

Development, Validation, and Two-Year Application of Rapid and Simple LC-MS/MS-Based Method for the Determination of K2MK-7 in Blood Samples


Abstract
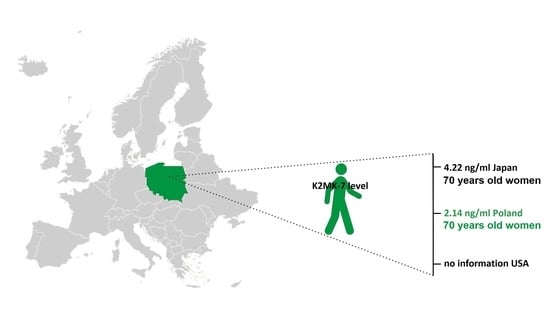
:
1. Introduction
2. Results and Discussion
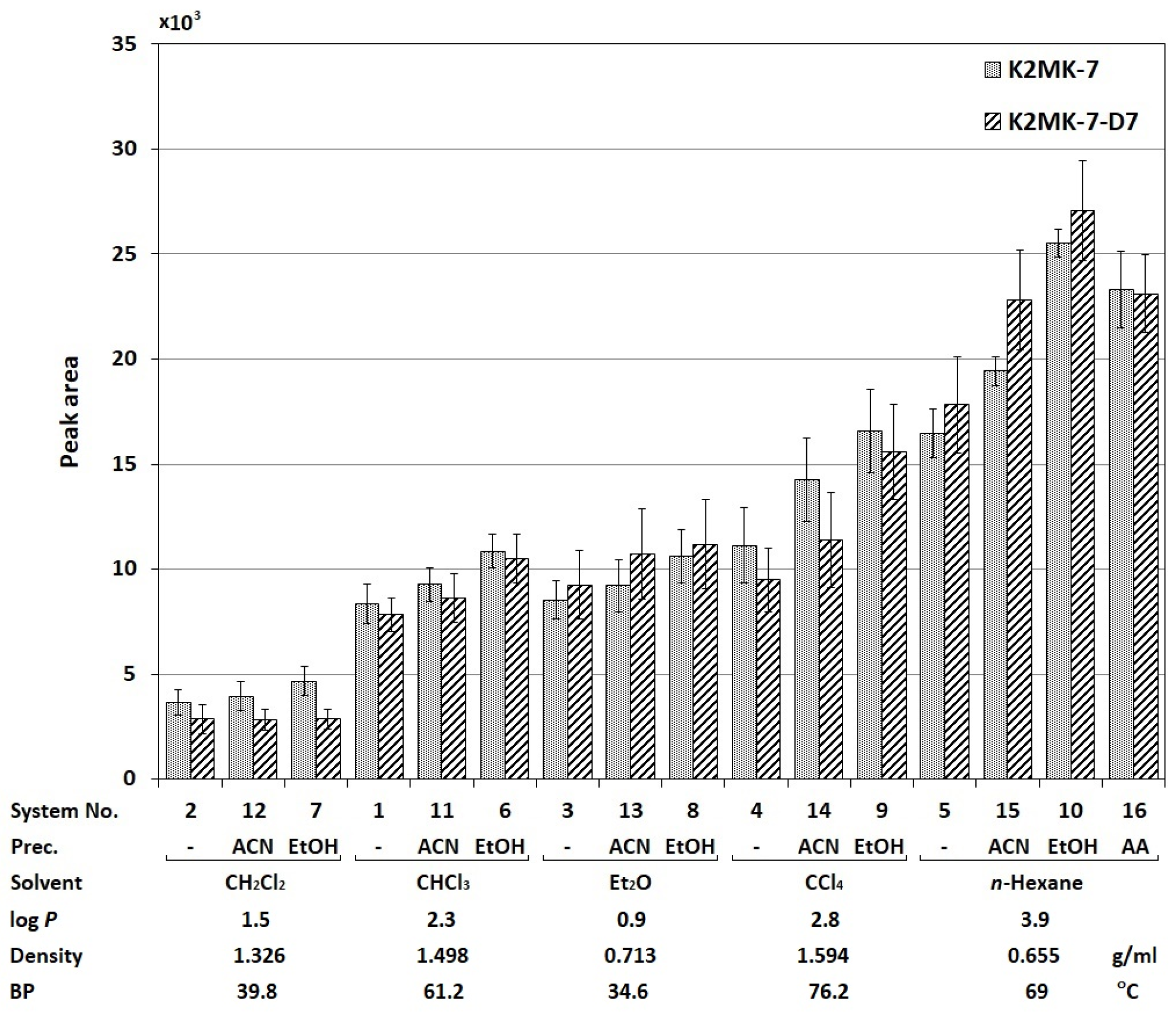
2.1. Optimization of the Sample Preparation Procedure
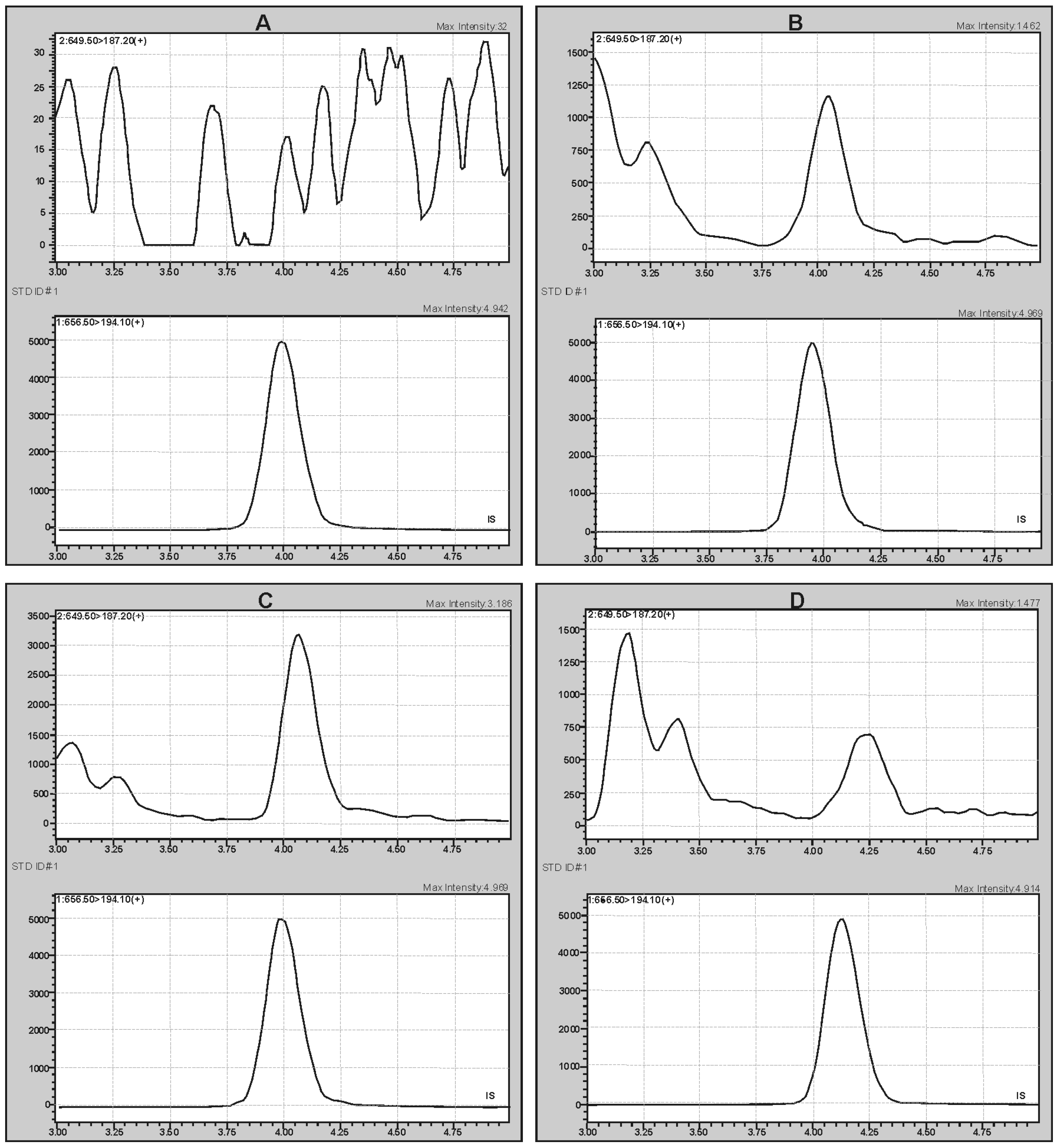
2.2. Method Validation
2.3. Practical Application
2.4. Comparison with the Published Data
3. Materials and Methods
3.1. Sample Preparation
3.2. Collection and Storage of Serum Samples
3.3. Sample Preparation
3.4. LC-APCI-MS Analysis and Its Optimization
3.5. Method Validation
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sato, T.; Yamada, Y.; Ohtani, Y.; Mitsui, N.; Murasawa, H.; Araki, S. Production of menaquinone (vitamin K2)-7 by Bacillus subtilis. J. Biosci. Bioeng. 2001, 91, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Simes, D.C.; Viegas, C.S.B.; Araújo, N.; Marreiros, C. Vitamin K as a powerful micronutrient in aging and age-related diseases: Pros and cons from clinical studies. Int. J. Mol. Sci. 2019, 20, 4150. [Google Scholar] [CrossRef] [PubMed]
- Walther, B.; Karl, J.P.; Booth, S.L.; Boyaval, P. Menaquinones, bacteria, and the food supply: The relevance of dairy and fermented food products to vitamin K requirements. Adv. Nutr. 2013, 4, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.J.; Booth, S.L.; van den Heuvel, E.G.H.M.; Stoecklin, E.; Baka, A.; Vermeer, C. The role of menaquinones (vitamin K 2 ) in human health. Br. J. Nutr. 2013, 110, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Paprotny, Ł.; Wianowska, D.; Izdebska, M.; Celejewska, A.; Szewczak, D.; Solski, J. Analysis of serum homocysteine in the laboratory practice—Comparison of the direct chemiluminescence immonoassay and high performance liquid chromatography coupled with fluorescent detection. Biochem. Med. 2020, 30, 030703. [Google Scholar] [CrossRef]
- Fusaro, M.; Gallieni, M.; Porta, C.; Nickolas, T.L.; Khairallah, P. Vitamin K effects in human health: New insights beyond bone and cardiovascular health. J. Nephrol. 2020, 33, 239–249. [Google Scholar] [CrossRef]
- Nimptsch, K.; Rohrmann, S.; Linseisen, J. Dietary intake of vitamin K and risk of prostate cancer in the Heidelberg cohort of the European Prospective Investigation into Cancer and Nutrition (EPIC-Heidelberg). Am. J. Clin. Nutr. 2008, 87, 985–992. [Google Scholar] [CrossRef]
- Dofferhoff, A.S.M.; Piscaer, I.; Schurgers, L.J.; Visser, M.P.J.; van den Ouweland, J.M.W.; de Jong, P.A.; Gosens, R.; Hackeng, T.M.; van Daal, H.; Lux, P.; et al. Reduced Vitamin K Status as a Potentially Modifiable Risk Factor of Severe Coronavirus Disease. Clin. Infect. Dis. 2020, 73, e4039–e4046. [Google Scholar] [CrossRef]
- Wianowska, D.; Bryshten, I. New insights into vitamin K—From its natural sources through biological properties and chemical methods of quantitative determination. Crit. Rev. Anal. Chem. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Knapen, M.H.J.; Vermeer, C.; Braam, L.A.J.L.M.; Theuwissen, E. Pharmacokinetics of menaquinone-7 (vitamin K2) in healthy volunteers. Clin. Trials 2014, 4, 1000160. [Google Scholar] [CrossRef]
- Møller, M.; Fange Gjelstad, I.M.; Baksaas, I.; Grande, T.; Reidun Aukrust, I.; Drevon, C.A. Bioavailability and chemical/functional aspects of synthetic MK-7 vs fermentation-derived MK-7 in randomised controlled trials C.A. Int. J. Vitam. Nutr. Res. 2016, 87, 297–311. [Google Scholar] [CrossRef]
- Klapkova, E.; Cepova, J.; Dunovska, K.; Prusa, R. Determination of vitamins K 1, MK-4, and MK-7 in human serum of postmenopausal women by HPLC with fluorescence detection. J. Clin. Lab. Anal. 2018, 32, e22381. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.J.; Bots, M.L.; Atsma, F.; Bartelink, M.-L.E.L.; Prokop, M.; Geleijnse, J.M.; Witteman, J.C.M.; Grobbee, D.E.; van der Schouw, Y.T. High dietary menaquinone intake is associated with reduced coronary calcification. Atherosclerosis 2009, 203, 489–493. [Google Scholar] [CrossRef]
- Simes, D.C.; Viegas, C.S.B.; Araújo, N.; Marreiros, C. Vitamin K as a diet supplement with impact in human health: Current evidence in age-related diseases. Nutrients 2020, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Gentili, A.; Cafolla, A.; Gasperi, T.; Bellante, S.; Caretti, F.; Curini, R.; Fernández, V.P. Rapid, high performance method for the determination of vitamin K1, menaquinone-4 and vitamin K1 2,3-epoxide in human serum and plasma using liquid chromatography-hybrid quadrupole linear ion trap mass spectrometry. J. Chromatogr. A 2014, 1338, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Marinova, M.; Lütjohann, D.; Westhofen, P.; Watzka, M.; Breuer, O.; Oldenburg, J. A validated HPLC method for the determination of vitamin K in human serum-first application in a pharmacological study. Open Clin. Chem. J. 2011, 4, 17–27. [Google Scholar] [CrossRef]
- Kamao, M.; Suhara, Y.; Tsugawa, N.; Okano, T. Determination of plasma Vitamin K by high-performance liquid chromatography with fluorescence detection using Vitamin K analogs as internal standards. J. Chromatogr. B 2005, 816, 41–48. [Google Scholar] [CrossRef]
- Karl, J.P.; Fu, X.; Dolnikowski, G.G.; Saltzman, E.; Booth, S.L. Quantification of phylloquinone and menaquinones in feces, serum, and food by high-performance liquid chromatography–mass spectrometry. J. Chromatogr. B 2014, 963, 128–133. [Google Scholar] [CrossRef]
- Riphagen, I.J.; van der Molen, J.C.; van Faassen, M.; Navis, G.; de Borst, M.H.; Muskiet, F.A.J.; de Jong, W.H.A.; Bakker, S.J.L.; Kema, I.P. Measurement of plasma vitamin K1 (phylloquinone) and K2 (menaquinones-4 and -7) using HPLC-tandem mass spectrometry. Clin. Chem. Lab. Med. 2016, 54, 1201–1210. [Google Scholar] [CrossRef]
- Huang, B.; Wang, Z.; Yao, J.; Ke, X.; Xu, J.; Pan, X.-D.; Xu, X.; Lu, M.; Ren, Y. Quantitative analysis of vitamin K 1 in fruits and vegetables by isotope dilution LC-MS/MS. Anal. Methods 2016, 8, 5707–5711. [Google Scholar] [CrossRef]
- Paprotny, Ł.; Celejewska, A.; Frajberg, M.; Wianowska, D. Development and validation of GC–MS/MS method useful in diagnosing intestinal dysbiosis. J. Chromatogr. B 2019, 121822, 1130–1131. [Google Scholar] [CrossRef]
- Gouda, A.S.; Abdel-Megied, A.M.; Rezk, M.R.; Marzouk, H.M. LC-MS/MS-based metabolite quantitation of the antiviral prodrug baloxavir marboxil, a new therapy for acute uncomplicated influenza, in human plasma: Application to a human pharmacokinetic study. J. Pharm. Biomed. Anal. 2023, 223, 115165. [Google Scholar] [CrossRef] [PubMed]
- Rezk, M.R.; Badr, K.A. Development, optimization and validation of a highly sensitive UPLC–ESI-MS/MS method for simultaneous quantification of amlodipine, benazeprile and benazeprilat in human plasma: Application to a bioequivalence study. J. Pharm. Biomed. Anal. 2014, 98, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rezk, M.R.; Basalious, E.B.; Badr, K.A. Novel determination of sofosbuvir and velpatasvir in human plasma by UPLC–MS/MS method: Application to a bioequivalence study. Biomed. Chromatogr. 2018, 32, e4347. [Google Scholar] [CrossRef] [PubMed]
- Viñas, P.; Bravo-Bravo, M.; López-García, I.; Hernández-Córdoba, M. Dispersive liquid–liquid microextraction for the determination of vitamins D and K in foods by liquid chromatography with diode-array and atmospheric pressure chemical ionization-mass spectrometry detection. Talanta 2013, 115, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Rishipal, S.; Alka, P.; Mojeer, H.; Bibhu Prasad, P. Development of a Rapid HPLC-UV Method for Analysis of Menaquinone-7 in Soy Nutraceutical. Pharm. Anal. Acta 2016, 7, 7–12. [Google Scholar] [CrossRef]
- Wianowska, D.; Gil, M.; Olszowy, M. Miniaturized methods of sample preparation. In Handbook on Miniaturization in Analytical Chemistry: Application of Nanotechnology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 99–125. [Google Scholar] [CrossRef]
- Agilent Technical Overview: Making Your LC Method Compatible with Mass Spectrometry, Edgar Naegale, Waldbronn, Germany. Available online: https://community.agilent.com/cfs-file/__key/docpreview-s/00-00-00-85-46/5990_2D002D00_7413EN.pdf (accessed on 12 August 2022).
- Colizza, K.; Mahoney, K.E.; Yevdokimov, A.V.; Smith, J.L.; Oxley, J.C. Acetonitrile ion suppression in atmospheric pressure ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 1796–1804. [Google Scholar] [CrossRef]
- Szterk, A.; Bus, K.; Zmysłowski, A.; Ofiara, K. Analysis of Menaquinone-7 Content and Impurities in Oil and Non-Oil Dietary Supplements. Molecules 2018, 23, 1056. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services, Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation; FDA: Silver Spring, MD, USA, 2001; pp. 4–10. Available online: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf (accessed on 12 August 2022).
- Vermeer, C.; Raes, J.; van ’t Hoofd, C.; Knapen, M.H.J.; Xanthoulea, S. Menaquinone content of cheese. Nutrients 2018, 10, 446. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC−MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- U.N. Department of Economic and Social Affairs. The Average Age in Global Comparison. Available online: www.worlddata.info/average-age.php (accessed on 12 August 2022).
- Hu, K.; Li, Y.; Ding, R.; Zhai, Y.; Chem, L.; Qian, W.; Yang, J. A simple, sensitive, and high-throughput LC-APCI-MS/MS method for simultaneous determination of vitamin K1, Vitamin K1 2,3-epoxide in human plasma and its application to a clinical pharmacodynamics study of warfarin. J. Pharm. Biomed. Anal. 2018, 159, 82–91. [Google Scholar] [CrossRef]
- Dunovska, K.; Klapkova, E.; Sopko, B.; Cepova, J.; Prusa, R. LC–MS/MS quantitative analysis of phylloquinone, menaquinone-4 and menaquinone-7 in the human serum of a healthy population. PeerJ 2019, 7, e7695. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effects | Fcal-Value | p-Value | Ftab-Value | |
|---|---|---|---|---|
| Effect of solvent type on K2MK-7 peak area | 48.45 | 1.64 × 10−6 | 3.48 | |
| Effect of EtOH adding to the extraction system on K2MK-7 peak area | 129.85 | 1.42 × 10−8 | 3.48 | |
| Effect of ACN adding to the extraction system on K2MK-7 peak area | 72.18 | 2.45 × 10−7 | 3.48 | |
| Effect of AA adding to the extraction system on K2MK-7 peak area | 3.93 | 0.12 | 7.71 | |
| Effect of CH2Cl2 volume | 1.82 | 0.24 | 5.14 | |
| Effect of CHCl3 volume | 6.48 | 0.03 | 5.14 | |
| Effect of Et2O volume | 2.57 | 0.16 | 5.14 | |
| Effect of CCl4 volume | 6.01 | 0.04 | 5.14 | |
| Effect of n-hexane volume | 83.97 | 4.10 × 10−5 | 5.14 | |
| Effect of age on K2MK7 concentration in female | 4.18 | 2.18 × 10−4 | 2.05 | |
| Effect of age on K2MK7 concentration in male | 3.85 | 5.45 × 10−4 | 2.05 | |
| Effect of sex on K2MK-7 concentration in individual age-groups: | <10 | 0.19 | 0.66 | 4.26 |
| 11–20 | 1.26 | 0.27 | 4.06 | |
| 21–30 | 0.24 | 0.63 | 3.96 | |
| 31–40 | 1.25 | 0.26 | 3.92 | |
| 41–50 | 3.97 | 4.8 × 10−2 | 3.94 | |
| 51–60 | 0.14 | 0.71 | 3.96 | |
| 61–70 | 2.71 | 0.10 | 4.04 | |
| >71 | 0.06 | 0.81 | 4.41 | |
| rho-value | p-value | |||
| Correlation between age and K2MK-7 concentration in female | 0.23 | 2.34 × 10−4 | ||
| Correlation between age and K2MK-7 concentration in male | 0.27 | 2.68 × 10−5 | ||
| Resultant correlation between age and K2MK7 concentration | 0.27 | 2.46 × 10−9 | ||
| Nominal Concentration (ng/mL) | Measured Concentration (mean ± SD), (ng/mL) | Imprecision (% CV) | Inaccuracy (% BIAS) |
|---|---|---|---|
| Intra-day (n = 5) | |||
| 0.10 | 0.116 ± 0.004 | 3.98 | 16.16 |
| 0.32 | 0.352 ± 0.022 | 6.40 | 10.10 |
| 0.64 | 0.728 ± 0.069 | 9.49 | −13.80 |
| 0.96 | 0.939 ± 0.030 | 3.29 | −2.12 |
| Inter-day (n = 5) | |||
| 0.10 | 0.108 ± 0.005 | 4.51 | 8.63 |
| 0.32 | 0.317 ± 0.030 | 9.48 | 0.98 |
| 0.64 | 0.649 ± 0.072 | 11.04 | −1.38 |
| 0.96 | 1.007 ± 0.073 | 7.22 | −4.88 |
| Autosampler at 15 °C (24 h) (n = 3) | |||
| 0.32 | 0.352 ± 0.01 | 2.8 | −10.0 |
| 0.64 | 0.621 ± 0.084 | 13.52 | 3.06 |
| 0.96 | 1.031 ± 0.052 | 5.04 | −12.81 |
| Refrigerator at 4 °C (24 h) (n = 3) | |||
| 0.32 | 0.360 ± 0.06 | 13.76 | 12.50 |
| 0.64 | 0.622 ± 0.06 | 8.59 | 2.82 |
| 0.96 | 1.014 ± 0.046 | 4.44 | −5.62 |
| Freezer at −18 °C (24 h) (n = 3) | |||
| 0.32 | 0.325 ± 0.042 | 12.92 | 1.56 |
| 0.64 | 0.619 ± 0.033 | 5.33 | 3.28 |
| 0.96 | 1.014 ± 0.046 | 5.80 | 6.66 |
| Exposed to light at room temp. (1 h) (n = 3) | |||
| 0.32 | 0.167 ± 0.004 | 2.39 | 47.81 |
| 0.64 | 0.250 ± 0.188 | 75.08 | 60.88 |
| 0.96 | 0.344 ± 0.240 | 69.79 | 64.18 |
| Exposed to light at room temp. (2 h) (n = 3) | |||
| 0.32 | 0.125 ± 0.060 | 47.83 | 60.80 |
| 0.64 | 0.299 ± 0.092 | 30.91 | 53.19 |
| 0.96 | 0.508 ± 0.12 | 23.60 | 47.03 |
| Freeze-thaw cycles (cycle number) (n = 3) | |||
| 0.64 (1) | 0.600 ± 0.014 | 2.33 | 6.20 |
| 0.64 (2) | 0.716 ± 0.041 | 5.74 | −11.94 |
| 0.64 (3) | 0.657 ± 0.058 | 8.83 | −2.65 |
| 0.64 (4) | 0.379 ± 0.052 | 13.71 | 40.66 |
| Nominal Concentration (ng/mL) | Mean Peak Area (% CV) | ME (%) | RE (%) | PE (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K2MK-7 | IS | |||||||||||
| Set 1 | Set 2 | Set 3 | Set 1 | Set 2 | Set 3 | K2MK-7 | IS | K2MK-7 | IS | K2MK-7 | IS | |
| 0.32 | 16,954 (5.21) | 19,281 (1.34) | 15,280 (9.54) | 35,768 (2.55) | 38,317 (2.25) | 32,313 (9.14) | 113.73 | 107.13 | 79.25 | 84.33 | 90.13 | 90.34 |
| 0.64 | 33,288 (10.87) | 36,584 (9.12) | 30,011 (9.20) | 32,235 (7.76) | 36,701 (1.28) | 31,833 (15.70) | 109.90 | 113.85 | 82.03 | 86.74 | 90.15 | 98.75 |
| 0.96 | 48,747 (4.08) | 49,094 (0.72) | 41,721 (2.58) | 33,729 (11.95) | 34,329 (2.13) | 30,039 (10.51) | 100.71 | 101.78 | 84.98 | 87.50 | 85.59 | 89.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paprotny, Ł.; Szewczak, D.; Bryshten, I.; Wianowska, D. Development, Validation, and Two-Year Application of Rapid and Simple LC-MS/MS-Based Method for the Determination of K2MK-7 in Blood Samples. Molecules 2023, 28, 6523. https://doi.org/10.3390/molecules28186523
Paprotny Ł, Szewczak D, Bryshten I, Wianowska D. Development, Validation, and Two-Year Application of Rapid and Simple LC-MS/MS-Based Method for the Determination of K2MK-7 in Blood Samples. Molecules. 2023; 28(18):6523. https://doi.org/10.3390/molecules28186523
Chicago/Turabian StylePaprotny, Łukasz, Dorota Szewczak, Iryna Bryshten, and Dorota Wianowska. 2023. "Development, Validation, and Two-Year Application of Rapid and Simple LC-MS/MS-Based Method for the Determination of K2MK-7 in Blood Samples" Molecules 28, no. 18: 6523. https://doi.org/10.3390/molecules28186523
APA StylePaprotny, Ł., Szewczak, D., Bryshten, I., & Wianowska, D. (2023). Development, Validation, and Two-Year Application of Rapid and Simple LC-MS/MS-Based Method for the Determination of K2MK-7 in Blood Samples. Molecules, 28(18), 6523. https://doi.org/10.3390/molecules28186523