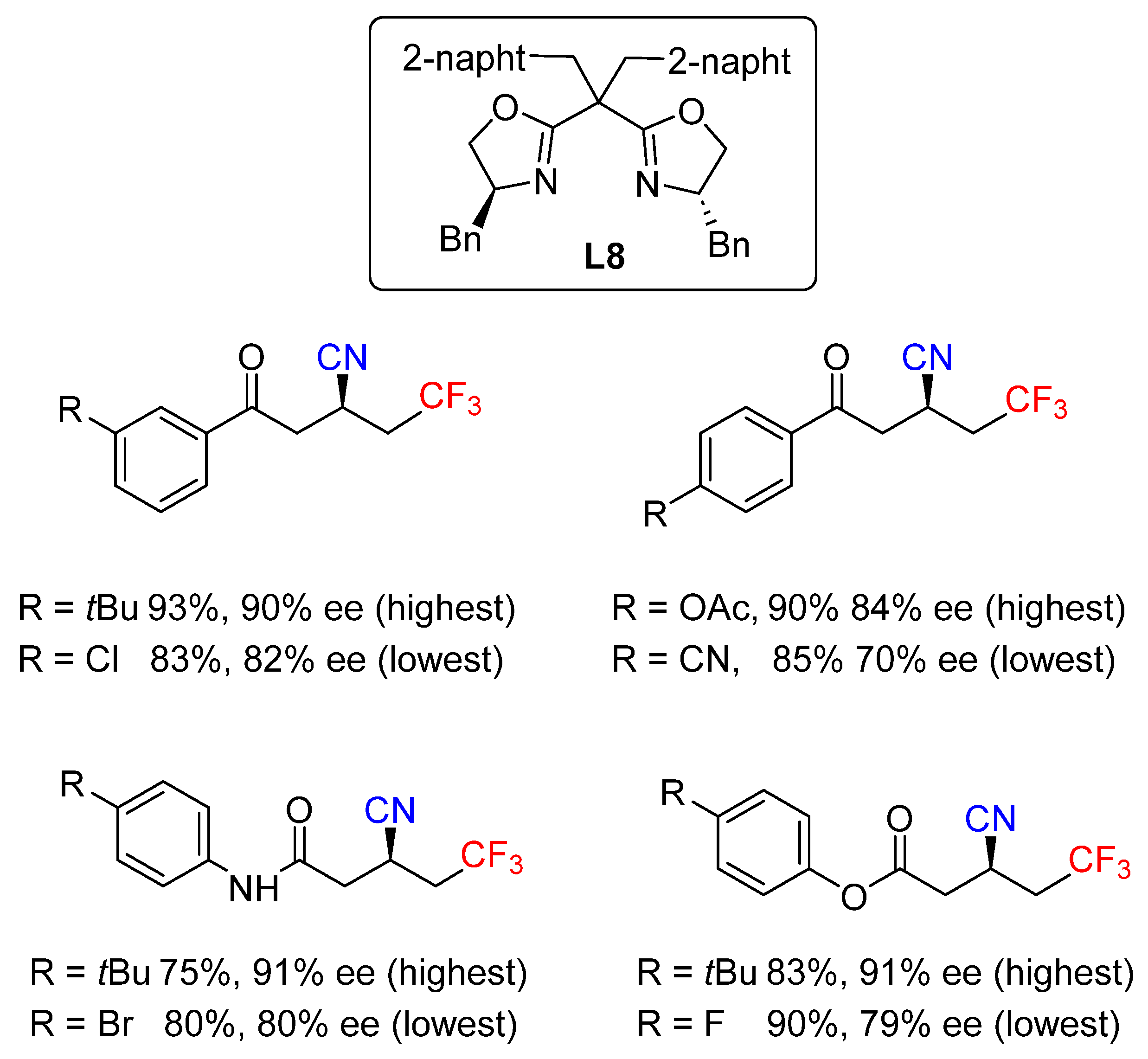
- Calvo, R.; Comas-Vives, A.; Togni, A.; Katayev, D. Taming Radical Intermediates for the Construction of Enantioenriched Trifluoromethylated Quaternary Carbon Centers. Angew. Chem. Int. Ed. 2019, 58, 1447–1452. [Google Scholar] [CrossRef]
- Xue, S.; Limburg, B.; Ghorai, D.; Benet-Buchholz, J.; Kleij, A.W. Asymmetric Synthesis of Homoallylic Alcohols Featuring Vicinal Tetrasubstituted Carbon Centers via Dual Pd/Photoredox Catalysis. Org. Lett. 2021, 23, 4447–4451. [Google Scholar] [CrossRef]
- Cheng, Y.-F.; Dong, X.-Y.; Gu, Q.-S.; Yu, Z.-L.; Liu, X.-Y. Achiral Pyridine Ligand-Enabled Enantioselective Radical Oxytrifluoromethylation of Alkenes with Alcohols. Angew. Chem. Int. Ed. 2017, 56, 8883–8886. [Google Scholar] [CrossRef]
- Lin, J.-S.; Wang, F.-L.; Dong, X.-Y.; He, W.-W.; Yuan, Y.; Chen, S.; Liu, X.-Y. Catalytic asymmetric radical aminoperfluoroalkyla-tion and aminodifluoromethylation of alkenes to versatile enantioenriched-fluoroalkyl amines. Nat. Comm. 2017, 8, 14841. [Google Scholar] [CrossRef]
- Wang, F.-L.; Dong, X.-Y.; Lin, J.-S.; Zeng, Y.; Jiao, G.-Y.; Gu, Q.-S.; Guo, X.-Q.; Ma, C.-L.; Liu, X.-Y. Catalytic Asymmetric Radical Diamination of Alkenes. Chem 2017, 3, 979–990. [Google Scholar] [CrossRef]
- Zeng, Y.; Liu, X.-D.; Guo, X.-Q.; Gu, Q.-S.; Li, Z.-L.; Chang, X.-Y.; Liu, X.-Y. Cu/Chiral Phosphoric Acid-Catalyzed Radical-Initiated Asymmetric Aminosilylation of Alkene with Hydrosilane. Sci. China Chem. 2019, 62, 1529–1536. [Google Scholar] [CrossRef]
- Zheng, D.; Studer, A. Asymmetric Synthesis of Heterocyclic γ-Amino-Acid and Diamine Derivatives by Three-Component Radical Cascade Reactions. Angew. Chem. Int. Ed. 2019, 58, 15803–15807. [Google Scholar] [CrossRef]
- Lin, J.S.; Li, T.T.; Liu, J.R.; Jiao, G.Y.; Gu, Q.S.; Cheng, J.T.; Guo, Y.L.; Hong, X.; Liu, X.Y. Cu/Chiral Phosphoric Acid-Catalyzed Asymmetric Three-Component Radical-Initiated 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc. 2019, 141, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, X.; He, F.; Yang, X. Asymmetric synthesis of oxazolines bearing α-stereocenters through radical addition–enantioselective protonation enabled by cooperative catalysis. Org. Chem. Front. 2021, 8, 5804–5809. [Google Scholar] [CrossRef]
- Xu, L.; Zhong, S.; Yang, Q.; Wei, J.; Zou, J.; Li, H.; Cai, Y. Catalytic Asymmetric Radical-Mediated Three-Component Piancatelli-Type Rearrangement of Furylalkenes. ACS Catal. 2021, 11, 10198–10207. [Google Scholar] [CrossRef]
- Wang, Z.; Cheng, J.T.; Shi, Z.; Wang, N.; Zhan, F.; Jiang, S.P.; Lin, J.S.; Jiang, Y.; Liu, X.Y. Catalytic Asymmetric Intermolec-ular Radical Aminotrifluoromethylation of Alkenes with Hydrazines by Cu(I)/CPA Cooperative Catalysis. ChemCatChem 2021, 13, 185–190. [Google Scholar] [CrossRef]
- Dai, Z.-Y.; Nong, Z.-S.; Wang, P.-S. Light-Mediated Asymmetric Aliphatic C–H Alkylation with Hydrogen Atom Transfer Catalyst and Chiral Phosphoric Acid. ACS Catal. 2020, 10, 4786–4790. [Google Scholar] [CrossRef]
- Dai, Z.-Y.; Nong, Z.-S.; Song, S.; Wang, P.-S. Asymmetric Photocatalytic C(sp3)–H Bond Addition to α-Substituted Acrylates. Org. Lett. 2021, 23, 3157–3161. [Google Scholar] [CrossRef]
- Ye, L.; Tian, Y.; Meng, X.; Gu, Q.S.; Liu, X.Y. Enantioselective Copper(I)/Chiral Phosphoric Acid Catalyzed Intramolecular Amination of Allylic and Benzylic C–H Bonds. Angew. Chem. Int. Ed. 2020, 59, 1129–1133. [Google Scholar] [CrossRef]
- Ye, L.; Gu, Q.-S.; Tian, Y.; Meng, X.; Chen, G.-C.; Liu, X.-Y. Radical asymmetric intramolecular α-cyclopropanation of aldehydes towards bicyclo[3.1.0]hexanes containing vicinal all-carbon quaternary stereocenters. Nat. Commun. 2018, 9, 227. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, L.; Luo, S. Asymmetric C–H Dehydrogenative Allylic Alkylation by Ternary Photoredox-Cobalt-Chiral Primary Amine Catalysis under Visible Light. J. Am. Chem. Soc. 2022, 144, 10705–10710. [Google Scholar] [CrossRef]
- Yin, Y.; Dai, Y.; Jia, H.; Li, J.; Bu, L.; Qiao, B.; Zhao, X.; Jiang, Z. Conjugate Addition–Enantioselective Protonation of N-Aryl Gly-cines to α-Branched 2-Vinylazaarenes via Cooperative Photoredox and Asymmetric Catalysis. J. Am. Chem. Soc. 2018, 140, 6083–6087. [Google Scholar] [CrossRef]
- Chai, X.; Hu, X.; Zhao, X.; Yin, Y.; Cao, S.; Jiang, Z. Asymmetric Hydroaminoalkylation of Alkenylazaarenes via Coopera-tive Photoredox and Chiral Hydrogen-Bonding Catalysis. Angew. Chem. Int. Ed. 2022, 61, e202115110. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Qiao, B.; Lee, R.; Zhao, X.; Jiang, Z. Formal enantioconvergent substitution of alkyl halides via catalytic asymmetric photoredox radical coupling. Nat. Commun. 2018, 9, 2445. [Google Scholar] [CrossRef]
- Zeng, G.; Li, Y.; Qiao, B.; Zhao, X.; Jiang, Z. Photoredox asymmetric catalytic enantioconvergent substitution of 3-chlorooxindoles. Chem. Commun. 2019, 55, 11362–11365. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, L.; Luo, S. All-Carbon Quaternary Stereocenters α to Azaarenes via Radical-Based Asymmetric Olefin Difunc-tionalization. J. Am. Chem. Soc. 2020, 142, 19451–19456. [Google Scholar] [CrossRef]
- Dai, Y.; Liang, S.; Zeng, G.; Huang, H.; Zhao, X.; Cao, S.; Jiang, Z. Asymmetric [3+2] photocycloadditions of cyclopropylamines with electron-rich and electron-neutral olefins. Chem. Sci. 2022, 13, 3787–3795. [Google Scholar] [CrossRef]
- Liang, D.; Chen, J.-R.; Tan, L.-P.; He, Z.-W.; Xiao, W.-J. Catalytic Asymmetric Construction of Axially and Centrally Chiral Het-erobiaryls by Minisci Reaction. J. Am. Chem. Soc. 2022, 144, 6040–6049. [Google Scholar] [CrossRef]
- Colgan, A.C.; Proctor, R.S.J.; Gibson, D.C.; Chuentragool, P.; Lahdenperä, A.S.K.; Ermanis, K.; Phipps, R.J. Hydrogen Atom Transfer Driven Enantioselective Minisci Reaction of Alcohols. Angew. Chem. Int. Ed. 2022, 61, e202200266. [Google Scholar] [CrossRef]
- Li, Y.; Han, C.; Wang, Y.; Huang, X.; Zhao, X.; Qiao, B.; Jiang, Z. Catalytic Asymmetric Reductive Azaarylation of Olefins via Enantioselective Radical Coupling. J. Am. Chem. Soc. 2022, 144, 7805–7814. [Google Scholar] [CrossRef]
- He, F.-S.; Zhang, C.; Jiang, M.; Lou, L.; Wu, J.; Ye, S. Access to chiral β-sulfonyl carbonyl compounds via photoinduced organocatalytic asymmetric radical sulfonylation with sulfur dioxide. Chem. Sci. 2022, 13, 8834–8839. [Google Scholar] [CrossRef]
- Mazzarella, D.; Crisenza, G.E.M.; Melchiorre, P. Asymmetric Photocatalytic C–H Functionalization of Toluene and Deriva-tives. J. Am. Chem. Soc. 2018, 140, 8439–8443. [Google Scholar] [CrossRef]
- Bonilla, P.; Rey, Y.P.; Holden, C.M.; Melchiorre, P. Photo-Organocatalytic Enantioselective Radical Cascade Reactions of Unactivated Olefins. Angew. Chem. Int. Ed. 2018, 57, 12819–12823. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, E.; Ma, D.; Bonilla, P.; Holden, C.M.; Lustosa, D.; Melchiorre, P. A General Organocatalytic System for Enantioselective Radical Conjugate Additions to Enals. Angew. Chem. Int. Ed. 2020, 60, 5357–5362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhu, Y.; Zhang, L.; Luo, S. Asymmetric α-Alkylation of β-Ketocarbonyls via Direct Phenacyl Bromide Photolysis by Chiral Primary Amine. Chin. J. Chem. 2018, 36, 716–722. [Google Scholar] [CrossRef]
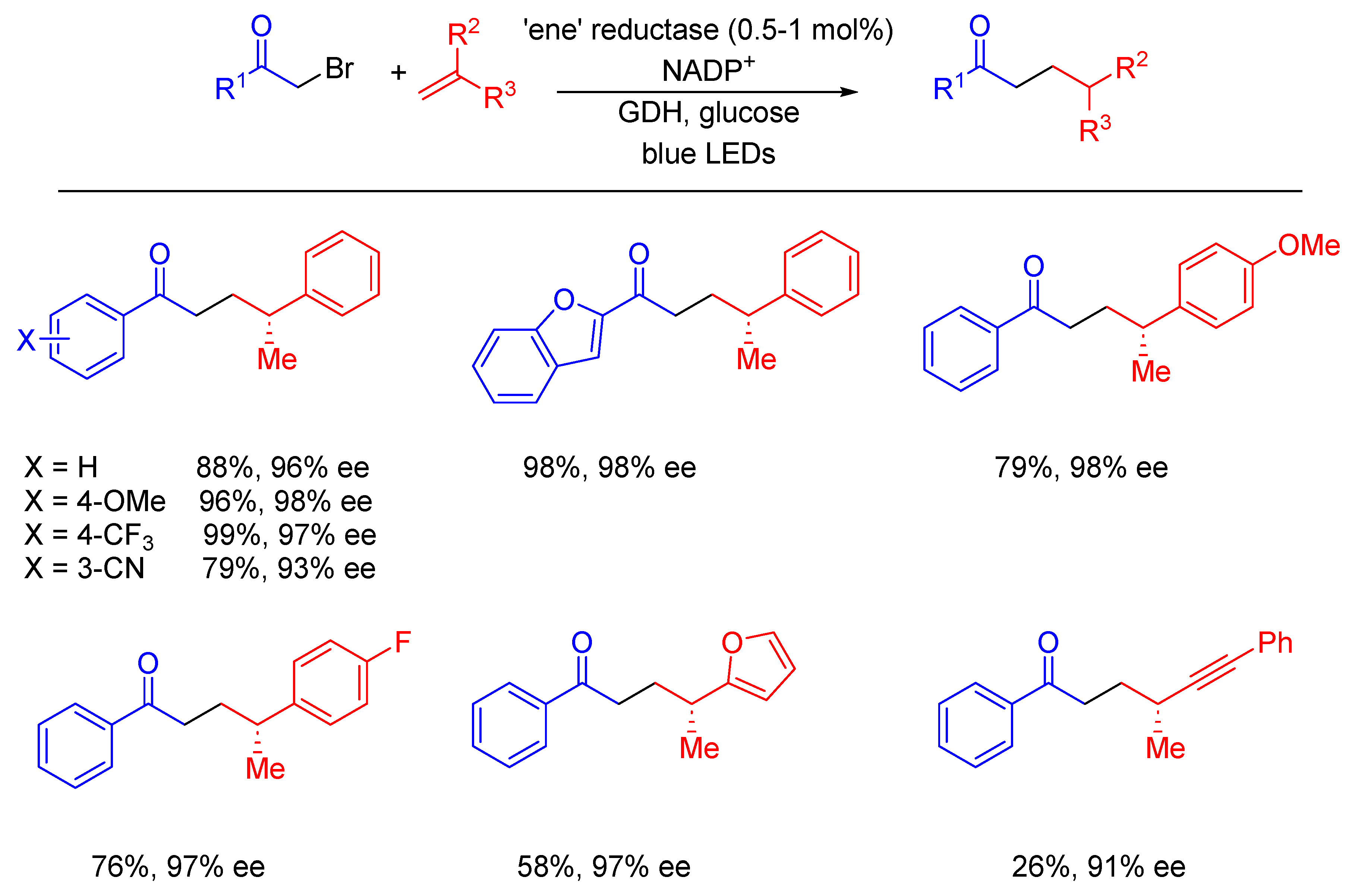
- Sandoval, B.A.; Meichan, A.J.; Hyster, T.K. Enantioselective Hydrogen Atom Transfer: Discovery of Catalytic Promiscuity in Flavin-Dependent ‘Ene’-Reductases. J. Am. Chem. Soc. 2017, 139, 11313–11316. [Google Scholar] [CrossRef] [PubMed]
- Clayman, P.D.; Hyster, T.K. Photoenzymatic Generation of Unstabilized Alkyl Radicals_An Asymmetric Reductive Cycliza-tion. J. Am. Chem. Soc. 2020, 142, 15673–15677. [Google Scholar] [CrossRef] [PubMed]
- Biegasiewicz, K.F.; Cooper, S.J.; Gao, X.; Oblinsky, D.G.; Kim, J.H.; Garfinkle, S.E.; Joyce, L.A.; Sandoval, B.A.; Scholes, G.D.; Hyster, T.K. Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 2019, 364, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Turek-Herman, J.R.; Choi, Y.J.; Cohen, R.D.; Hyster, T.K. Photoenzymatic Synthesis of α-Tertiary Amines by Engineered Flavin-Dependent “Ene”-Reductases. J. Am. Chem. Soc. 2021, 143, 19643–19647. [Google Scholar] [CrossRef]
- Black, M.J.; Biegasiewicz, K.F.; Meichan, A.J.; Oblinsky, D.G.; Kudisch, B.; Scholes, G.D.; Hyster, T.K. Asymmetric redox-neutral radical cyclization catalysed by flavin-dependent ‘ene’-reductases. Nat. Chem. 2020, 12, 71–75. [Google Scholar] [CrossRef]
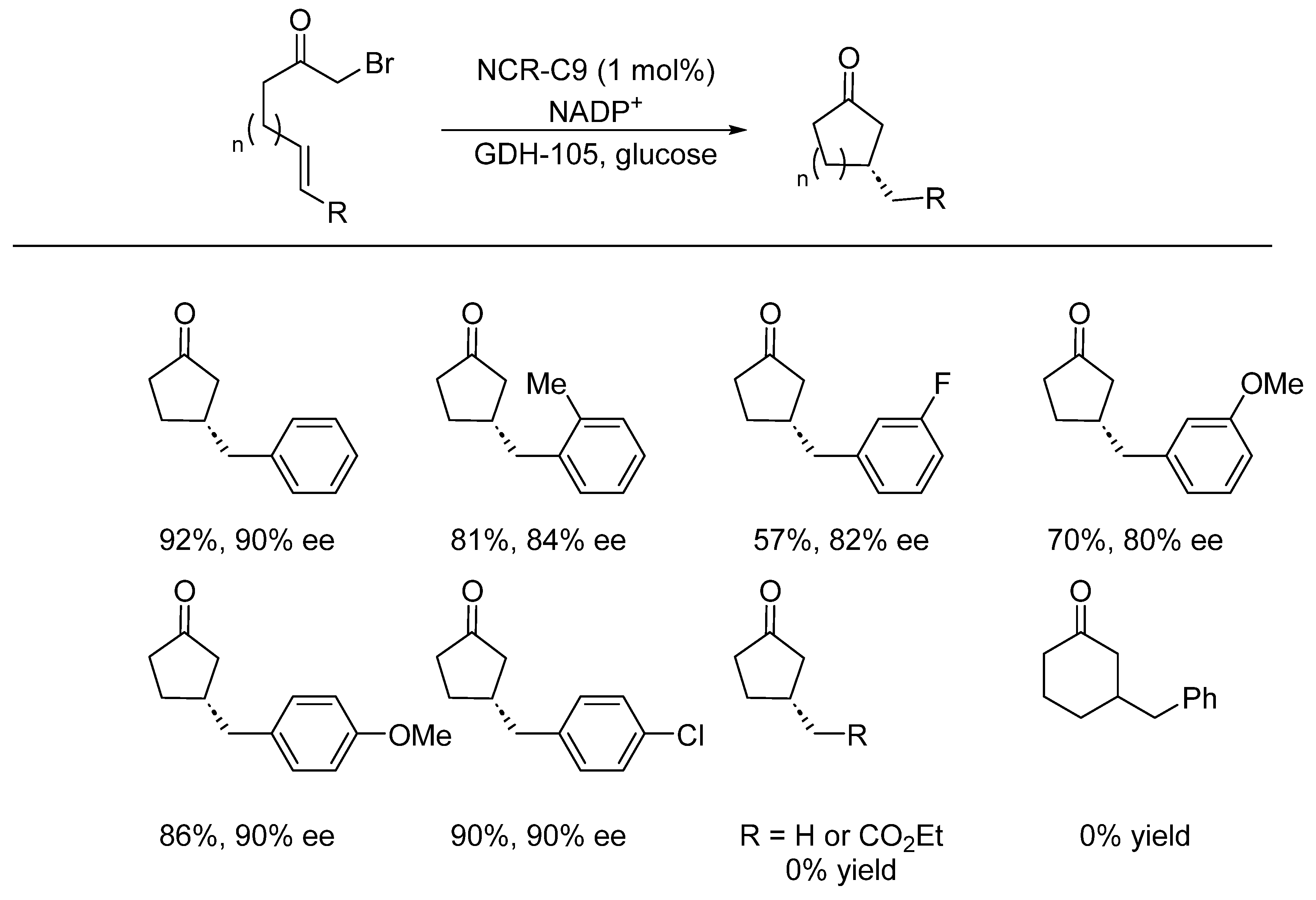
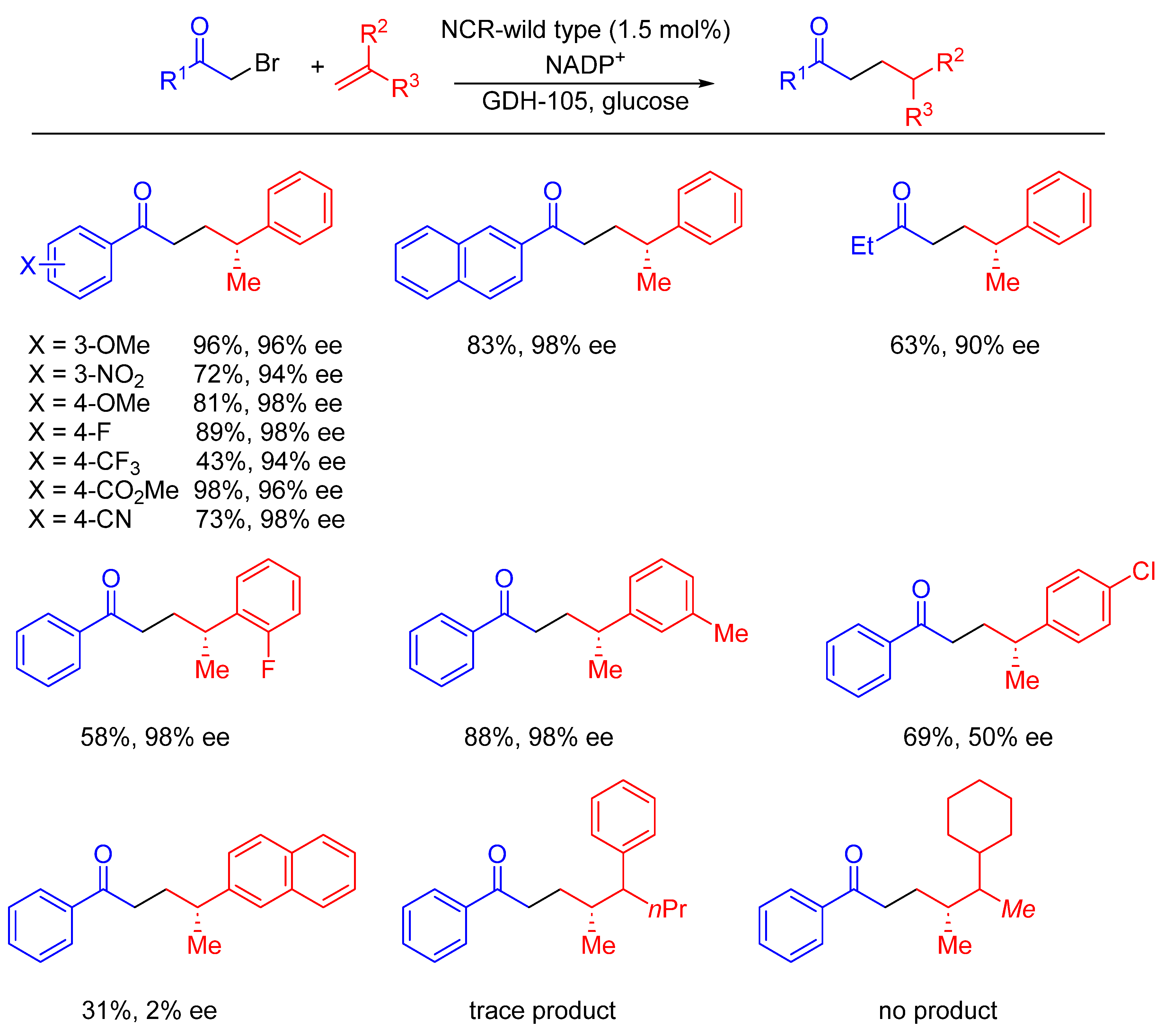
- Huang, X.; Wang, B.; Wang, Y.; Jiang, G.; Feng, J.; Zhao, H. Photoenzymatic enantioselective intermolecular radical hydroalkylation. Nature 2020, 584, 69–74. [Google Scholar] [CrossRef]
- Fu, H.; Cao, J.; Qiao, T.; Qi, Y.; Charnock, S.J.; Garfinkle, S.; Hyster, T.K. An asymmetric sp3–sp3 cross-electrophile coupling using ‘ene’-reductases. Nature 2022, 610, 302–307. [Google Scholar] [CrossRef]
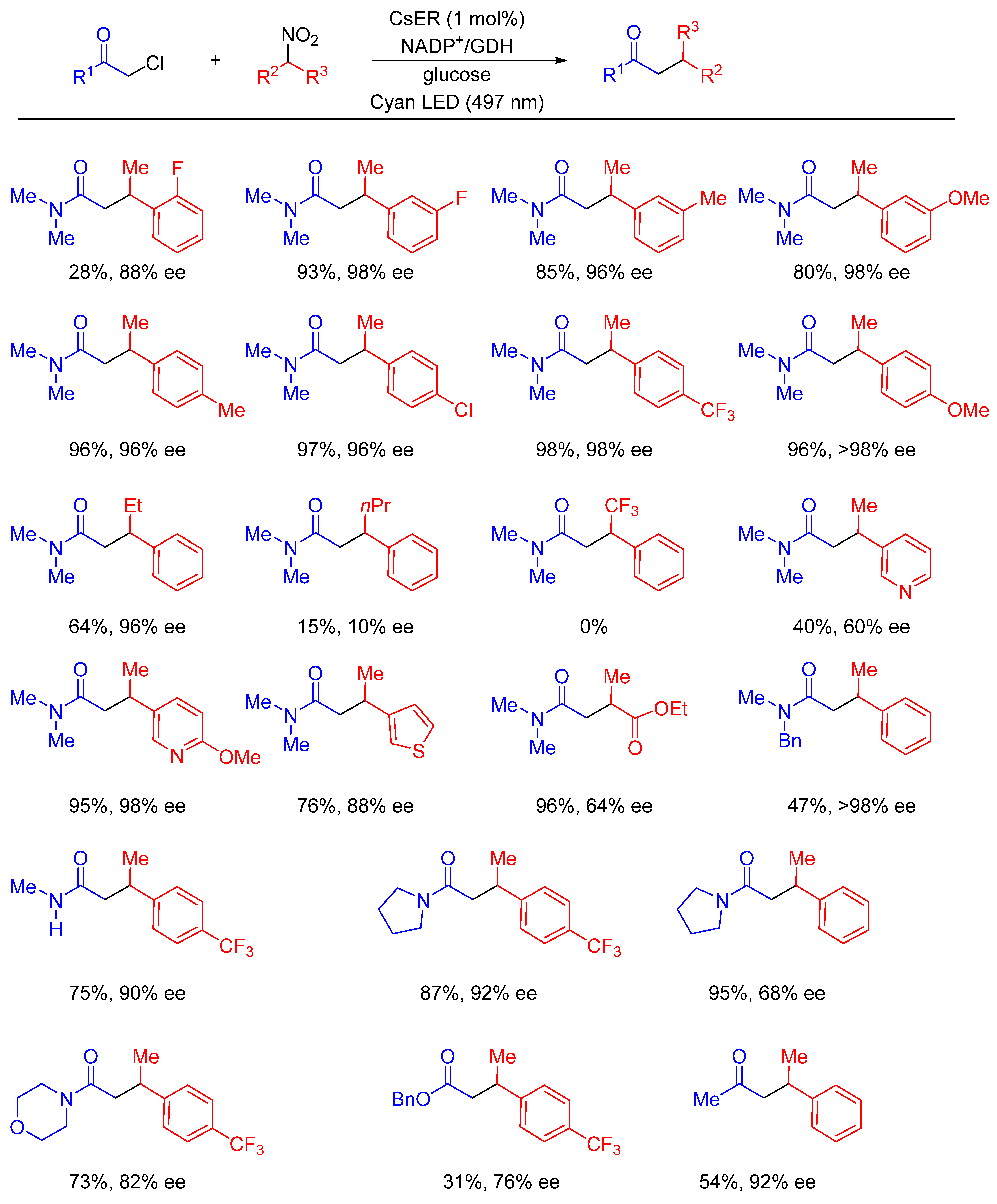
- Fu, H.; Qiao, T.; Carceller, J.M.; MacMillan, S.N.; Hyster, T.K. Asymmetric C-Alkylation of Nitroalkanes via Enzymatic Photoredox Catalysis. J. Am. Chem. Soc. 2023, 145, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Black, M.J.; Meichan, A.J.; Sandoval, B.A.; Chung, M.M.; Biegasiewicz, K.F.; Zhu, T.; Hyster, T.K. Photoenzymatic Hydrogenation of Heteroaromatic Olefins using ‘Ene’-Reductases with Photoredox Catalysts. Angew. Chem. Int. Ed. 2020, 59, 10484–11048. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cho, I.; Qi, X.; Liu, P.; Arnold, F.H. An enzymatic platform for the asymmetric amination of primary, secondary and tertiary C(sp3)–H bonds. Nat. Chem. 2019, 11, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Mai, B.K.; Neris, N.M.; Yang, Y.; Liu, P. C–N Bond Forming Radical Rebound Is the Enantioselectivity-Determining Step in P411-Catalyzed Enantioselective C(sp3)–H Amination: A Combined Computational and Experimental Investigation. J. Am. Chem. Soc. 2022, 144, 11215–11225. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chen, H.; Fu, W.; Garcia-Borràs, M.; Yang, Y.; Liu, P. Engineered P450 Atom-Transfer Radical Cyclases are Bifunctional Biocatalysts: Reaction Mechanism and Origin of Enantioselectivity. J. Am. Chem. Soc. 2022, 144, 13344–13355. [Google Scholar] [CrossRef]
- Coleman, T.; Kirk, A.M.; Chao, R.R.; Podgorski, M.N.; Harbort, J.S.; Churchman, L.R.; Bruning, J.B.; Bernhardt, P.V.; Harmer, J.R.; Krenske, E.H.; et al. Understanding the Mechanistic Requirements for Efficient and Stereoselective Alkene Epoxidation by a Cytochrome P450 Enzyme. ACS Catal. 2021, 11, 1995–2010. [Google Scholar] [CrossRef]
- Zhou, Q.; Chin, M.; Fu, Y.; Liu, P.; Yang, Y. Stereodivergent atom-transfer radical cyclization by engineered cytochromes P450. Science 2021, 374, 1612–1616. [Google Scholar] [CrossRef]
- Biegasiewicz, K.F.; Cooper, S.J.; Emmanuel, M.A.; Miller, D.C.; Hyster, T.K. Catalytic promiscuity enabled by photoredox catalysis in nicotinamide-dependent oxidoreductases. Nat. Chem. 2018, 10, 770–775. [Google Scholar] [CrossRef]
- Albarrán-Velo, J.; Lavandera, I.; Gotor-Fernández, V. Sequential Two-Step Stereoselective Amination of Allylic Alcohols through Combination of Laccases and Amine Transaminases. ChemBioChem 2020, 21, 200–211. [Google Scholar] [CrossRef]
- Carminati, D.M.; Fasan, R. Stereoselective Cyclopropanation of Electron-Deficient Olefins with a Cofactor Redesigned Carbene Transferase Featuring Radical Reactivity. ACS Catal. 2019, 9, 9683–9697. [Google Scholar] [CrossRef]
- Cheng, L.; Li, D.; Mai, B.K.; Bo, Z.; Cheng, L.; Liu, P.; Yang, Y. Stereoselective amino acid synthesis by synergistic photoredox-pyridoxal radical biocatalysis. Science 2023, 381, 444–451. [Google Scholar] [CrossRef] [PubMed]



























































































































































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, T.; Hakim, Y.Z.; Morawska, P. Recent Advances in the Enantioselective Radical Reactions. Molecules 2023, 28, 6252. https://doi.org/10.3390/molecules28176252
Bauer T, Hakim YZ, Morawska P. Recent Advances in the Enantioselective Radical Reactions. Molecules. 2023; 28(17):6252. https://doi.org/10.3390/molecules28176252
Chicago/Turabian StyleBauer, Tomasz, Yusuf Zaim Hakim, and Paulina Morawska. 2023. "Recent Advances in the Enantioselective Radical Reactions" Molecules 28, no. 17: 6252. https://doi.org/10.3390/molecules28176252
APA StyleBauer, T., Hakim, Y. Z., & Morawska, P. (2023). Recent Advances in the Enantioselective Radical Reactions. Molecules, 28(17), 6252. https://doi.org/10.3390/molecules28176252