
Ab Initio Approach to the Structure, Vibrational Properties, and Electron Binding Energies of H2S∙∙∙SO2



Abstract
:
1. Introduction
2. Results and Discussion
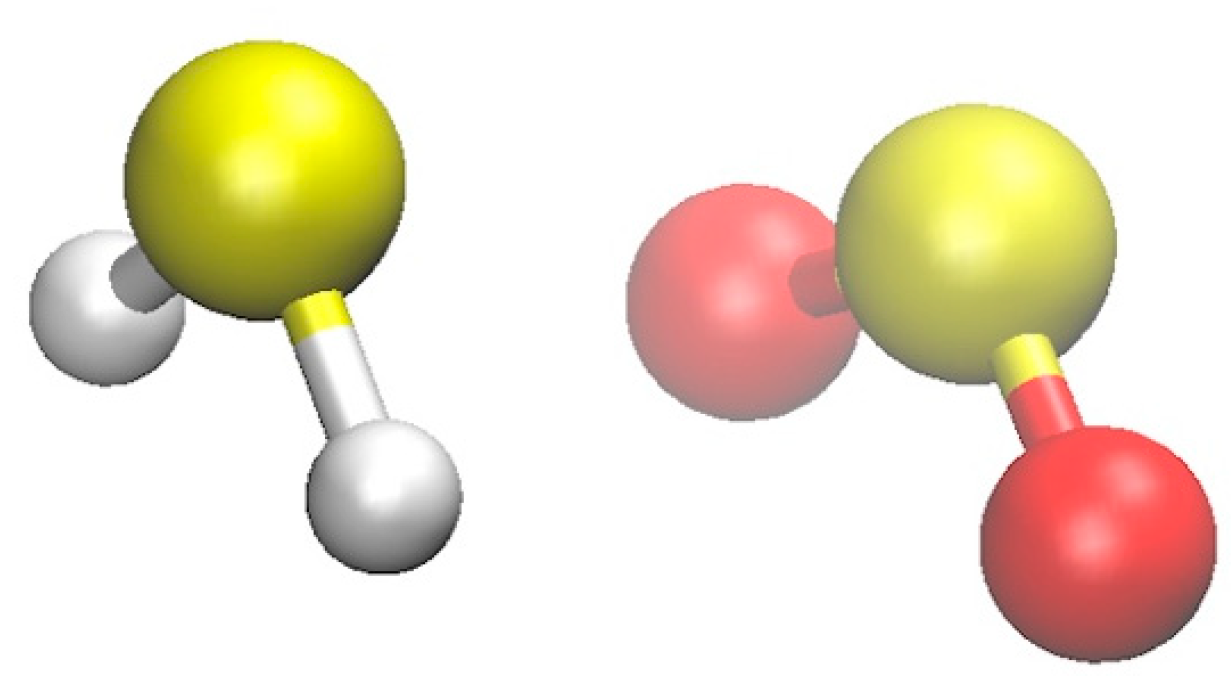
2.1. Structure, Vibrational Frequencies, and Rotational Constants
2.1.1. Structure
2.1.2. Vibrational Frequencies
2.2. Interaction Energies and Electronic PropertiesInteraction Energies
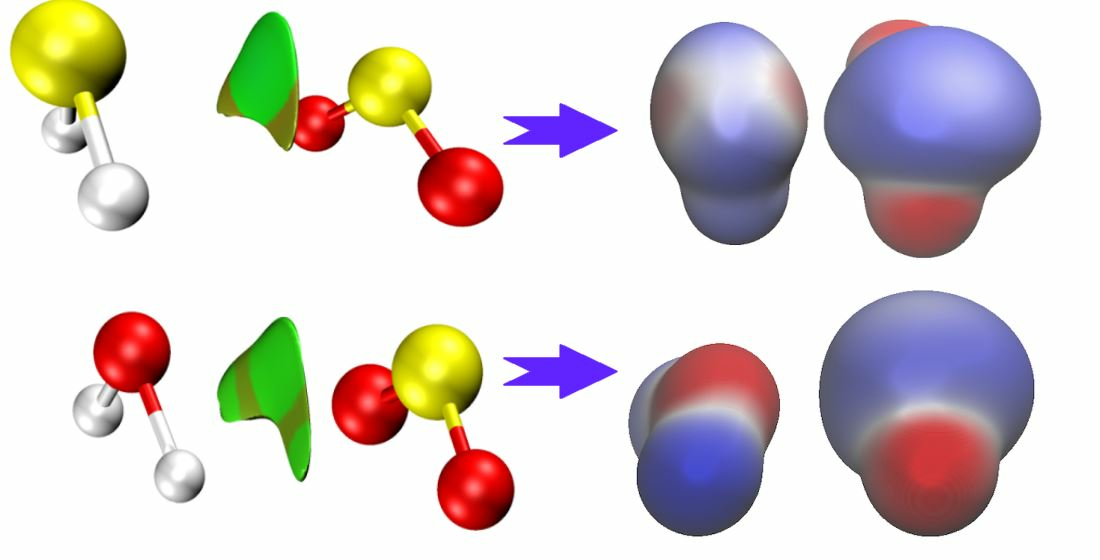
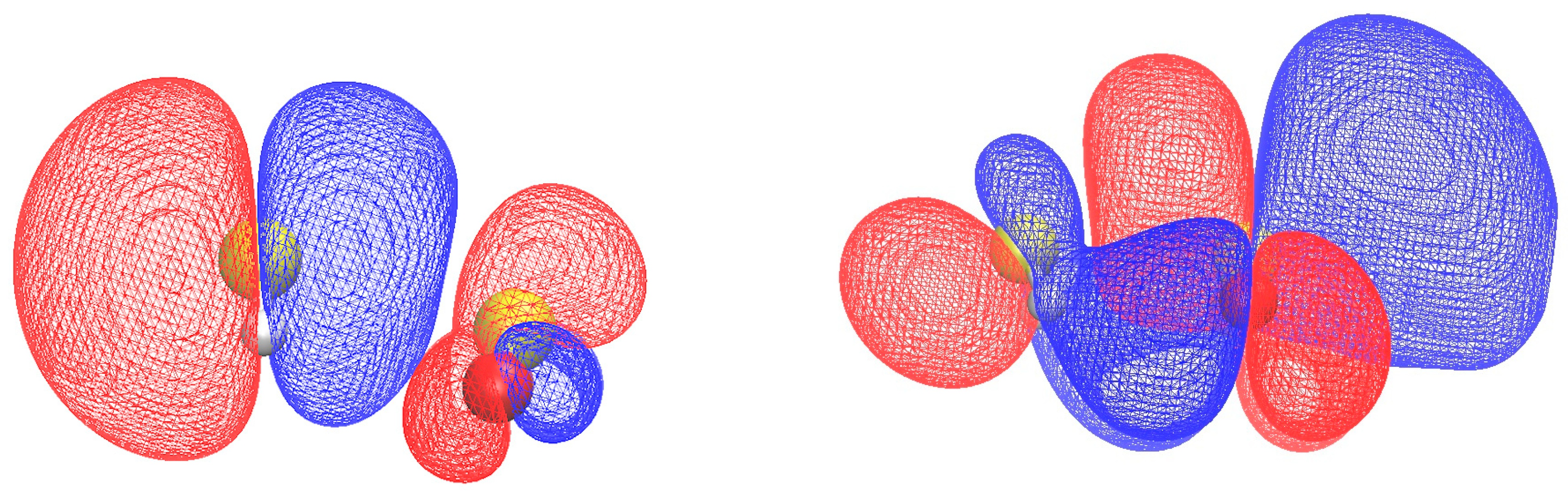
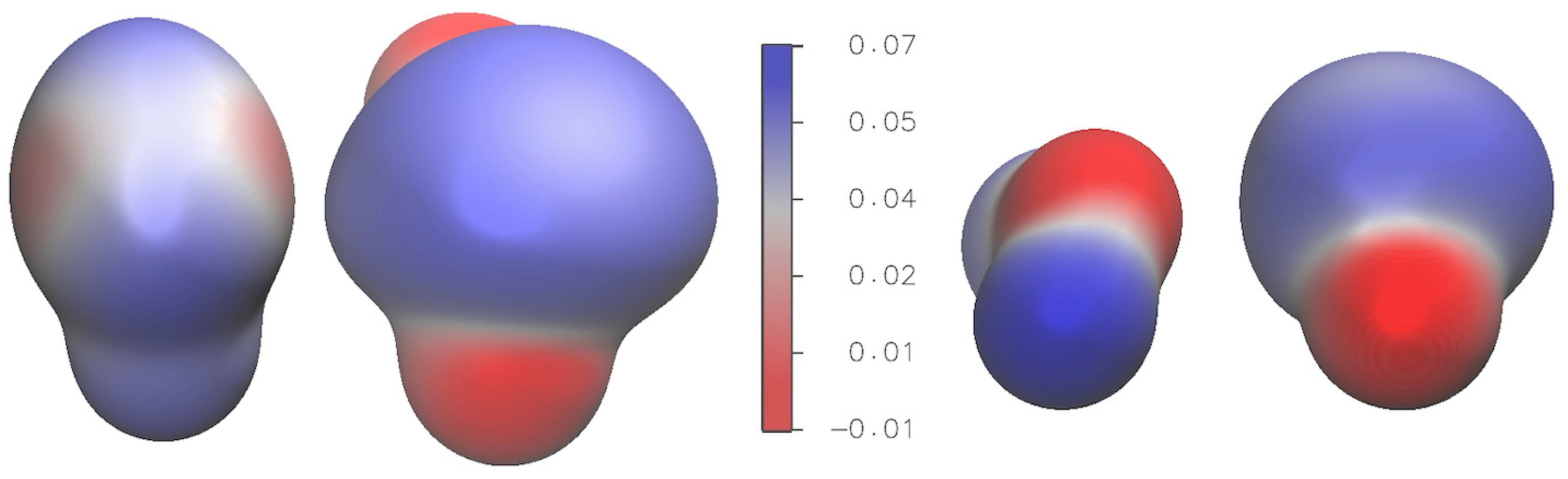
2.3. Electronic Properties
2.3.1. Electron Binding Energies and Reactivity Parameters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vertical | Adiabatic | |||
|---|---|---|---|---|
| KT | OVGF | P3+ | [CCSD] | |
| IE [EA] | IE [EA] | IE [EA] | IE [EA] | |
| H2S (a) | 10.487 [−0.682] | 10.448 [−0.507] | 10.328 [−0.485] | 10.343 [−0.468] |
| SO2 (b) | 13.560 [−0.069] | 12.619 [0.730] | 12.751 [0.792] | 12.537 [1.267] |
| H2S-SO2 | 10.144 [0.111] | 10.129 [0.979] | 9.929 [1.043] | 9.873 [1.637] |
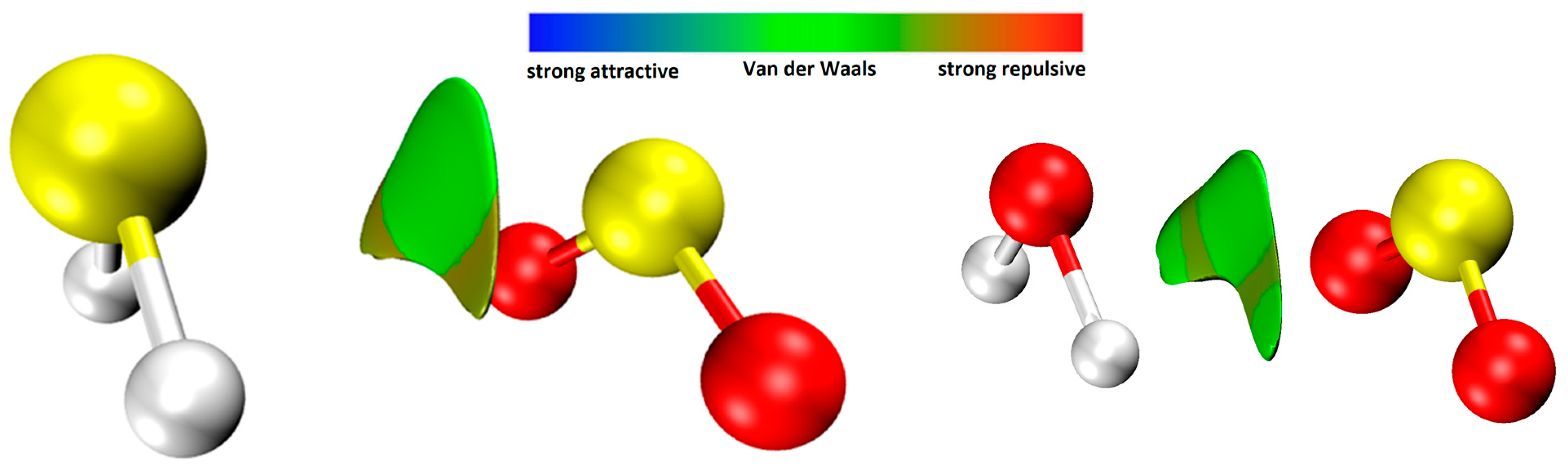
2.3.2. Electrostatic Potential and Non-Covalent Interactions (NCI) Analysis
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Taieb, D.; Ben Brahim, A. Electrochemical Method for Sulphur Dioxide Removal from Flue Gases: Application on Sulphuric Acid Plant in Tunisia. Comptes Rendus Chim. 2013, 16, 39–50. [Google Scholar] [CrossRef]
- Gerhard, E.R.; Johnstone, H.F. Air Pollution Studies-Photochemical Oxidation of Sulfur Dioxide in Air. Ind. Eng. Chem. 1955, 47, 972–976. [Google Scholar] [CrossRef]
- Tewari, A.; Shukla, N.P. Air Pollution—Adverse Effects of Sulfur Dioxide. Rev. Environ. Health 1991, 9, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.L. Sulfur Dioxide Initiates Global Climate Change in Four Ways. Thin Solid Films 2009, 517, 3188–3203. [Google Scholar] [CrossRef]
- Johnstone, H.F.; Moll, A.J. Air Pollution by Formation of Sulfuric Acid in Fogs. Ind. Eng. Chem. 1960, 52, 861–863. [Google Scholar] [CrossRef]
- Hoesly, R.M.; Smith, S.J.; Feng, L.; Klimont, Z.; Janssens-Maenhout, G.; Pitkanen, T.; Seibert, J.J.; Vu, L.; Andres, R.J.; Bolt, R.M.; et al. Historical (1750–2014) Anthropogenic Emissions of Reactive Gases and Aerosols from the Community Emissions Data System (CEDS). Geosci. Model Dev. 2018, 11, 369–408. [Google Scholar] [CrossRef]
- Dignon, J.; Hameed, S. Global Emissions of Nitrogen and Sulfur Oxides from 1860 to 1980. JAPCA 1989, 39, 180–186. [Google Scholar] [CrossRef]
- Clarke, A.G.; Radojevic, M. Oxidation of SO2 in Rainwater and Its Role in Acid Rain Chemistry. Atmos. Environ. 1987, 21, 1115–1123. [Google Scholar] [CrossRef]
- Speight, J.G. 8—Gas Cleaning Processes, 2nd ed.; Gulf Professional Publishing: Boston, MA, USA, 2019; pp. 277–324. ISBN 978-0-12-809570-6. [Google Scholar]
- Maslin, M.; Van Heerde, L.; Day, S. Sulfur: A Potential Resource Crisis That Could Stifle Green Technology and Threaten Food Security as the World Decarbonises. Geogr. J. 2022, 188, 498–505. [Google Scholar] [CrossRef]
- Kato, N.; Akimoto, H. Anthropogenic Emissions of SO2 and NOx in Asia: Emission Inventories. Atmos. Environ. Part A Gen. Top. 1992, 26, 2997–3017. [Google Scholar] [CrossRef]
- Klimont, Z.; Cofala, J.; Schöpp, W.; Amann, M.; Streets, D.G.; Ichikawa, Y.; Fujita, S. Projections of SO2, NOx, NH3 and VOC Emissions in East Asia Up to 2030. Water. Air. Soil Pollut. 2001, 130, 193–198. [Google Scholar] [CrossRef]
- Möller, D. Estimation of the Global Man-Made Sulphur Emission. Atmos. Environ. 1984, 18, 19–27. [Google Scholar] [CrossRef]
- Fioletov, V.E.; McLinden, C.A.; Krotkov, N.; Li, C.; Joiner, J.; Theys, N.; Carn, S.; Moran, M.D. A Global Catalogue of Large SO2 Sources and Emissions Derived from the Ozone Monitoring Instrument. Atmos. Chem. Phys. Discuss. 2016, 16, 11497–11519. [Google Scholar] [CrossRef]
- Robinson, E.; Robbins, R.C. Gaseous Sulfur Pollutants from Urban and Natural Sources. J. Air Pollut. Control Assoc. 1970, 20, 233–235. [Google Scholar] [CrossRef]
- Malone Rubright, S.L.; Pearce, L.L.; Peterson, J. Environmental Toxicology of Hydrogen Sulfide. Nitric Oxide Biol. Chem. 2017, 71, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sendt, K.; Haynes, B.S. Role of the Direct Reaction H2S + S2O in the Homogeneous Claus Reaction. J. Phys. Chem. A 2005, 109, 8180–8186. [Google Scholar] [CrossRef]
- Cunha, W.F.; Gargano, R.; Garcia, E.; Politi, J.R.S.; Albernaz, A.F.; Martins, J.B.L. Rovibrational Energy and Spectroscopic Constant Calculations of CH4 Center Dot Center Dot Center Dot CH4, CH4 Center Dot Center Dot Center Dot H2O, CH4 Center Dot Center Dot Center Dot CHF3, and H2O Center Dot Center Dot Center Dot CHF3 Dimers. J. Mol. Model. 2014, 20, 2298. [Google Scholar] [CrossRef]
- de Menezes, R.F.; de Macedo, L.G.M.; Martins, J.B.L.; Pirani, F.; Gargano, R. Investigation of Strength and Nature of the Weak Intermolecular Bond in NH2 Radical-Noble Gas Atom Adducts and Evaluation of Their Basic Spectroscopic Features. Chem. Phys. Lett. 2021, 769, 138386. [Google Scholar] [CrossRef]
- Paura, E.N.C.; da Cunha, W.F.; Lopes Martins, J.B.; Magela e Silva, G.; Roncaratti, L.F.; Gargano, R. Carbon Dioxide Adsorption on Doped Boron Nitride Nanotubes. RSC Adv. 2014, 4, 28249–28258. [Google Scholar] [CrossRef]
- Martins, J.B.L.; Quintino, R.P.; Politi, J.R.d.S.; Sethio, D.; Gargano, R.; Kraka, E. Computational Analysis of Vibrational Frequencies and Rovibrational Spectroscopic Constants of Hydrogen Sulfide Dimer Using MP2 and CCSD(T). Spectrochim. Acta-Part A Mol. Biomol. Spectrosc. 2020, 239, 118540. [Google Scholar] [CrossRef]
- Politi, J.R.S.; Martins, J.B.L.; Cabral, B.J.C. Born-Oppenheimer Molecular Dynamics and Electronic Properties of Liquid H2S: The Importance of a Non-Local Approach to Dispersion Interactions. J. Mol. Liq. 2022, 366, 120252. [Google Scholar] [CrossRef]
- Ford, T.A. Ab Initio Molecular Orbital Calculations of the Structures and Vibrational Spectra of Some Molecular Complexes Containing Sulphur Dioxide. J. Mol. Struct. 2009, 924–926, 466–472. [Google Scholar] [CrossRef]
- Pauley, D.J.; Bumgarner, R.E.; Kukolich, S.G. Microwave Spectrum of the SO2-H2S Complex. Chem. Phys. Lett. 1986, 132, 67–68. [Google Scholar] [CrossRef]
- Bumgarner, R.E.; Pauley, D.J.; Kukolich, S.G. Microwave Spectra and Structure for SO2∙∙∙H2S, SO2∙∙∙HDS, and SO2∙∙∙D2S Complexes. J. Chem. Phys. 1987, 87, 3749–3752. [Google Scholar] [CrossRef]
- Kukolich, S.G.; Pauley, D.J. Comment on: Structure of H2S-SO2. J. Chem. Phys. 1990, 93, 871–872. [Google Scholar] [CrossRef]
- Plummer, P.L.M. Quantum-Mechanical Studies of Weakly-Bound Molecular Clusters. J. Mol. Struct. 1994, 113, 119–133. [Google Scholar] [CrossRef]
- Lemke, K.H. Structure and Binding Energy of the H2S Dimer at the CCSD (T) Complete Basis Set Limit. J. Chem. Phys. 2017, 146, 234301. [Google Scholar] [CrossRef]
- Ortiz, J.V. Electron Propagator Theory: An Approach to Prediction and Interpretation in Quantum Chemistry. WIREs Comput. Mol. Sci. 2013, 3, 123–142. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.v.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Matsumura, K.; Lovas, F.J.; Suenram, R.D. The Microwave Spectrum and Structure of the H2S-SO2 Complex. J. Chem. Phys. 1989, 91, 5887–5894. [Google Scholar] [CrossRef]
- Wilson, A.K.; Dunning, T.H., Jr. SO2 Revisited: Impact of Tight d Augmented Correlation Consistent Basis Sets on Structure and Energetics. J. Chem. Phys. 2003, 119, 11712–11714. [Google Scholar] [CrossRef]
- Wilson, A.K.; Dunning, T.H. The HSO-SOH Isomers Revisited: The Effect of Tight d Functions. J. Phys. Chem. A 2004, 108, 3129–3133. [Google Scholar] [CrossRef]
- Wang, N.X.; Wilson, A.K. Effects of Basis Set Choice upon the Atomization Energy of the Second-Row Compounds SO2, CCl, and ClO2 for B3LYP and B3PW91. J. Phys. Chem. A 2003, 107, 6720–6724. [Google Scholar] [CrossRef]
- Bell, R.D.; Wilson, A.K. SO3 Revisited: Impact of Tight d Augmented Correlation Consistent Basis Sets on Atomization Energy and Structure. Chem. Phys. Lett. 2004, 394, 105–109. [Google Scholar] [CrossRef]
- Lee, T.J.; Taylor, P.R. A Diagnostic for Determining the Quality of Single-Reference Electron Correlation Methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef]
- Karton, A.; Rabinovich, E.; Martin, J.M.L.; Ruscic, B. W4 Theory for Computational Thermochemistry: In Pursuit of Confident Sub-KJ/Mol Predictions. J. Chem. Phys. 2006, 125, 144108. [Google Scholar] [CrossRef]
- Wang, J.; Manivasagam, S.; Wilson, A.K. Multireference Character for 4d Transition Metal-Containing Molecules. J. Chem. Theory Comput. 2015, 11, 5865–5872. [Google Scholar] [CrossRef]
- Karton, A.; Daon, S.; Martin, J.M.L. W4-11: A High-Confidence Benchmark Dataset for Computational Thermochemistry Derived from First-Principles W4 Data. Chem. Phys. Lett. 2011, 510, 165–178. [Google Scholar] [CrossRef]
- Uzer, T.; Miller, W.H. Theories of Intramolecular Vibrational Energy Transfer. Phys. Rep. 1991, 199, 73–146. [Google Scholar] [CrossRef]
- Tardy, D.C.; Rabinovitch, B.S. Intermolecular Vibrational Energy Transfer in Thermal Unimolecular Systems. Chem. Rev. 1977, 77, 369–408. [Google Scholar] [CrossRef]
- Venkataramanan, N.S. Electronic Structure, Stability, and Cooperativity of Chalcogen Bonding in Sulfur Dioxide and Hydrated Sulfur Dioxide Clusters: A DFT Study and Wave Functional Analysis. Struct. Chem. 2022, 33, 179–193. [Google Scholar] [CrossRef]
- Pawłowski, F.; Ortiz, J.V. Ionization Energies and Dyson Orbitals of the Iso-Electronic SO2, O3, and S3 Molecules from Electron Propagator Calculations. J. Phys. Chem. A 2021, 125, 3664–3680. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lee, Y.T.; Shirley, D.A. Molecular Beam Photoelectron Spectroscopy of SO2: Geometry, Spectroscopy, and Dynamics of SO+2. J. Chem. Phys. 1987, 87, 2489–2497. [Google Scholar] [CrossRef]
- Rothe, E.W.; Tang, S.Y.; Reck, G.P. Measurement of Electron Affinities of O3, SO2, and SO3 by Collisional Ionization. J. Chem. Phys. 1975, 62, 3829–3831. [Google Scholar] [CrossRef]
- Nimlos, M.R.; Ellison, G.B. Photoelectron Spectroscopy of Sulfur-Containing Anions (SO2-, S3-, and S2O-). J. Phys. Chem. 1986, 90, 2574–2580. [Google Scholar] [CrossRef]
- Walters, E.A.; Blais, N.C. Molecular Beam Photoionization and Fragmentation of D2S, (H2S)2, (D2S)2, and H2S⋅H2O. J. Chem. Phys. 1984, 80, 3501–3502. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute Hardness: Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Pearson, R.G. The Principle of Maximum Hardness. Acc. Chem. Res. 1993, 26, 250–255. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Head-Gordon, T. Analytic MP2 Frequencies without Fifth-Order Storage. Theory and Application to Bifurcated Hydrogen Bonds in the Water Hexamer. Chem. Phys. Lett. 1994, 220, 122–128. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Schaefer III, H.F. Is Coupled Cluster Singles and Doubles (CCSD) More Computationally Intensive than Quadratic Configuration Interaction (QCISD)? J. Chem. Phys. 1989, 90, 3700–3703. [Google Scholar] [CrossRef]
- Hirata, S.; Podeszwa, R.; Tobita, M.; Bartlett, R.J. Coupled-Cluster Singles and Doubles for Extended Systems. J. Chem. Phys. 2004, 120, 2581–2592. [Google Scholar] [CrossRef] [PubMed]
- Purvis III, G.D.; Bartlett, R.J. A Full Coupled-cluster Singles and Doubles Model: The Inclusion of Disconnected Triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Stanton, J.F.; Gauss, J.; Cheng, L.; Harding, M.E.; Matthews, D.A.; Szalay, P.G. CFOUR, Coupled-Cluster Techniques for Computational Chemistry, a Quantum-Chemical Program Package. J. Chem. Phys. 2020, 152, 214108. [Google Scholar]
- Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Mentel, Ł.M.; Baerends, E.J. Can the Counterpoise Correction for Basis Set Superposition Effect Be Justified? J. Chem. Theory Comput. 2014, 10, 252–267. [Google Scholar] [CrossRef]




| S∙∙∙S | H∙∙∙O | OS∙∙∙S | HS∙∙∙S | |||
|---|---|---|---|---|---|---|
| MP2/AVTZ | 3.414 | 3.107 | 89.27 | 76.78 | 88.623 | 70.768 |
| MP2/AVQZ | 3.387 | 3.137 | 90.11 | 78.40 | 90.215 | 73.140 |
| MP2/AV5Z | 3.382 | 3.156 | 90.33 | 79.24 | 90.615 | 74.288 |
| CCSD/AVTZ | 3.499 | 3.315 | 90.48 | 81.83 | 90.619 | 77.391 |
| CCSD/AVQZ | 3.481 | 3.335 | 90.70 | 82.37 | 91.324 | 78.983 |
| CCSDT/AVTZ | 3.454 | 3.181 | 89.25 | 78.43 | 88.558 | 73.168 |
| CCSDT/AVQZ | 3.427 | 3.221 | 90.12 | 80.33 | 90.248 | 75.960 |
| Other Values | ||||||
| Matsumura (a) | 3.520 | 3.145 | 99.0(13) | 56.8(11) | ||
| Kukolich (b) | 3.45(1) | 103(1) | 71(3) | |||
| Ford (c) | 3.802 | 72.88 | 67.28 | |||
| Plumer (d) | 3.577 | 3.07 |
| AVTZ | AVQZ | AV5Z | |
|---|---|---|---|
| H2S | |||
| MP2 | 8.92 | 8.57 | 8.21 |
| CCSD | 4.19 | 3.56 | |
| CCSD(T) | 5.40 | 5.07 | |
| SO2 | |||
| MP2 | −4.59 | 2.39 | 8.15 |
| CCSD | −17.96 | −10.98 | |
| CCSD(T) | −15.13 | −7.40 |
| MP2 | CCSD | CCSD (T) | |||||
|---|---|---|---|---|---|---|---|
| AVTZ | AVQZ | AV5Z | AVTZ | AVQZ | AVTZ | AVQZ | |
| HS | |||||||
| −14.60 | −16.05 | −11.76 | −3.74 | −5.86 | −7.06 | −6.71 | |
| −13.88 | −15.43 | −11.29 | −3.31 | −5.93 | −6.79 | −6.62 | |
| SO | |||||||
| −1.60 | −3.02 | −3.76 | −6.12 | −5.83 | −5.50 | −5.86 | |
| 5.80 | 5.20 | 4.47 | 1.34 | 1.68 | 1.34 | 1.68 | |
| S∙∙∙S | BSSE | |||
|---|---|---|---|---|
| MP2/AVTZ | 3.4142 | 3.2268 | 0.4757 | 2.7600 |
| MP2/AVQZ | 3.3866 | 3.1661 | 0.2333 | 2.9500 |
| MP2/AV5Z | 3.3822 | 3.1021 | 0.1205 | 3.0000 |
| CCSD/AVTZ | 3.4993 | 2.7071 | 0.3788 | 2.3400 |
| CCSD/AVQZ | 3.4805 | 2.6006 | 0.1558 | 2.4600 |
| CCSD(T)/AVTZ | 3.4543 | 3.0396 | 0.4473 | 2.6000 |
| CCSD(T)/AVQZ | 3.4275 | 2.9537 | 0.1831 | 2.7800 |
| H2S | −4.937 | 5.406 | 2.255 |
| SO2 | −6.902 | 5.635 | 4.227 [4.027] |
| H2S-SO2 | −5.755 | 4.118 | 4.021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magalhães, I.O.M.; Cabral, B.J.C.; Martins, J.B.L. Ab Initio Approach to the Structure, Vibrational Properties, and Electron Binding Energies of H2S∙∙∙SO2. Molecules 2023, 28, 6656. https://doi.org/10.3390/molecules28186656
Magalhães IOM, Cabral BJC, Martins JBL. Ab Initio Approach to the Structure, Vibrational Properties, and Electron Binding Energies of H2S∙∙∙SO2. Molecules. 2023; 28(18):6656. https://doi.org/10.3390/molecules28186656
Chicago/Turabian StyleMagalhães, Isaac O. M., Benedito J. C. Cabral, and João B. L. Martins. 2023. "Ab Initio Approach to the Structure, Vibrational Properties, and Electron Binding Energies of H2S∙∙∙SO2" Molecules 28, no. 18: 6656. https://doi.org/10.3390/molecules28186656
APA StyleMagalhães, I. O. M., Cabral, B. J. C., & Martins, J. B. L. (2023). Ab Initio Approach to the Structure, Vibrational Properties, and Electron Binding Energies of H2S∙∙∙SO2. Molecules, 28(18), 6656. https://doi.org/10.3390/molecules28186656