

Synthesis of Spirocyclopropane-Containing 4H-Pyrazolo[1,5-a]indoles via Alkylative Dearomatization and Intramolecular N-Imination of an Indole–O-(Methylsulfonyl)oxime

 ,
,
Abstract
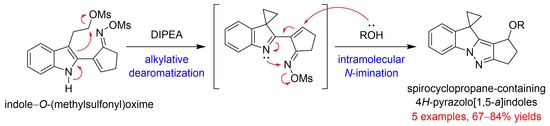
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure
3.2. Synthesis of Compounds 9–11
3.2.1. 1,2,3,5,6,11-Hexahydrocyclopenta[2,3]oxepino[4,5-b]indole (9)
3.2.2. (E)-2-[3-(2-Hydroxyethyl)-1H-indol-2-yl]cyclopent-2-en-1-one Oxime (10)
3.2.3. 2-[3-(2-Hydroxyethyl)-1H-indol-2-yl]cyclopent-2-en-1-one (13)
3.2.4. (E)-2-[2-(5-{[(Methylsulfonyl)oxy]imino}cyclopent-1-en-1-yl)-1H-indol-3-yl]ethyl Methanesulfonate (11)
3.3. Synthesis of 1-Alkoxy-2,3-dihydro-1H-spiro[cyclopenta[3,4]pyrazolo[1,5-a]indole-10,1′-cyclopropanes] 6a–e
3.3.1. Standard Procedure
3.3.2. 1-Ethoxy-2,3-dihydro-1H-spiro[cyclopenta[3,4]pyrazolo[1,5-a]indole-10,1′-cyclopropane] (6a)
3.3.3. 1-Propoxy-2,3-dihydro-1H-spiro[cyclopenta[3,4]pyrazolo[1,5-a]indole-10,1′-cyclopropane] (6b)
3.3.4. 1-(Tert-butoxy)-2,3-dihydro-1H-spiro[cyclopenta[3,4]pyrazolo[1,5-a]indole-10,1′-cyclopropane] (6c)
3.3.5. 1-(Benzyloxy)-2,3-dihydro-1H-spiro[cyclopenta[3,4]pyrazolo[1,5-a]indole-10,1′-cyclopropane] (6d)
3.3.6. (E)-1-(But-2-en-1-yloxy)-2,3-dihydro-1H-spiro[cyclopenta[3,4]pyrazolo[1,5-a]indole-10,1′-cyclopropane] (6e)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Shen, J.-K.; Katayama, H. 1H-Pyrazolo[l,5-a]indoles: Isoelectronic analogues of azulene (pseudoazulene). Chem. Pharm. Bull. 1992, 40, 2879–2881. [Google Scholar] [CrossRef]
- Shen, J.-K.; Katayama, H. Preparation and reaction of 1H-pyrazolo[l,5-a]indoles as isoelectronic analogues of azulene (pseudoazulene). J. Chem. Soc. Perkin Trans. 1 1994, 1871–1877. [Google Scholar] [CrossRef]
- Shen, J.-K.; Katayama, H. Preparation and reactions of 3H-pyrazolo[l,5-a]indole derivatives. Chem. Pharm. Bull. 1994, 42, 214–221. [Google Scholar] [CrossRef]
- Katayama, H.; Tagawa, N.; Kawada, Y.; Shiobara, K.; Kaneko, K.; Honda, Y.; Kondo, N.; Ikeda, Y. Heterocyclic analogues of quinone methide: Preparation and cytotoxicity of 3-oxo-3H-pyrazolo[1,5-a]indole derivatives. Chem. Pharm. Bull. 1997, 45, 143–147. [Google Scholar] [CrossRef]
- Kaszynski, P.; Dougherty, D.A. Synthesis and properties of diethyl 5,10-dihetera-5,10-dihydroindeno[2,1-a]indene-2,7-dicarboxylates. J. Org. Chem. 1993, 58, 5209–5220. [Google Scholar] [CrossRef]
- Katayama, H.; Kawada, Y.; Kaneko, K.; Oshiyama, T.; Takatsu, N. Synthetic inhibitors of DNA topoisomerase I and II. Chem. Pharm. Bull. 1999, 47, 48–53. [Google Scholar] [CrossRef]
- Katayama, H.; Kiryu, Y.; Kaneko, K.; Ohshima, R. Anti-cancer activities of pyrazolo[1,5-a]indole derivatives. Chem. Pharm. Bull. 2000, 48, 1628–1633. [Google Scholar] [CrossRef]
- Ji, Y.Y.; Zhu, Y.M.; Wang, J.W. GS-2, a pyrazolo[1,5-a]indole derivative with inhibitory activity of topoisomerases, exerts its potent cytotoxic activity by ROS generation. Environ. Toxicol. Pharmacol. 2013, 36, 1186–1196. [Google Scholar] [CrossRef]
- Guo, C.; Li, B.; Liu, H.; Zhang, X.; Zhang, X.; Fan, X. Synthesis of fused or spiro polyheterocyclic compounds via the dehydrogenative annulation reactions of 2-arylindazoles with maleimides. Org. Lett. 2019, 21, 7189–7193. [Google Scholar] [CrossRef]
- Vogel, A.; Dechert, S.; John, M.; Brückner, C.; Meyer, F. Siamese-twin porphyrin origami: Oxidative fusing and folding. Chem. Eur. J. 2016, 22, 2307–2316. [Google Scholar] [CrossRef]
- Katayama, H.; Takatsu, N.; Kitano, H.; Shimaya, Y. Intramolecular cycloaddition of 2-allylphenylhydrazones. Chem. Pharm. Bull. 1990, 38, 1129–1135. [Google Scholar] [CrossRef]
- Katayama, H.; Sakurada, M.; Herath, W.H.H.; Takatsu, N.; Shen, J.-K. Preparation of 4H-pyrazolo[1,5-a]indole. Chem. Pharm. Bull. 1992, 40, 2267–2269. [Google Scholar] [CrossRef]
- Shen, J.-K.; Katayama, H. Preparation of pyrazole and pyrazoline derivatives by intramolecular reaction of hydrazones. Chem. Lett. 1992, 21, 451–452. [Google Scholar] [CrossRef]
- Shen, J.-K.; Katayama, H.; Takatsu, N.; Shiro, I. Intramolecular reaction of the hydrazonyl group with formyl and oxo groups: Preparation of pyrazolo[1,5-a]indoles and related pyrazolo compounds. J. Chem. Soc. Perkin Trans. 1 1993, 2087–2097. [Google Scholar] [CrossRef]
- Katayama, H.; Takatsu, N.; Sakurada, M.; Kawada, Y. Preparation of 2-amino-4H-pyrazolo[1,5-a]indole derivatives by Boulton-Katritzky rearrangement. Heterocycles 1993, 35, 453–459. [Google Scholar] [CrossRef]
- Zhu, Y.-m.; Kiryu, Y.; Katayama, H. Intramolecular aromatic amination by a hydrazino group for the synthesis of indolo[1,2-b]indazole derivatives. Tetrahedron Lett. 2002, 43, 3577–3580. [Google Scholar] [CrossRef]
- Chi, J.; Hang, C.; Zhu, Y.; Katayama, H. Synthesis of indolo[1,2-b]indazole derivatives via copper(I)-catalyzed intramolecular amination reaction. Synth. Commun. 2010, 40, 1123–1133. [Google Scholar] [CrossRef]
- Zhu, Y.-M.; Qin, L.-N.; Liu, R.; Ji, S.-J.; Katayama, H. Synthesis of pyrazolo[1,5-a]indoles via copper(I)-catalyzed intramolecular amination. Tetrahedron Lett. 2007, 48, 6262–6266. [Google Scholar] [CrossRef]
- Hang, C.; Li, Q.; Zhu, Y.; Katayama, H. Copper(I)-catalyzed tandem cyclization/condensation reaction to novel 4,5-dihydropyrazolo[1,5-a]quinolines and pyrazolo[1,5-a]indoles. Synth. Commun. 2011, 41, 3318–3324. [Google Scholar] [CrossRef]
- Taylor, A.P.; Robinson, R.P.; Fobian, Y.M.; Blakemore, D.C.; Jones, L.H.; Fadeyi, O. Modern advances in heterocyclic chemistry in drug discovery. Org. Biomol. Chem. 2016, 14, 6611–6637. [Google Scholar] [CrossRef]
- Hsueh, W.-Y.; Lee, Y.-S.E.; Huang, M.-S.; Lai, C.-H.; Gao, Y.-S.; Lin, J.-C.; Chen, Y.-F.; Chang, C.-L.; Chou, S.-Y.; Chen, S.-F.; et al. Copper(I)-catalyzed nitrile-addition/N-arylation ring-closure cascade: Synthesis of 5,11-dihydro-6H-indolo[3,2-c]quinolin-6-ones as potent topoisomerase-I inhibitors. J. Med. Chem. 2021, 64, 1435–1453. [Google Scholar] [CrossRef] [PubMed]
- Talele, T.T. The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug molecules. J. Med. Chem. 2016, 59, 8712–8756. [Google Scholar] [CrossRef] [PubMed]
- Johansen, J.E.; Christie, B.D.; Rapoport, H. Iminium salts from α-amino acid decarbonylation. Application to the synthesis of octahydroindolo[2,3-a]quinolizines. J. Org. Chem. 1981, 46, 4914–4920. [Google Scholar] [CrossRef]
- Arumugam, S.; Verkade, J.G. P(CH3NCH2CH2)3N: A nonionic superbase for efficient dehydrohalogenation. J. Org. Chem. 1997, 62, 4827–4828. [Google Scholar] [CrossRef]
- Counceller, C.M.; Eichman, C.C.; Wray, B.C.; Stambuli, J.P. A practical, metal-free synthesis of 1H-indazoles. Org. Lett. 2008, 10, 1021–1023. [Google Scholar] [CrossRef] [PubMed]
- Wray, B.C.; Stambuli, J.P. Synthesis of N-arylindazoles and benzimidazoles from a common intermediate. Org. Lett. 2010, 12, 4576–4579. [Google Scholar] [CrossRef]
- Yan, X.; Yang, X.; Xi, C. Recent progress in copper-catalyzed electrophilic amination. Catal. Sci. Technol. 2014, 4, 4169–4177. [Google Scholar] [CrossRef]
- Sączewski, J.; Gdaniec, M. Synthesis of heterocycles by intramolecular nucleophilic substitution at an electron-deficient sp2 nitrogen atom. Eur. J. Org. Chem. 2010, 2387–2394. [Google Scholar] [CrossRef]
- Hassner, A.; Michelson, M.J. The formation of the N–N bond in pyrazolines. J. Org. Chem. 1962, 27, 298–301. [Google Scholar] [CrossRef]
- Stankevicius, A.P.; Janushene, L.N.; Terentiev, P.B.; Vitkevicius, K.T. Cleavage of 9,10-phenanthrenequinone monooxime O-arenesulfonates in the presence of amines. Russ. J. Org. Chem. 2006, 42, 1725–1726. [Google Scholar] [CrossRef]
- Stankjavicius, A.P.; Yanusiene, L.N.; Zablockaite, D.P.; Pechura, R.B. Synthesis of aliphatic amides of 2-(2(-cyanophenyl)benzoic acid. Pharm. Chem. J. 2007, 41, 646–647. [Google Scholar] [CrossRef]
- Tang, X.; Gao, H.; Yang, J.; Wu, W.; Jiang, H. Efficient access to 1H-indazoles via copper-catalyzed cross-coupling/cyclization of 2-bromoaryl oxime acetates and amines. Org. Chem. Front. 2014, 1, 1295–1298. [Google Scholar] [CrossRef]
- Meshram, H.M.; Reddy, P.N.; Sadashiv, K.; Yadav, J.S. Amberlyst-15®-promoted efficient 2-halogenation of 1,3-keto-esters and cyclic ketones using N-halosuccinimides. Tetrahedron Lett. 2005, 46, 623–626. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | X | Acid (mol %) | Solvent | T (°C) b | Time (h) | Yield (%) c |
|---|---|---|---|---|---|---|
| 1 | Cl | PPTS (20) d | toluene | reflux | 2.0 | Trace e |
| 2 | Br | PPTS (20) d | toluene | reflux | 2.0 | Trace e |
| 3 | Cl | TsOH (20) | toluene | reflux | 2.0 | 0 f |
| 4 | Br | TsOH (20) | toluene | reflux | 2.0 | 0 f |
| 5 | Cl | BF3·OEt2 (150) | CH2Cl2 | 0 | 3.0 | 20 |
| 6 | Br | BF3·OEt2 (150) | CH2Cl2 | 0 | 3.0 | 26 |
| 7 | Cl | HBF4·OEt2 (150) | CH2Cl2 | 0 | 10 | 56 |
| 8 | Br | HBF4·OEt2 (150) | CH2Cl2 | 0 | 10 | 63 |
| 9 | Cl | HBF4·OEt2 (75) | CH2Cl2 | 0 | 10 | 25 |
| 10 | Br | HBF4·OEt2 (75) | CH2Cl2 | 0 | 10 | 30 |

| Entry | Equivalent of NH2OH·HCl | T (°C) b | Time (h) | Yield (%) c | |
|---|---|---|---|---|---|
| 10 | 13 | ||||
| 1 | 2.2 | r.t. | 15 | 0 d | 0 d |
| 2 | 2.2 | 55 | 15 | 63 | 27 |
| 3 | 2.2 | reflux | 15 | 65 | 25 |
| 4 | 3.2 | reflux | 30 | 62 | 20 |
| 5 | 3.2 | reflux | 45 | 63 | 18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.-J.; Liao, H.-C.; Hsu, C.-E.; Liu, Y.-R.; Chang, Y.-F.; Chou, S.-Y. Synthesis of Spirocyclopropane-Containing 4H-Pyrazolo[1,5-a]indoles via Alkylative Dearomatization and Intramolecular N-Imination of an Indole–O-(Methylsulfonyl)oxime. Molecules 2023, 28, 6374. https://doi.org/10.3390/molecules28176374
Huang J-J, Liao H-C, Hsu C-E, Liu Y-R, Chang Y-F, Chou S-Y. Synthesis of Spirocyclopropane-Containing 4H-Pyrazolo[1,5-a]indoles via Alkylative Dearomatization and Intramolecular N-Imination of an Indole–O-(Methylsulfonyl)oxime. Molecules. 2023; 28(17):6374. https://doi.org/10.3390/molecules28176374
Chicago/Turabian StyleHuang, Jiann-Jyh, Hung-Chun Liao, Cheng-En Hsu, Yan-Ru Liu, Yi-Fu Chang, and Shan-Yen Chou. 2023. "Synthesis of Spirocyclopropane-Containing 4H-Pyrazolo[1,5-a]indoles via Alkylative Dearomatization and Intramolecular N-Imination of an Indole–O-(Methylsulfonyl)oxime" Molecules 28, no. 17: 6374. https://doi.org/10.3390/molecules28176374
APA StyleHuang, J. -J., Liao, H. -C., Hsu, C. -E., Liu, Y. -R., Chang, Y. -F., & Chou, S. -Y. (2023). Synthesis of Spirocyclopropane-Containing 4H-Pyrazolo[1,5-a]indoles via Alkylative Dearomatization and Intramolecular N-Imination of an Indole–O-(Methylsulfonyl)oxime. Molecules, 28(17), 6374. https://doi.org/10.3390/molecules28176374