
A Theoretical Analysis of the Differential Chemical Reaction Results Caused by Chirality Induction



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
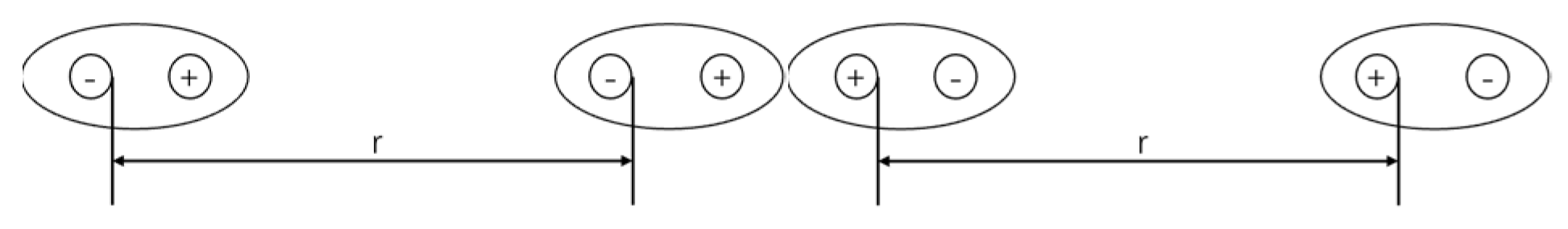
2. Chiral-Induced Spin Selectivity (CISS)
3. Results
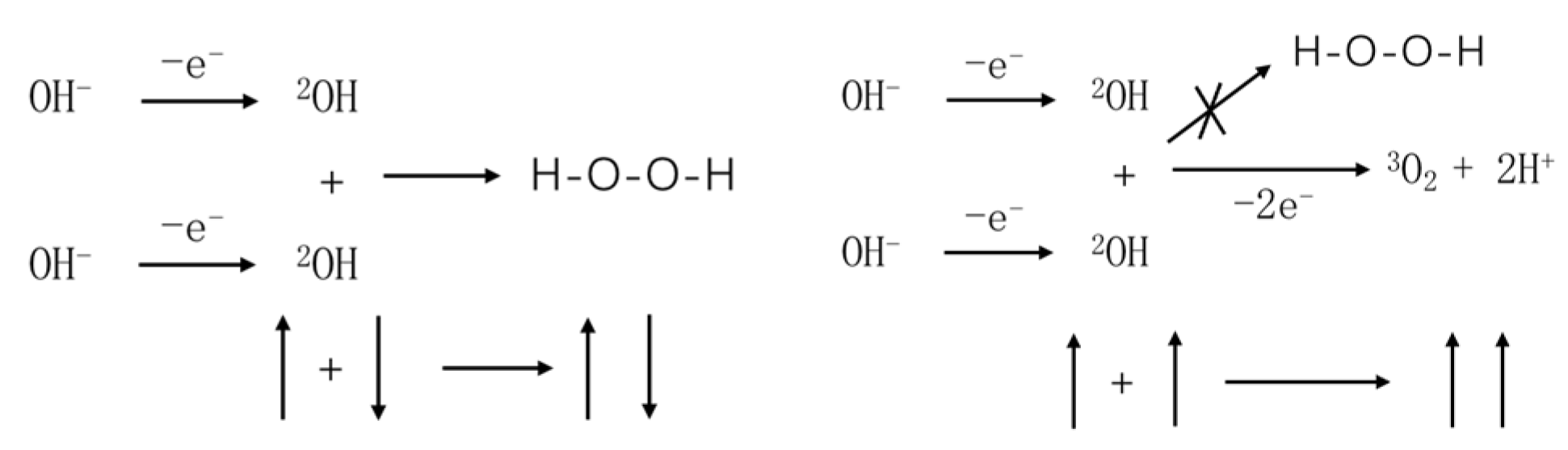
3.1. Electron Spin and Chemical Reactions
3.2. Chiral Selectivity of the AADC Enzyme
3.3. L-DOPA and D-DOPA
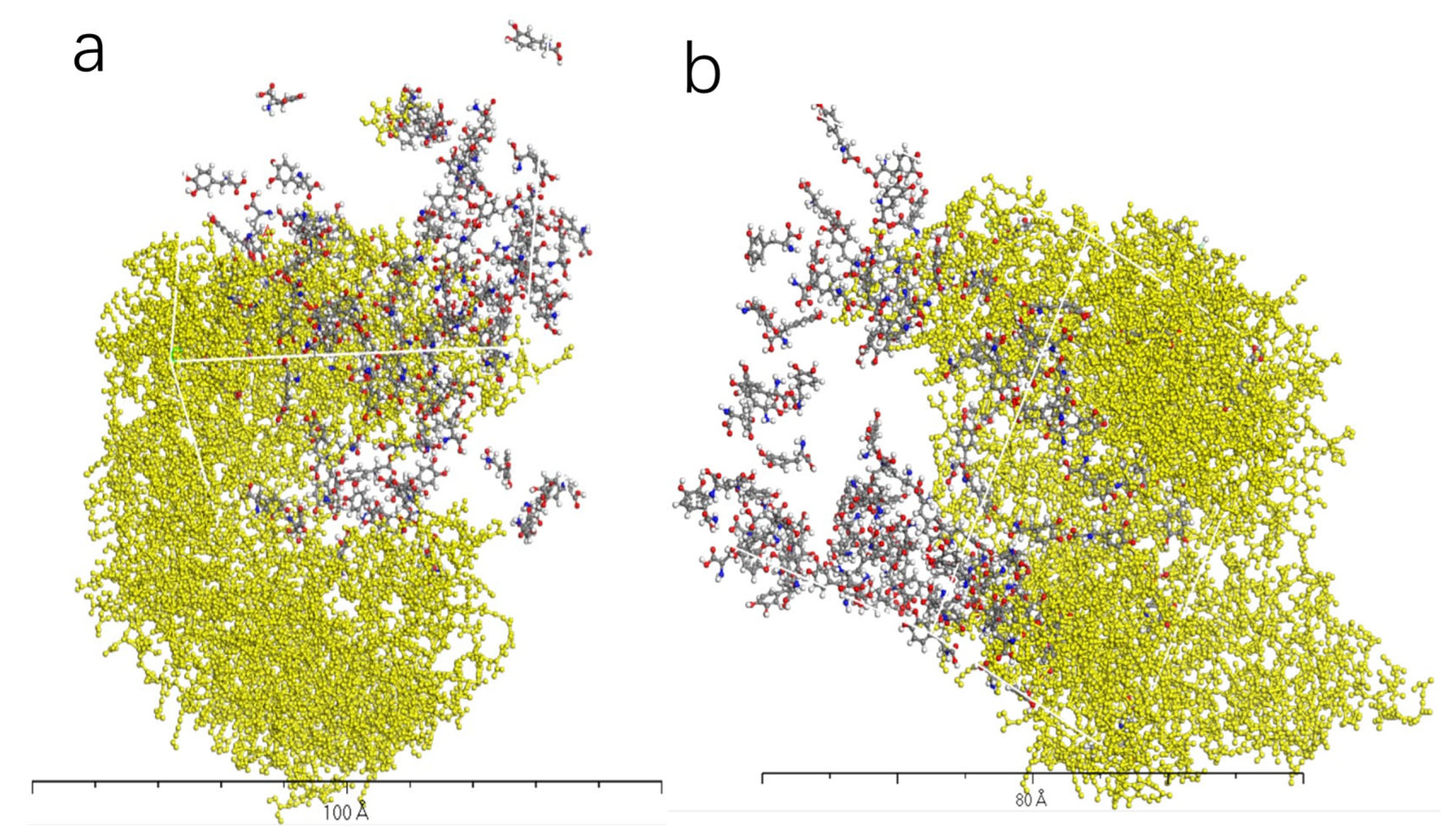
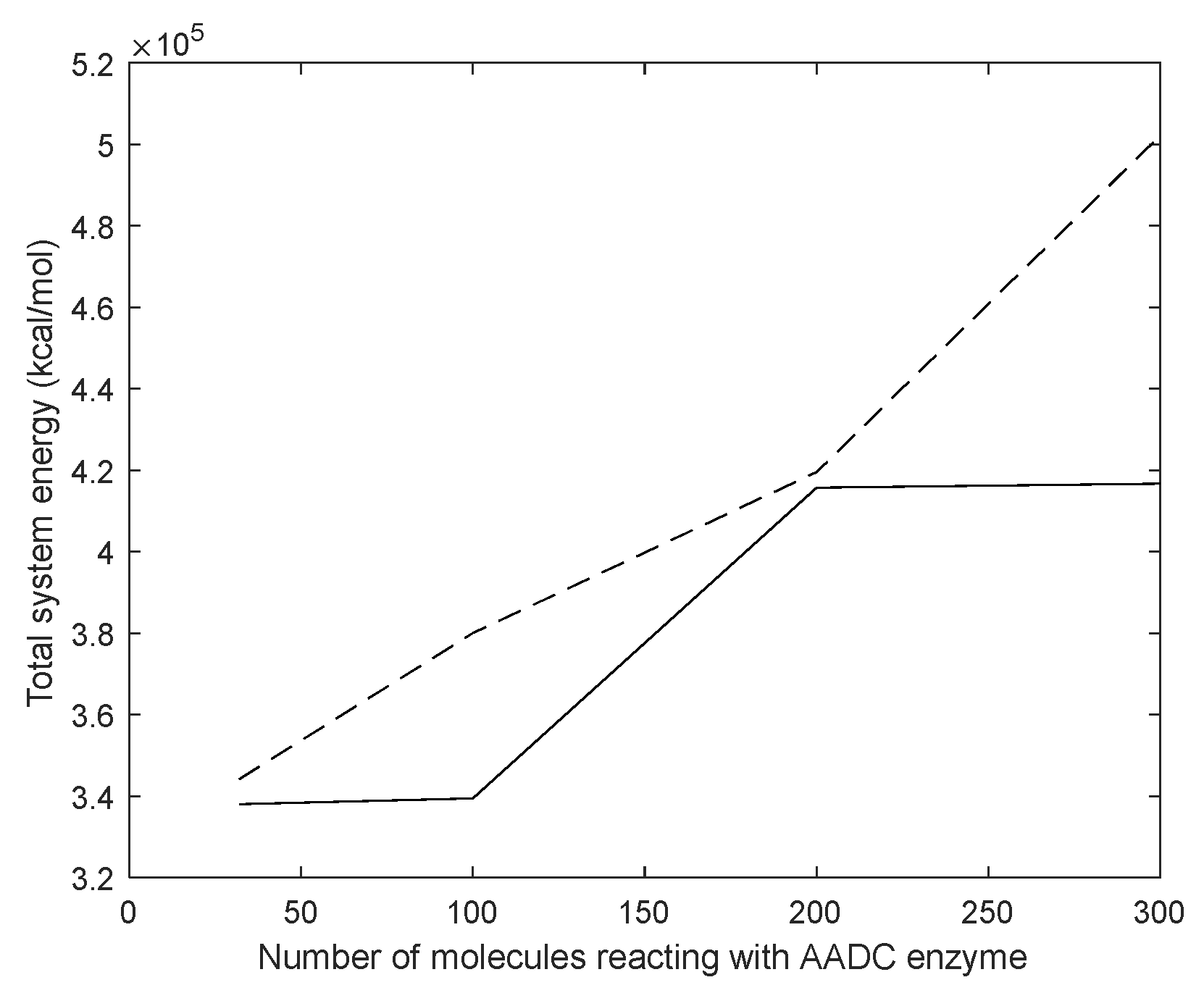
3.4. Molecular Dynamics Simulation of Dopamine Molecules with AADC Enzyme
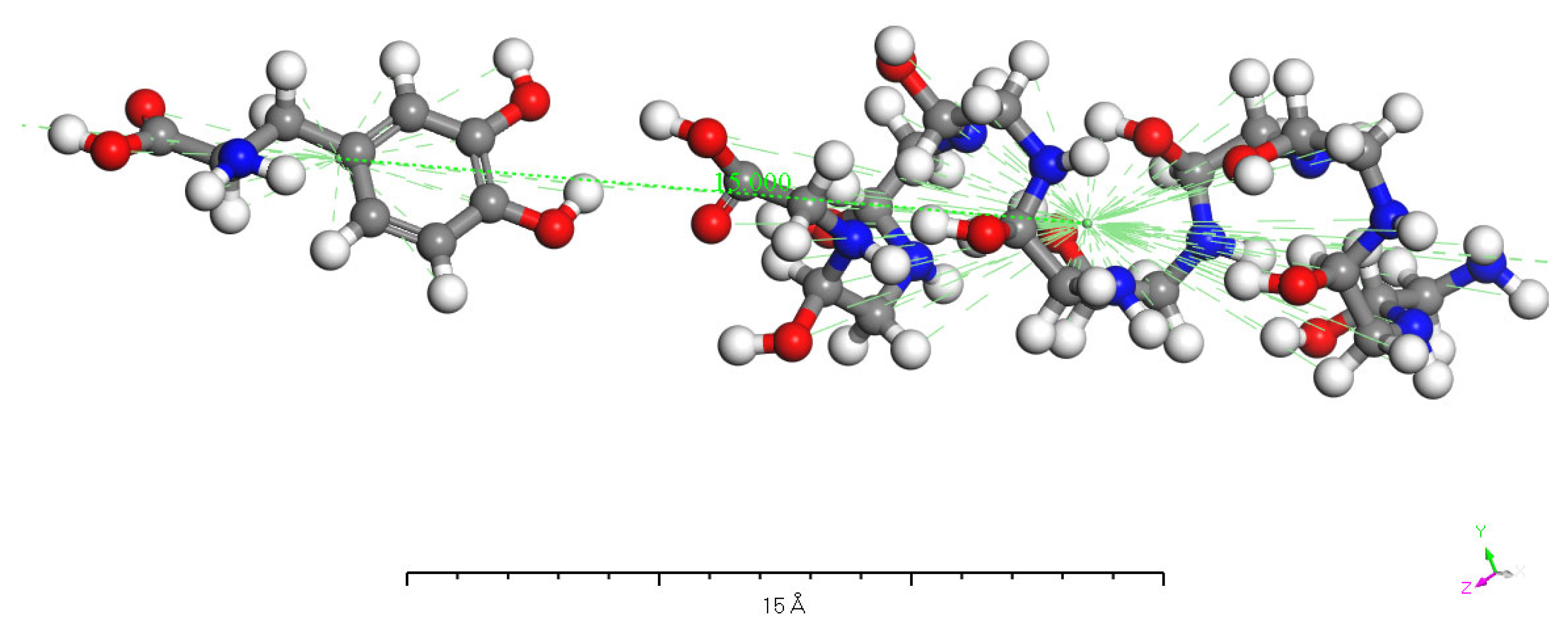
3.5. Charge Transfer in the Interaction of L-DOPA and D-DOPA with Peptide Chains
3.6. Reaction Trends and CISS Effect
4. Discussion
4.1. Dynamic Simulation Analysis
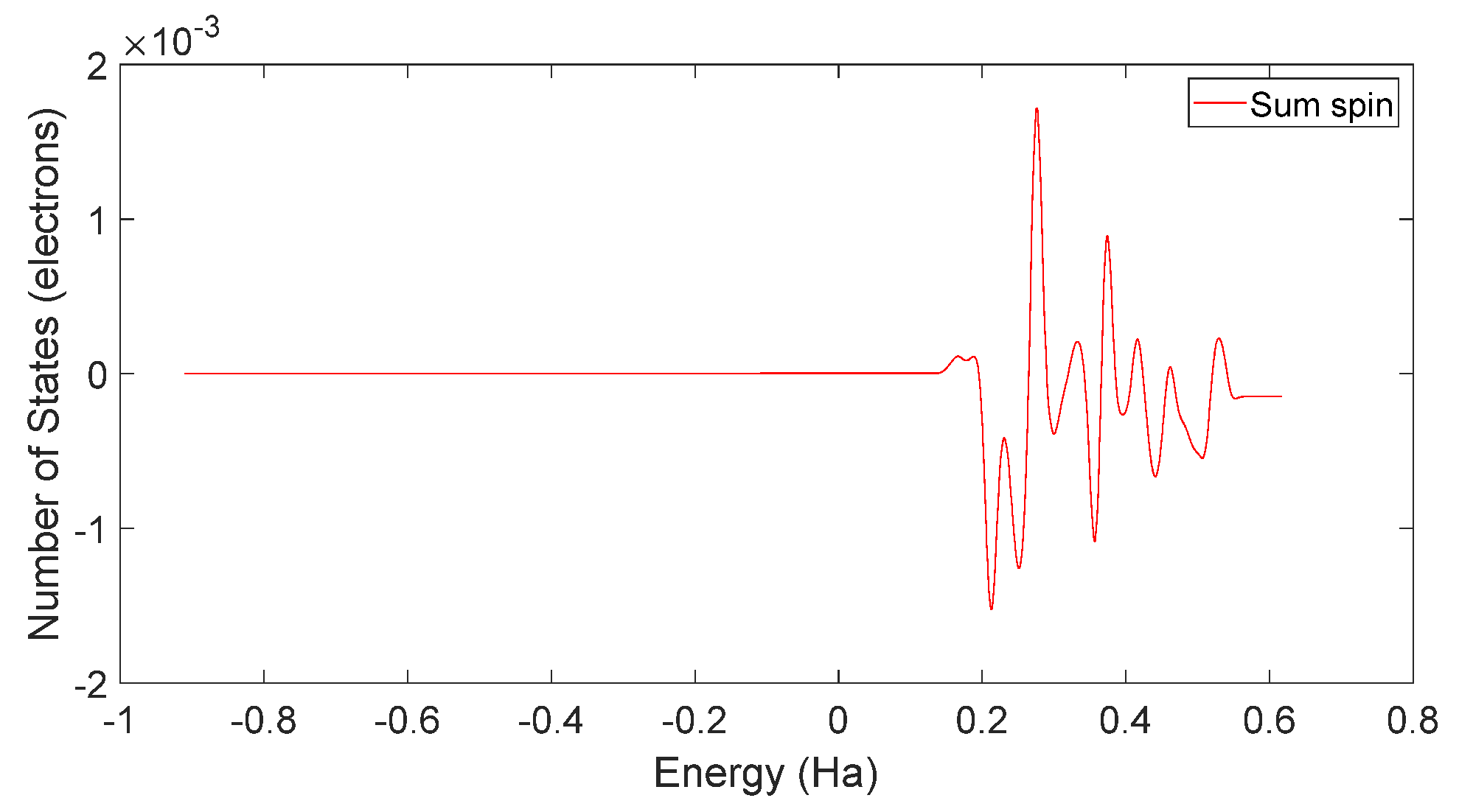
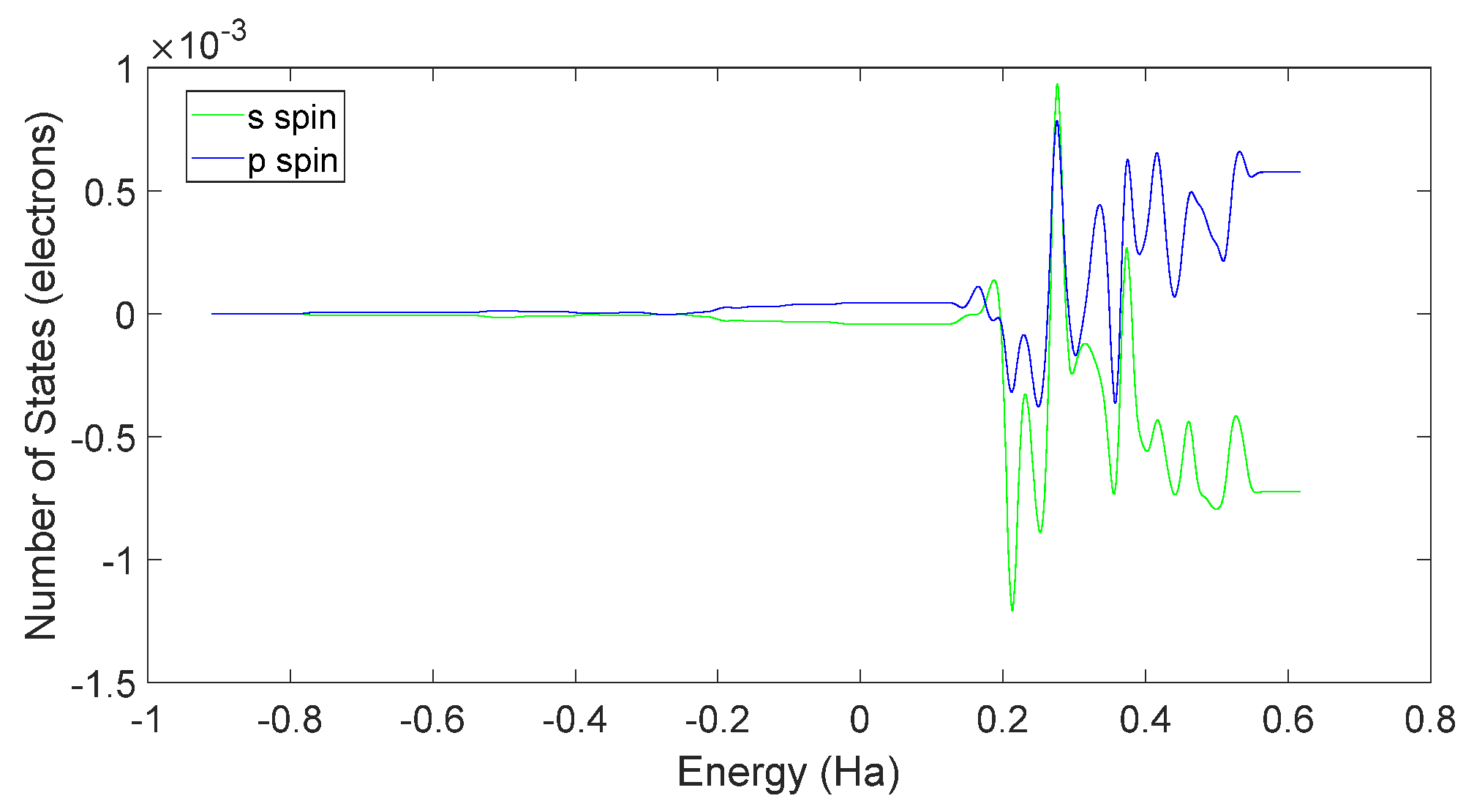
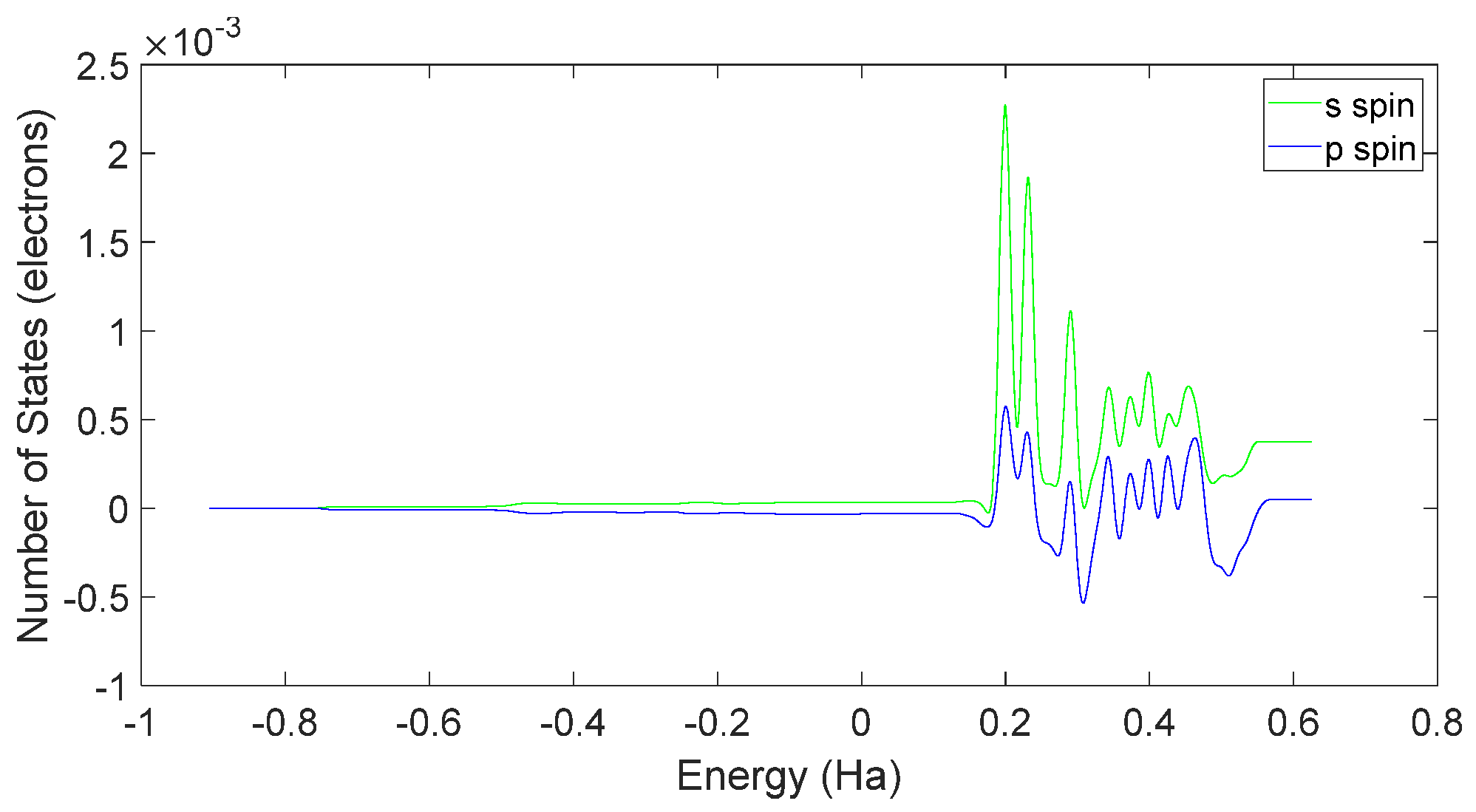
4.2. Spin Theoretical Analysis and Verification of L-DOPA and D-DOPA
5. Method
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Corey, E.J. The logic of chemical synthesis: Multistep synthesis of complex carbogenic molecules (Nobel lecture). Angew. Chem. Int. Ed. 1991, 30, 455–465. [Google Scholar] [CrossRef]
- Noyori, R.; Kitamura, M. The asymmetric hydrogenation of β-keto carboxylic esters catalyzed by chiral ruthenium complexes. J. Am. Chem. Soc. 1991, 113, 9885–9886. [Google Scholar]
- Naaman, R.; Waldeck, D.H. Spintronics and Chirality: Spin Selectivity in Electron Transport through Chiral Molecules. Annu. Rev. Phys. Chem. 2015, 66, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Michaeli, K.; Kantor-Uriel, N.; Naaman, R.; Waldeck, D.H. The electron’s spin and molecular chirality—How are they related and how do they affect life processes? Chem. Soc. Rev. 2016, 45, 6478–6487. [Google Scholar] [CrossRef]
- Mtangi, W.; Kiran, V.; Fontanesi, C.; Naaman, R. Role of the Electron Spin Polarization in Water Splitting. J. Phys. Chem. Lett. 2015, 6, 4916–4922. [Google Scholar] [CrossRef]
- Simon, Y.C.; Srebro, M.; Jortner, J.; Naaman, R. Chiral-Induced Spin Selectivity: A Magnetic Property of Chiral Molecules. Nature 2013, 493, 216–220. [Google Scholar]
- Srebro, M.; Simon, Y.C.; Naaman, R. Chiral-Induced Spin Selectivity: On the Role of Symmetry. J. Am. Chem. Soc. 2014, 136, 17682–17686. [Google Scholar]
- Capua, L.; Capua, M.; Naaman, R. Chiral-Induced Spin Selectivity in Electron Transport through Multilayered Chiral Structures. J. Phys. Chem. Lett. 2018, 9, 4344–4349. [Google Scholar]
- Naaman, R.; Paltiel, Y.; Waldeck, D.H. Chiral induced spin selectivity and its implications for biological functions. Annu. Rev. Biophys. 2022, 51, 99–114. [Google Scholar] [CrossRef]
- Kettner, M.; Artner, C.; Winkler, T.; Hohenester, U. Chiral Induced Spin Selectivity for the Identification and Classification of Biomolecules. J. Phys. Chem. B 2019, 123, 4404–4411. [Google Scholar]
- Wolf, Y.; Liu, Y.; Xiao, J.; Park, N.; Yan, B. Unusual Spin Polarization in the ChiralityInduced Spin Selectivity. ACS Nano 2022, 16, 18601–18607. [Google Scholar] [CrossRef]
- Zöllner, M.S.; Varela, S.; Medina, E.; Mujica, V.; Herrmann, C. Insight into the Origin of Chiral-Induced Spin Selectivity from a Symmetry Analysis of Electronic Transmission. J. Chem. Theory Comput. 2020, 16, 2914–2929. [Google Scholar] [CrossRef]
- Qi, D.; Kenaan, A.; Cui, D.; Song, J. Novel insights into the selection to electron’s spin of chiral structure. Nano Energy 2018, 52, 142–152. [Google Scholar] [CrossRef]
- Dednam, W.; García-Blázquez, M.A.; Zotti, L.A.; Lombardi, E.B.; Sabater, C.; Pakdel, S.; Palacios, J.J. A Group-Theoretic Approach to the Origin of Chirality-Induced Spin-Selectivity in Nonmagnetic Molecular Junctions. ACS Nano 2023, 17, 6452–6465. [Google Scholar] [CrossRef] [PubMed]
- Dalum, S.; Hedegård, P. Theory of Chiral Induced Spin Selectivity. Nano Lett. 2019, 19, 5253–5259. [Google Scholar] [CrossRef]
- Goudsmit, S.; Uhlenbeck, G.E. The Quantum Theory of the Motion of Electrons. Phys. Rev. 1925, 27, 359–364. [Google Scholar]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Rahman, M.K.; Nagatsu, T. Demonstration of aromatic L-amino acid decarboxylase activity in human brain with L-DOPA and L-5-hydroxytryptophan as substrates by high-performance liquid chromatography with electrochemical detection. Neurochem. Int. 1982, 4, 1–6. [Google Scholar] [CrossRef]
- Garcia, A.M.; Martínez, G.; Ruiz-Carretero, A. The Importance of Spin State in Chiral Supramolecular Electronics. Front. Chem. 2021, 9, 722727. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Pecul, M.; Ruud, K. The Ab Initio Calculation of Optical Rotation and Electronic Circular Dichroism. Adv. Quantum Chem. 2005, 50, 186–210. [Google Scholar] [CrossRef]








































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.-Y.; Liu, S.; Huang, A.; Li, Y.-N.; Liu, X.-Y.; Zhang, P. A Theoretical Analysis of the Differential Chemical Reaction Results Caused by Chirality Induction. Molecules 2023, 28, 6286. https://doi.org/10.3390/molecules28176286
Zhang F-Y, Liu S, Huang A, Li Y-N, Liu X-Y, Zhang P. A Theoretical Analysis of the Differential Chemical Reaction Results Caused by Chirality Induction. Molecules. 2023; 28(17):6286. https://doi.org/10.3390/molecules28176286
Chicago/Turabian StyleZhang, Feng-Yu, Sicheng Liu, Anwei Huang, Yi-Ning Li, Xiao-Yan Liu, and Peng Zhang. 2023. "A Theoretical Analysis of the Differential Chemical Reaction Results Caused by Chirality Induction" Molecules 28, no. 17: 6286. https://doi.org/10.3390/molecules28176286
APA StyleZhang, F. -Y., Liu, S., Huang, A., Li, Y. -N., Liu, X. -Y., & Zhang, P. (2023). A Theoretical Analysis of the Differential Chemical Reaction Results Caused by Chirality Induction. Molecules, 28(17), 6286. https://doi.org/10.3390/molecules28176286