
Structure-Based Identification of Natural-Product-Derived Compounds with Potential to Inhibit HIV-1 Entry

 , , , ,
, , , ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. In Silico High-Throughput Screening of Natural Compound Mimetics of VRC01
2.1.1. Protein Structure Extraction
2.1.2. Determination of gp120 Core Residues of Interaction with VRC01
2.1.3. Molecular Dynamics (MD) Simulation
2.1.4. Ligand Generation, Preparation, and Molecular Docking
2.1.5. Protein–Ligand Interaction Profiling
2.1.6. High-Throughput Virtual Screening (vHTS) and Analysis
2.1.7. Determination of HIV HXB2 Envelope Protein Structure
2.1.8. Virtual Screening and Analysis of HIV HXB2 Envelope Protein
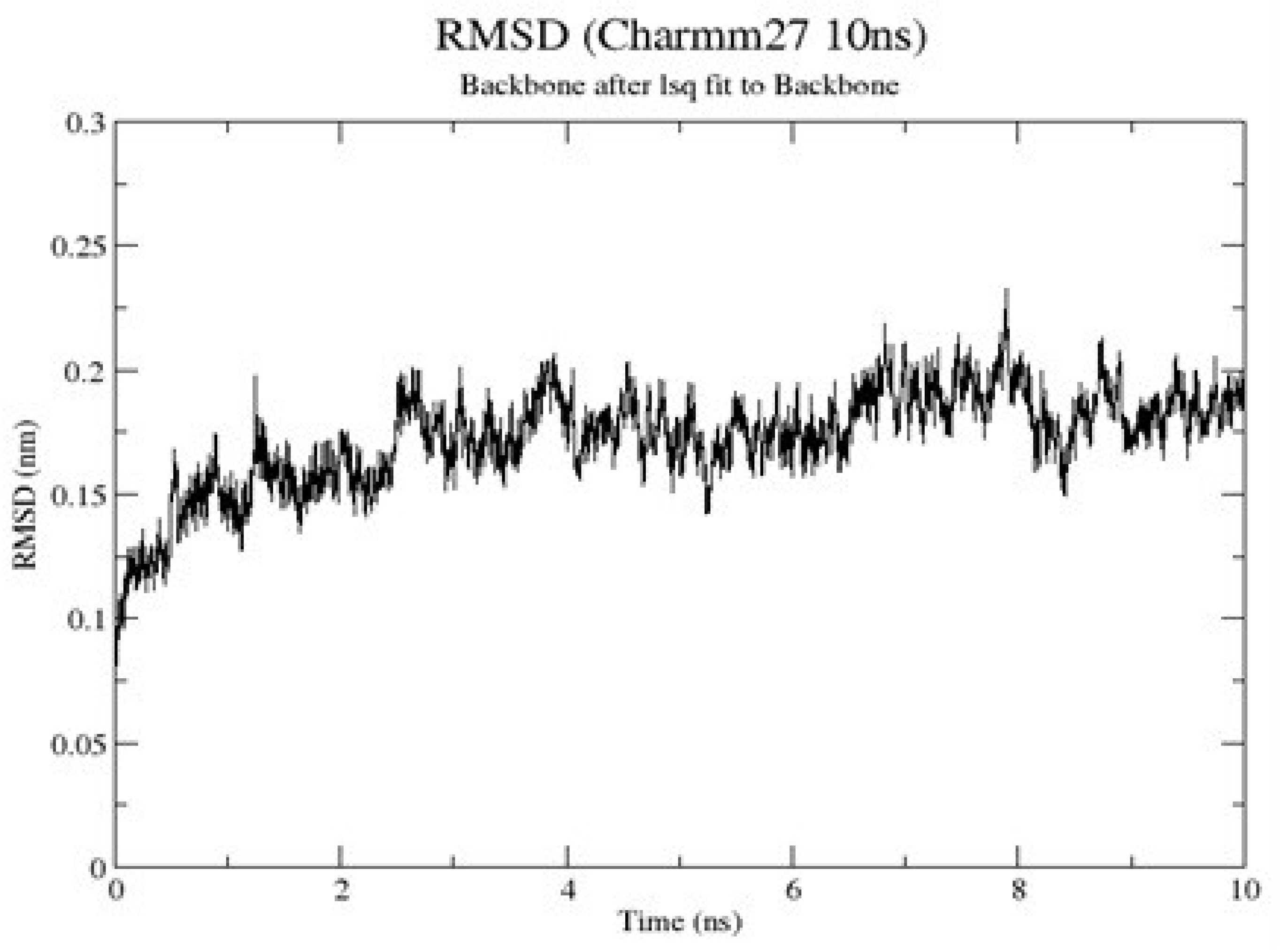
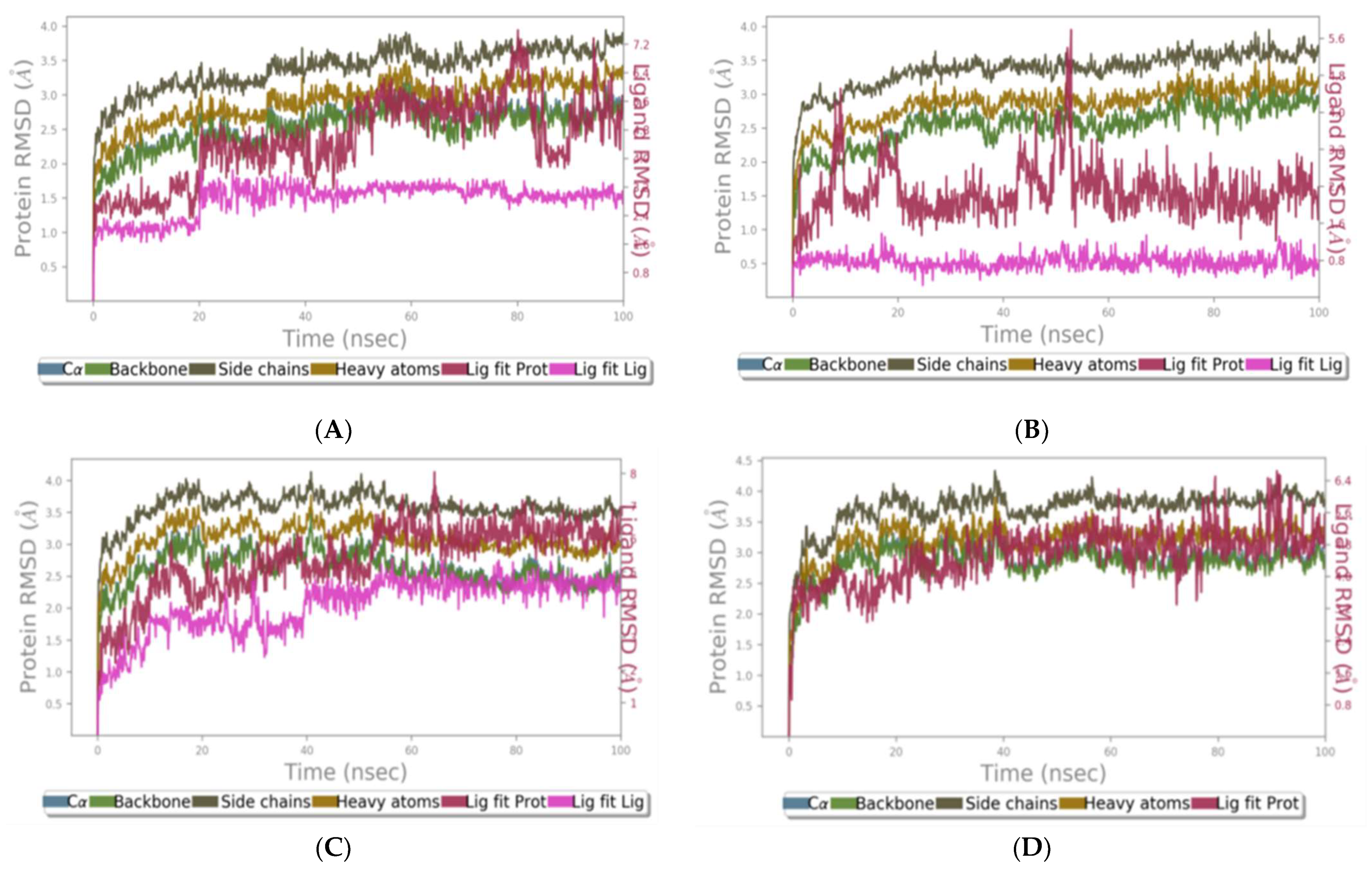
2.1.9. RMSD of Clade A/E-Ligand Complexes
2.1.10. RMSD of HXB2-Ligand Complexes
2.1.11. RMSF of Clade A/E-Ligand Complexes
2.1.12. RMSF of HXB2-Ligand Complexes
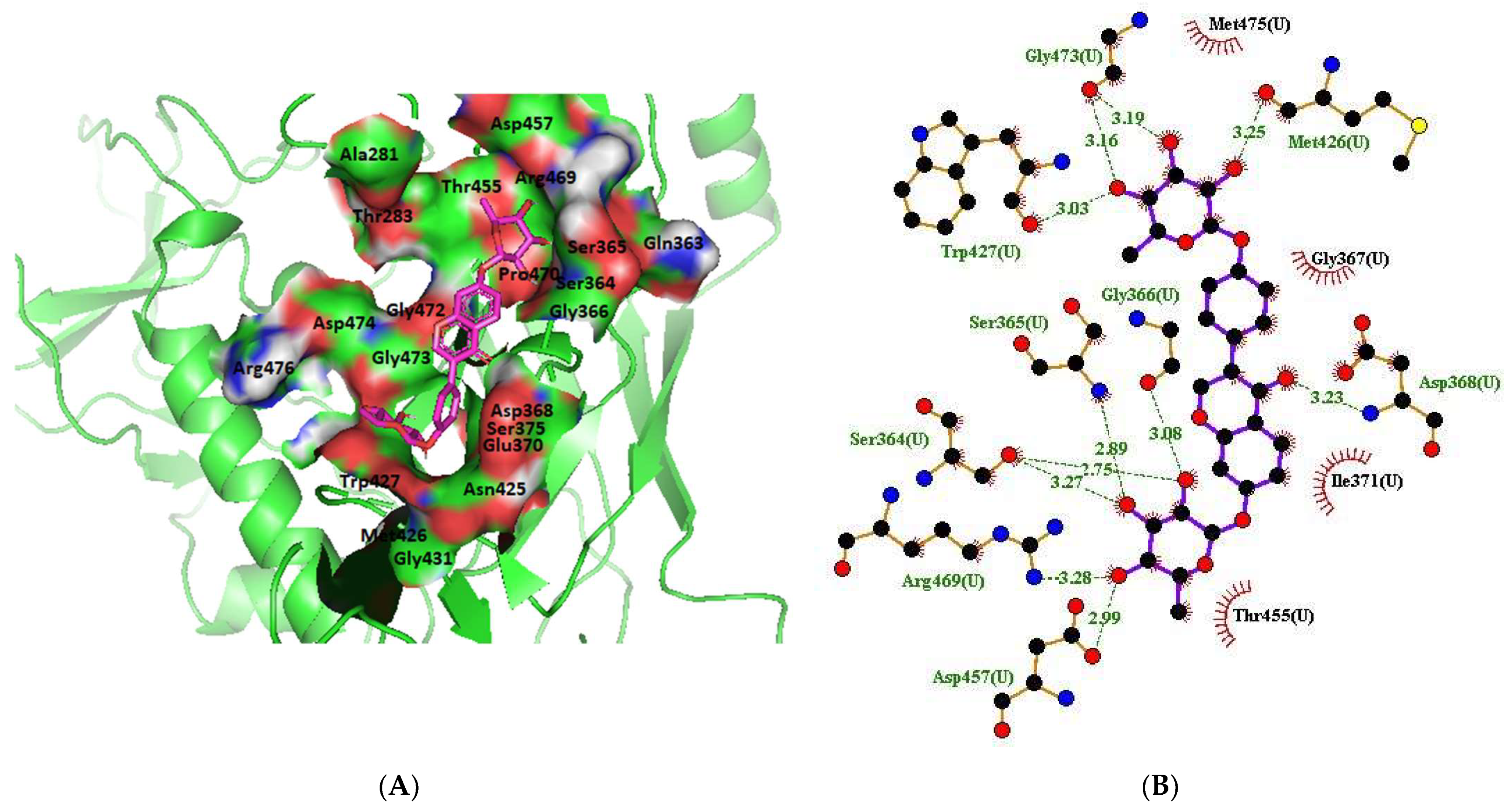
2.1.13. Clade A/E-Ligand Complex Molecular Interaction
2.1.14. HXB2-Ligand Complex Molecular Interaction
2.1.15. Origin and Sources of Selected Compounds
2.1.16. Pharmacological Profiling
2.1.17. Toxicity Profiling of the Selected Compounds
2.2. Cell-Based Viral Infectivity Inhibition Assay
2.2.1. In Vitro Cell Cytotoxicity Assessment
2.2.2. Determination of 50% Cytotoxicity Concentration (CC50)
2.2.3. Pseudotype Virus (PV) Titration and Determination of 50% Tissue Culture Infectivity Dose (TCID50)
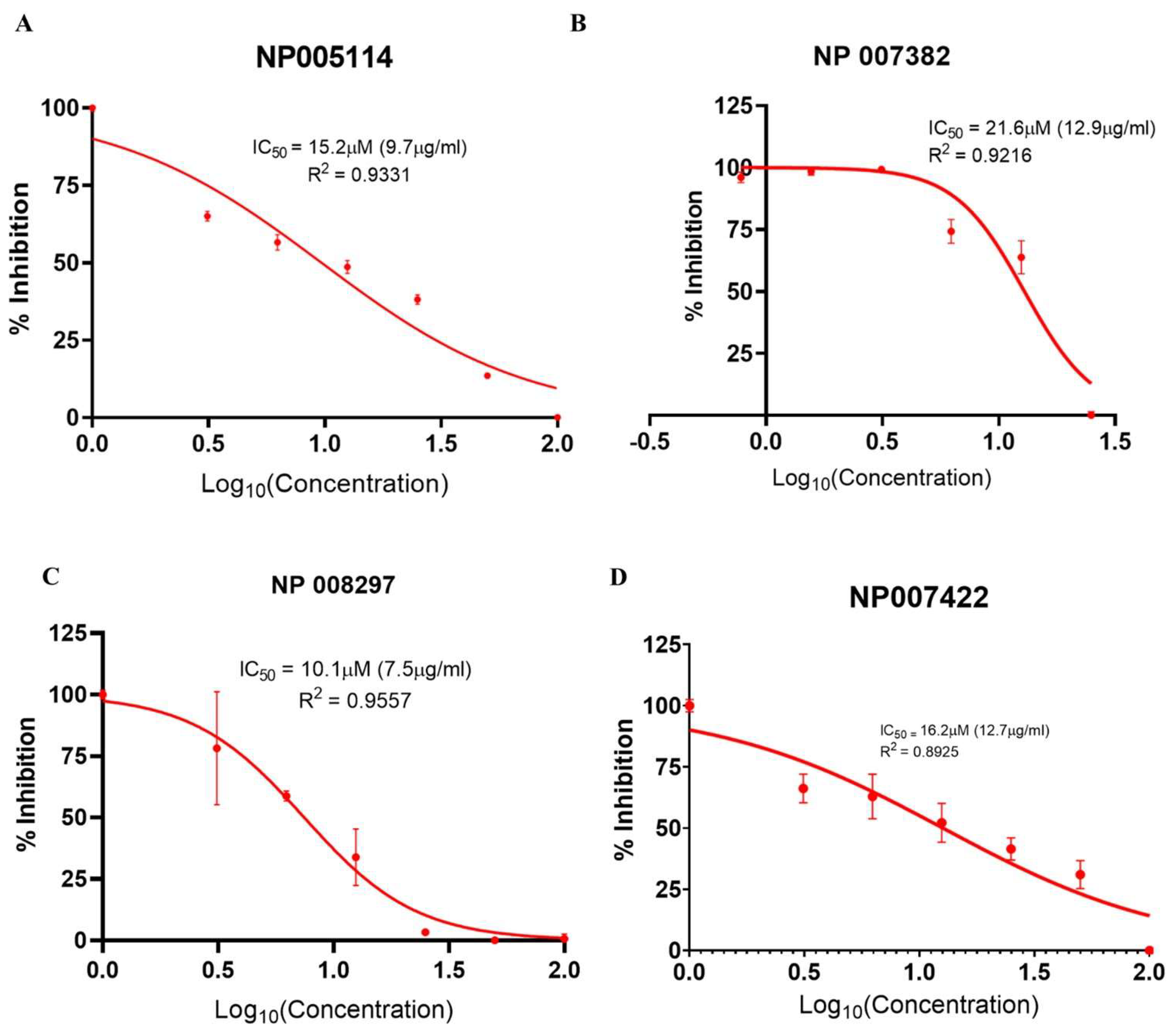
2.2.4. Viral Infectivity Inhibition Assay
2.2.5. Selectivity Index
3. Discussion
4. Materials and Methods
4.1. In Silico High-Throughput Screening of Natural Compound Mimetics of Vrc01
4.1.1. Protein Structure Extraction
4.1.2. Determination of gp120 Core Residues of Interaction with VRC01
4.1.3. Receptor Molecular Dynamics (MD) Simulation
4.1.4. Receptor Preparation
4.1.5. Ligand Compound Library Generation
4.1.6. Ligands Preparation
4.1.7. High-Throughput Virtual Screening
4.1.8. Virtual Screening Result Analysis
4.1.9. Protein–Ligand Interaction Profiling
4.1.10. HIV-1 HXB2 Envelope Protein
4.1.11. Determination of HIV-1 HXB2 Envelope Protein Structure
4.1.12. Protein Structure Extraction and Energy Minimization of HXB2 gp120
4.1.13. Receptor Preparation
4.1.14. The Root-Mean-Square Deviation (RMSD) of the Complexes
4.1.15. The Root-Mean-Square Fluctuation (RMSF) of the Complexes
4.1.16. Molecular Interactions under Dynamic Simulation
4.1.17. Physicochemical, Pharmacokinetics, and Drug-Likeness Properties Prediction
4.1.18. Toxicity Profile Prediction
4.2. Cell-Based Viral Infectivity Inhibition Assay
4.2.1. Reagents
4.2.2. Plasmids
4.2.3. Cell Lines
4.2.4. Cell Culture
4.2.5. Production of HIV Pseudotype Viruses (PV)
4.2.6. Determination of Tissue Culture Infectivity Dose (TCID50) of Harvested Pseudotype Virus (PV)
4.2.7. Preparation of Test Compounds
4.2.8. Evaluation of Compounds Cytotoxicity
4.2.9. Viral Infectivity Inhibition Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef]
- Henrich, T.J.; Kuritzkes, D.R. HIV-1 entry inhibitors: Recent development and clinical use. Curr. Opin. Virol. 2013, 3, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Haqqani, A.A.; Tilton, J.C. Entry inhibitors and their use in the treatment of HIV-1 infection. Antivir. Res. 2013, 98, 158–170. [Google Scholar] [CrossRef]
- Gulick, R.M.; Lalezari, J.; Goodrich, J.; Clumeck, N.; DeJesus, E.; Horban, A.; Nadler, J.; Clotet, B.; Karlsson, A.; Wohlfeiler, M.; et al. Maraviroc for Previously Treated Patients with R5 HIV-1 Infection. N. Engl. J. Med. 2008, 359, 1429–1441. [Google Scholar] [CrossRef] [Green Version]
- Fätkenheuer, G.; Nelson, M.; Lazzarin, A.; Konourina, I.; Hoepelman, A.I.M.; Lampiris, H.; Hirschel, B.; Tebas, P.; Raffi, F.; Trottier, B.; et al. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N. Engl. J. Med. 2008, 359, 1442–1455. [Google Scholar] [CrossRef]
- Lazzarin, A.; Clotet, B.; Cooper, D.; Reynes, J.; Arastéh, K.; Nelson, M.; Katlama, C.; Stellbrink, H.-J.; Delfraissy, J.-F.; Lange, J.; et al. Efficacy of Enfuvirtide in Patients Infected with Drug-Resistant HIV-1 in Europe and Australia. N. Engl. J. Med. 2003, 348, 2186–2195. [Google Scholar] [CrossRef] [Green Version]
- Wibmer, C.K.; Moore, P.L.; Morris, L. HIV broadly neutralizing antibody targets. Curr. Opin. HIV AIDS 2015, 10, 135–143. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Z.-Y.; Li, Y.; Hogerkorp, C.-M.; Schief, W.R.; Seaman, M.S.; Zhou, T.; Schmidt, S.D.; Wu, L.; Xu, L.; et al. Rational Design of Envelope Identifies Broadly Neutralizing Human Monoclonal Antibodies to HIV-1. Science 2010, 329, 856–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binley, J.M.; Wrin, T.; Korber, B.; Zwick, M.B.; Wang, M.; Chappey, C.; Stiegler, G.; Kunert, R.; Zolla-Pazner, S.; Katinger, H.; et al. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 2004, 78, 13232–13252. [Google Scholar] [CrossRef] [Green Version]
- Hessell, A.J.; Rakasz, E.G.; Poignard, P.; Hangartner, L.; Landucci, G.; Forthal, D.N.; Koff, W.C.; Watkins, D.I.; Burton, D.R. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 2009, 5, e1000433. [Google Scholar] [CrossRef]
- Caskey, M.; Klein, F.; Lorenzi, J.C.C.; Seaman, M.S.; West, A.P.; Buckley, N.; Kremer, G.; Nogueira, L.; Braunschweig, M.; Scheid, J.F.; et al. Corrigendum: Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 2016, 535, 580. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, J.A.; Halper-Stromberg, A.; Mouquet, H.; Gitlin, A.D.; Tretiakova, A.; Eisenreich, T.R.; Malbec, M.; Gravemann, S.; Billerbeck, E.; Dorner, M.; et al. HIV-1 suppression and durable control by combining single broadly neutralizing antibodies and antiretroviral drugs in humanized mice. Proc. Natl. Acad. Sci. USA 2013, 110, 16538–16543. [Google Scholar] [CrossRef] [Green Version]
- Klein, F.; Halper-Stromberg, A.; Horwitz, J.A.; Gruell, H.; Scheid, J.F.; Bournazos, S.; Mouquet, H.; Spatz, L.A.; Diskin, R.; Abadir, A.; et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature 2012, 492, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Halper-Stromberg, A.; Lu, C.L.; Klein, F.; Horwitz, J.A.; Bournazos, S.; Nogueira, L.; Eisenreich, T.R.; Liu, C.; Gazumyan, A.; Schaefer, U.; et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell 2014, 158, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Scheid, J.F.; Mouquet, H.; Ueberheide, B.; Diskin, R.; Klein, F.; Oliveira, T.Y.K.; Pietzsch, J.; Fenyo, D.; Abadir, A.; Velinzon, K.; et al. Sequence and Structural Convergence of Broad and Potent HIV Antibodies That Mimic CD4 Binding. Science 2011, 333, 1633–1637. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.H.; Su, D.; Torabi, J.; Fennessey, C.M.; Shiakolas, A.; Lynch, R.; Chun, T.W.; Doria-Rose, N.; Alter, G.; Seaman, M.S.; et al. Predicting the broadly neutralizing antibody susceptibility of the HIV reservoir. JCI Insight 2019, 4, e130153. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Georgiev, I.; Wu, X.; Yang, Z.Y.; Dai, K.; Finzi, A.; Do Kwon, Y.; Scheid, J.F.; Shi, W.; Xu, L.; et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 2010, 329, 811–817. [Google Scholar] [CrossRef] [Green Version]
- Bar, K.J.; Sneller, M.C.; Harrison, L.J.; Justement, J.S.; Overton, E.T.; Petrone, M.E.; Salantes, D.B.; Seamon, C.A.; Scheinfeld, B.; Kwan, R.W.; et al. Effect of HIV Antibody VRC01 on Viral Rebound after Treatment Interruption. N. Engl. J. Med. 2016, 375, 2037–2050. [Google Scholar] [CrossRef]
- Gautam, R.; Nishimura, Y.; Pegu, A.; Nason, M.C.; Klein, F.; Gazumyan, A.; Golijanin, J.; Buckler-White, A.; Sadjadpour, R.; Wang, K.; et al. A single injection of anti-HIV-1 antibodies protects against repeated SHIV challenges. Nature 2016, 533, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Mayer, K.H.; Seaton, K.E.; Huang, Y.; Grunenberg, N.; Isaacs, A.; Allen, M.; Ledgerwood, J.E.; Frank, I.; Sobieszczyk, M.E.; Baden, L.R.; et al. Safety, pharmacokinetics, and immunological activities of multiple intravenous or subcutaneous doses of an anti-HIV monoclonal antibody, VRC01, administered to HIV-uninfected adults: Results of a phase 1 randomized trial. PLoS Med. 2017, 14, e1002435. [Google Scholar] [CrossRef]
- Cale, E.M.; Bai, H.; Bose, M.; Messina, M.A.; Colby, D.J.; Sanders-Buell, E.; Dearlove, B.; Li, Y.; Engeman, E.; Silas, D.; et al. Neutralizing antibody VRC01 failed to select for HIV-1 mutations upon viral rebound. J. Clin. Investig. 2020, 130, 3299–3304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awi, N.J.; Teow, S.-Y. Antibody-Mediated Therapy against HIV/AIDS: Where Are We Standing Now? J. Pathog. 2018, 2018, 8724549. [Google Scholar] [CrossRef] [PubMed]
- Arantes, I.; Ribeiro-Alves, M.; de Azevedo, S.S.D.; Delatorre, E.; Bello, G. Few amino acid signatures distinguish HIV-1 subtype B pandemic and non-pandemic strains. PLoS ONE 2020, 15, e0238995. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, E.C.; Kumar, R.; Davis, R.A. The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat. Prod. Rep. 2016, 33, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, A.D.G.; Maccoss, M.; Heer, J.P. Importance of Rigidity in Designing Small Molecule Drugs to Tackle Protein-Protein Interactions (PPIs) through Stabilization of Desired Conformers. J. Med. Chem. 2018, 61, 4383–4389. [Google Scholar] [CrossRef] [PubMed]
- Cary, D.C.; Peterlin, B.M. Natural Products and HIV/AIDS. AIDS Res. Hum. Retroviruses 2018, 34, 31–38. [Google Scholar] [CrossRef]
- Chinsembu, K.C. Chemical diversity and activity profiles of HIV-1 reverse transcriptase inhibitors from plants. Rev. Bras. Farmacogn. 2019, 29, 504–528. [Google Scholar] [CrossRef]
- Fang, J.; Liu, C.; Wang, Q.; Lin, P.; Cheng, F. In silico polypharmacology of natural products. Brief. Bioinform. 2017, 19, 1153–1171. [Google Scholar] [CrossRef]
- Yi, F.; Li, L.; Xu, L.J.; Meng, H.; Dong, Y.M.; Liu, H.B.; Xiao, P.G. In silico approach in reveal traditional medicine plants pharmacological material basis. Chin. Med. 2018, 13, 33. [Google Scholar] [CrossRef]
- Crentsil, J.A.; Yamthe, L.R.T.; Anibea, B.Z.; Broni, E.; Kwofie, S.K.; Tetteh, J.K.A.; Osei-Safo, D. Leishmanicidal Potential of Hardwickiic Acid Isolated From Croton sylvaticus. Front. Pharmacol. 2020, 11, 753. [Google Scholar] [CrossRef] [PubMed]
- Stevanovic, S.; Sencanski, M.; Danel, M.; Menendez, C.; Belguedj, R.; Bouraiou, A.; Nikolic, K.; Cojean, S.; Loiseau, P.M.; Glisic, S.; et al. Synthesis, in silico, and in vitro evaluation of anti-leishmanial activity of oxadiazoles and indolizine containing compounds flagged against anti-targets. Molecules 2019, 24, 1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panigrahi, D.; Mishra, A.; Sahu, S.K. Rational in silico drug design of HIV-RT inhibitors through G-QSAR and molecular docking study of 4-arylthio and 4-aryloxy-3-iodopyridine-2(1-H)-one derivative. Beni-Suef Univ. J. Basic Appl. Sci. 2020, 9, 48. [Google Scholar] [CrossRef]
- Sinha, C.; Nischal, A.; Bandaru, S.; Kasera, P.; Rajput, A.; Nayarisseri, A.; Khattri, S. An In silico Approach for Identification of Novel Inhibitors as a Potential Therapeutics Targeting HIV-1 Viral Infectivity Factor. Curr. Top. Med. Chem. 2015, 15, 65–72. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, Y.; Gao, Y.; Wu, F.; Luo, X.; Ju, X.; Liu, G. In silico Design of Novel HIV-1 NNRTIs Based on Combined Modeling Studies of Dihydrofuro[3,4-d]pyrimidines. Front. Chem. 2020, 8, 164. [Google Scholar] [CrossRef] [Green Version]
- Kirchmair, J.; Distinto, S.; Roman Liedl, K.; Markt, P.; Maria Rollinger, J.; Schuster, D.; Maria Spitzer, G.; Wolber, G. Development of Anti-Viral Agents Using Molecular Modeling and Virtual Screening Techniques. Infect. Disord.—Drug Targets 2012, 11, 64–93. [Google Scholar] [CrossRef]
- Bonsignori, M.; Scott, E.; Wiehe, K.; Easterhoff, D.; Munir Alam, S.; Hwang, K.-K.; Cooper, M.; Xia, S.-M.; Zhang, R.; Montefiori, D.C.; et al. Inference of the HIV-1 VRC01 Antibody Lineage Unmutated Common Ancestor Reveals Alternative Pathways to Overcome a Key Glycan Barrier. Immunity 2018, 49, 1162–1174. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of Protein Structure Comparison. In Methods in Molecular Biology; NIH Public Access: Clifton, NJ, USA, 2011; Volume 857, pp. 231–257. [Google Scholar]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Choudhary, M.I.; Shaikh, M.; tul-Wahab, A.; ur-Rahman, A. In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation. PLoS ONE 2020, 15, e0235030. [Google Scholar] [CrossRef]
- Wen, C.C.; Kuo, Y.H.; Jan, J.T.; Liang, P.H.; Wang, S.Y.; Liu, H.G.; Lee, C.K.; Chang, S.T.; Kuo, C.J.; Lee, S.S.; et al. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Kumar, R.; Planey, S.L. Lipophilicity Indices for Drug Development. J. Appl. Biopharm. Pharmacokinet. 2013, 1, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.; Gattass, C.R. Topological polar surface area defines substrate transport by multidrug resistance associated protein 1 (MRP1/ABCC1). J. Med. Chem. 2009, 52, 1214–1218. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Hitchcock, S.A.; Pennington, L.D. Structure−Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef] [Green Version]
- Lei, C.; Yang, J.; Hu, J.; Sun, X. On the Calculation of TCID50 for Quantitation of Virus Infectivity. Virol. Sin. 2021, 36, 141–144. [Google Scholar] [CrossRef]
- Péret-Almeida, L.; Cherubino, A.P.F.; Alves, R.J.; Dufossé, L.; Glória, M.B.A. Separation and determination of the physico-chemical characteristics of curcumin, demethoxycurcumin and bisdemethoxycurcumin. Food Res. Int. 2005, 38, 1039–1044. [Google Scholar] [CrossRef]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef]
- Wood, N.T.; Fadda, E.; Davis, R.; Grant, O.C.; Martin, J.C.; Woods, R.J.; Travers, S.A. The influence of N-linked glycans on the molecular dynamics of the HIV-1 gp120 V3 loop. PLoS ONE 2013, 8, e80301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moebius, U.; Clayton, L.K.; Abraham, S.; Harrison, S.C.; Reinherz, E.L. The human inmaunodeficiency virus gpl20 binding site on CD4: Delineation by quantitative equilibrium and kinetic binding studies of mutants in conjunction with a high-resohtion cd4 atomic structure. J. Exp. Med. 1992, 176, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, M.; Potz, J.; Basiripour, L.; Dorfman, T.; Goh, W.C.; Terwilliger, E.; Dayton, A.; Rosen, C.; Haseltine, W.; Sodroski, J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science 1987, 237, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Olshevsky, U.; Helseth, E.; Furman, C.; Li, J.; Haseltine, W.; Sodroski, J. Identification of individual human immunodeficiency virus type 1 gp120 amino acids important for CD4 receptor binding. J. Virol. 1990, 64, 5701–5707. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Yu, F.; Cai, L.; Debnath, A.K.; Jiang, S. Development of Small-molecule HIV Entry Inhibitors Specifically Targeting gp120 or gp41. Curr. Top. Med. Chem. 2016, 16, 1074–1090. [Google Scholar] [CrossRef] [Green Version]
- Dey, B.; Berger, E.A. Blocking HIV-1 gp120 at the Phe43 cavity: If the extension fits. Structure 2013, 21, 871–872. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Xu, L.; Dey, B.; Hessell, A.J.; Van Ryk, D.; Xiang, S.H.; Yang, X.; Zhang, M.Y.; Zwick, M.B.; Arthos, J.; et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 2007, 445, 732–737. [Google Scholar] [CrossRef] [Green Version]
- Barret, R. Lipinski’s Rule of Five. In Therapeutical Chemistry; Elsevier Ltd.: Oxford, UK, 2018; pp. 97–100. [Google Scholar] [CrossRef]
- Dahan, A.; Miller, J.M. The solubility-permeability interplay and its implications in formulation design and development for poorly soluble drugs. AAPS J. 2012, 14, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRX 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Heller, F.; Voelker, S.; Wacharamanotham, C.; Borchers, J. Transporters. In Proceedings of the Proceedings of the 33rd Annual ACM Conference Extended Abstracts on Human Factors in Computing Systems—CHI EA ’15, Seoul, Republic of Korea, 18–23 April 2015. [Google Scholar]
- McDonnell, A.M.; Dang, C.H. Basic review of the cytochrome p450 system. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magaret, C.A.; Benkeser, D.C.; Williamson, B.D.; Borate, B.R.; Carpp, L.N.; Georgiev, I.S.; Setliff, I.; Dingens, A.S.; Simon, N.; Carone, M.; et al. Prediction of VRC01 neutralization sensitivity by HIV-1 gp160 sequence features. PLoS Comput. Biol. 2019, 15, e1006952. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.Z.; Lorenzi, J.C.C.; Krassnig, L.; Barton, J.P.; Burke, L.; Pai, J.; Lu, C.L.; Mendoza, P.; Oliveira, T.Y.; Sleckman, C.; et al. Relationship between latent and rebound viruses in a clinical trial of anti-HIV-1 antibody 3BNC117. J. Exp. Med. 2018, 215, 2311–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, L.M.; Huber, M.; Doores, K.J.; Falkowska, E.; Pejchal, R.; Julien, J.-P.; Wang, S.-K.; Ramos, A.; Chan-Hui, P.-Y.; Moyle, M.; et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 2011, 477, 466–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, N.; Hwang, C.; Healy, M.D.; Whitcomb, J.; Lataillade, M.; Wind-Rotolo, M.; Krystal, M.; Hanna, G.J. Prediction of Virological Response and Assessment of Resistance Emergence to the HIV-1 Attachment Inhibitor BMS-626529 During 8-Day Monotherapy With Its Prodrug BMS-663068. JAIDS J. Acquir. Immune Defic. Syndr. 2013, 64, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalezari, J.P.; Latiff, G.H.; Brinson, C.; Echevarría, J.; Treviño-Pérez, S.; Bogner, J.R.; Thompson, M.; Fourie, J.; Sussmann Pena, O.A.; Mendo Urbina, F.C.; et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of AI438011, a phase 2b, randomised controlled trial. Lancet HIV 2015, 2, e427–e437. [Google Scholar] [CrossRef] [PubMed]
- Meuser, M.E.; Murphy, M.B.; Rashad, A.A.; Cocklin, S. Kinetic Characterization of Novel HIV-1 Entry Inhibitors: Discovery of a Relationship between Off-Rate and Potency. Molecules 2018, 23, 1940. [Google Scholar] [CrossRef] [Green Version]
- Pancera, M.; Lai, Y.-T.; Bylund, T.; Druz, A.; Narpala, S.; O’Dell, S.; Schön, A.; Bailer, R.T.; Chuang, G.-Y.; Geng, H.; et al. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat. Chem. Biol. 2017, 13, 1115–1122. [Google Scholar] [CrossRef]
- Zhao, Q.; Ma, L.; Jiang, S.; Lu, H.; Liu, S.; He, Y.; Strick, N.; Neamati, N.; Debnath, A.K. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 2005, 339, 213–225. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, A.; Brijwal, M.; Choudhary, A.; Singh, K.; Singh, R.; Husain, M.; Dar, L. Intra-Clade C signature polymorphisms in HIV-1 LTR region: The Indian and African lookout. Virus Res. 2021, 297, 198370. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.J.; Chul, Y.K.; Ji, S.L.; Tae, G.K.; Seung, H.K.; Lee, C.K.; Lee, B.B.; Shin, C.G.; Huh, H.; Kim, J. Inhibition of HIV-1 integrase by galloyl glucoses from Terminalia chebula and flavonol glycoside gallates from Euphorbia pekinensis. Planta Med. 2002, 68, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Gambari, R.; Lampronti, I. Inhibition of immunodeficiency type-1 virus (HIV-1) life cycle by medicinal plant extracts and plant-derived compounds. Adv. Phytomed. 2006, 2, 299–311. [Google Scholar]
- Salehi, B.; Kumar, N.; Şener, B.; Sharifi-Rad, M.; Kılıç, M.; Mahady, G.; Vlaisavljevic, S.; Iriti, M.; Kobarfard, F.; Setzer, W.; et al. Medicinal Plants Used in the Treatment of Human Immunodeficiency Virus. Int. J. Mol. Sci. 2018, 19, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, I.P.; Bharate, S.B.; Bhutani, K.K. Anti-HIV natural products. Curr. Sci. 2005, 89, 269–290. [Google Scholar]
- Lee, J.; Hattori, M.; Kim, J. Inhibition of HIV-1 Protease and RNase H of HIV-1 Reverse Transcriptase Activities by Long Chain Phenols from the Sarcotestas of Ginkgo biloba. Planta Med. 2008, 74, 532–534. [Google Scholar] [CrossRef] [Green Version]
- Salve, J.; Pate, S.; Debnath, K.; Langade, D. Adaptogenic and Anxiolytic Effects of Ashwagandha Root Extract in Healthy Adults: A Double-blind, Randomized, Placebo-controlled Clinical Study. Cureus 2019, 11, e6466. [Google Scholar] [CrossRef] [Green Version]
- Kurapati, K.R.V.; Atluri, V.S.R.; Samikkannu, T.; Nair, M.P.N. Ashwagandha (Withania somnifera) Reverses β-Amyloid1-42 Induced Toxicity in Human Neuronal Cells: Implications in HIV-Associated Neurocognitive Disorders (HAND). PLoS ONE 2013, 8, e77624. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Turner, P. XMGRACE, Version 5.1. 19; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Methods in Molecular Biology; NIH Public Access: Clifton, NJ, USA, 2015; Volume 1263, pp. 243–250. ISBN 9781118435762. [Google Scholar] [CrossRef]
- Huang, S.-Y. Comprehensive assessment of flexible-ligand docking algorithms: Current effectiveness and challenges. Brief. Bioinform. 2017, 19, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheed, M.; Bettadapura, R.; Bajaj, C. Computational Refinement and Validation Protocol for Proteins with Large Variable Regions Applied to Model HIV Env Spike in CD4 and 17b Bound State. Structure 2015, 23, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, W.; Littlejohn, T.G. Swiss-PDB Viewer (Deep View). Brief. Bioinform. 2001, 2, 195–197. [Google Scholar] [CrossRef]
- Cheng, X.; Ivanov, I. Molecular Dynamics. In Methods in Molecular Biology; NIH Public Access: Clifton, NJ, USA, 2012; Volume 929, pp. 243–285. ISBN 9781627030496. [Google Scholar]
- Dong, Y.W.; Liao, M.L.; Meng, X.L.; Somero, G.N. Structural flexibility and protein adaptation to temperature: Molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc. Natl. Acad. Sci. USA 2018, 115, 1274–1279. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Longo, P.A.; Kavran, J.M.; Kim, M.S.; Leahy, D.J. Transient mammalian cell transfection with polyethylenimine (PEI). In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 529, pp. 227–240. [Google Scholar] [CrossRef]
- Ferrara, F.; Temperton, N.; Ferrara, F.; Temperton, N. Pseudotype Neutralization Assays: From Laboratory Bench to Data Analysis. Methods Protoc. 2018, 1, 8. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | Company | Library | Number of Distinct Compounds |
|---|---|---|---|
| 1 | AnalytiCon Discovery | Fragment from nature (FRGx) | 247 |
| 2 | Macrocyclic semi-synthetic compounds (MACROx) | 2040 | |
| 3 | Purified natural product screening compounds (MEGx) | 3781 | |
| 4 | SPECS | Pre-plated 20k diverse compound library | 20,636 |
| 5 | Pre-plated natural product library | 373 | |
| 6 | InterBioScreen (IBS) | IBS 2017_sep_Natual compound Libraries | 747 |
| Total | 27,824 | ||
| Recombinant Clade A/E | Clade B | |||
|---|---|---|---|---|
| Compound | Binding Energy (kcal/mol) | Amino Acid Residues | Binding Energy (kcal/mol) | Amino Acid Residues |
| NP-008297 | −10.3 | Glu370, Gln258, Pro470, Ile371, Pro363, Ser365, Gln362, Gly471, Thr455, Gly472, and Asn425. | −7.9 | Asp368, Ser365, Ser364, Gly366, Asn425, and Met426. |
| NP-000088 | −9.7 | Glu370, His375, Gln258, Gly472, Gln363, and Ser365 | −7.1 | Gly473, Thr283, Ser364, and Ser365. |
| NP-007382 | −9.6 | Pro470, Gly366, Ile371, Gly473, and Ile475. | −7.2 | Asp457, Arg469, Ser364, Ser365, and Gly472. |
| NP-005003 | −9.3 | Asp457, Asn280, Pro470, and Arg469. | −7.3 | Thr283 and Ala281. |
| NP-007422 | −9.3 | Gln258, Ile371, Thr257, Met373, Glu370, Asn425, Asp280, and Asp457. | −6.7 | Asp474, Arg476, Trp427, Gly473, Asn425, Asp368, and Glu370. |
| NP-001800 | −9.2 | Glu370, Trp427, Gly429, Gln105, and Asn474. | −7.8 | Ser365 and Gln363. |
| NP-005114 | −9.1 | Gln258, Ile371, Gln362, Pro363, Gly471, Asp457, Asn280, and Gly472. | −8.2 | Arg476, Gly472, Pro470, Gly366, Ser364, Glu370, Asn425, Trp427, and Met426. |
| NP-004255 | −9 | Trp427, Gly429, Ile371, Gln258, Pro470, and Gly472. | −7.3 | Arg476, Asp474, Thr455, and Pro470. |
| FRG-00075 | −7.4 | Glu370, Ile371, Ser365, Arg469, and Gly472. | −6.8 | Gly431 and Ser375. |
| Compound ID | Chemical Structure |
|---|---|
| NP-000088 |  |
| NP-004255 |  |
| NP-005003 |  |
| NP-005114 |  |
| NP-007382 |  |
| NP-007422 |  |
| NP-008297 |  |
| NP-001800 |  |
| FRG-00075 |  |
| Compound Structure | Organism | Name | Purity |
|---|---|---|---|
| NP-000088 | Plant | Mentha piperita | 79 |
| NP-004255 | Plant | Terminalia chebula | 92 |
| NP-005003 | Micro-organism | Aspergillus | 97 |
| NP-005114 | Plant | Terminalia chebula | 99 |
| NP-007382 | Micro-organism | Actinomycete | 98 |
| NP-007422 | Plant | Withania somnifera | 96 |
| NP-008297 | Plant | Ginkgo biloba | 94 |
| NP-001800 | Micro-organism | Fungi | 79 |
| FRG-00075 | Fragment | N/A | 70 |
| Compound Name | Molecular Weight (Dalton) | Number of Rotatable Bonds | Number of H-bond Acceptors | Number of H-bond Donors |
|---|---|---|---|---|
| NP-008297 | 740.66 | 9 | 16 | 9 |
| NP-004255 | 634.45 | 2 | 15 | 11 |
| NP-000088 | 610.56 | 7 | 15 | 8 |
| NP-007422 | 782.91 | 9 | 14 | 9 |
| NP-005114 | 636.46 | 5 | 15 | 11 |
| NP-007382 | 596.57 | 5 | 12 | 4 |
| NP-005003 | 723.78 | 5 | 9 | 9 |
| NP-001800 | 800.90 | 7 | 9 | 9 |
| FRG-00075 | 335.72 | 2 | 4 | 2 |
| Compound Name | TPSA * | Lipophilicity | Water Solubility | GI Absorption ¥ | BBB Permeant £ | Pgp Substrate § | CYP Inhibitor ȡ |
|---|---|---|---|---|---|---|---|
| NP-008297 | 275.5 | 1.79 | Soluble | Low | No | Yes | No |
| NP-004255 | 310.66 | −0.87 | Soluble | Low | No | Yes | No |
| NP-000088 | 234.29 | −0.43 | Soluble | Low | No | Yes | No |
| NP-007422 | 245.29 | 0.07 | Soluble | Low | No | Yes | No |
| NP-005114 | 310.66 | −3.12 | Soluble | Low | No | No | No |
| NP-007382 | 186.12 | 1.91 | Soluble | Low | No | Yes | No |
| NP-005003 | 251.16 | −0.98 | Soluble | Low | No | Yes | No |
| NP-001800 | 251.16 | 0.61 | Insoluble | Low | No | Yes | No |
| FRG-00075 | 71.18 | 1.3 | Soluble | High | No | Yes | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ugwu-Korie, N.; Quaye, O.; Wright, E.; Languon, S.; Agyapong, O.; Broni, E.; Gupta, Y.; Kempaiah, P.; Kwofie, S.K. Structure-Based Identification of Natural-Product-Derived Compounds with Potential to Inhibit HIV-1 Entry. Molecules 2023, 28, 474. https://doi.org/10.3390/molecules28020474
Ugwu-Korie N, Quaye O, Wright E, Languon S, Agyapong O, Broni E, Gupta Y, Kempaiah P, Kwofie SK. Structure-Based Identification of Natural-Product-Derived Compounds with Potential to Inhibit HIV-1 Entry. Molecules. 2023; 28(2):474. https://doi.org/10.3390/molecules28020474
Chicago/Turabian StyleUgwu-Korie, Nneka, Osbourne Quaye, Edward Wright, Sylvester Languon, Odame Agyapong, Emmanuel Broni, Yash Gupta, Prakasha Kempaiah, and Samuel K. Kwofie. 2023. "Structure-Based Identification of Natural-Product-Derived Compounds with Potential to Inhibit HIV-1 Entry" Molecules 28, no. 2: 474. https://doi.org/10.3390/molecules28020474
APA StyleUgwu-Korie, N., Quaye, O., Wright, E., Languon, S., Agyapong, O., Broni, E., Gupta, Y., Kempaiah, P., & Kwofie, S. K. (2023). Structure-Based Identification of Natural-Product-Derived Compounds with Potential to Inhibit HIV-1 Entry. Molecules, 28(2), 474. https://doi.org/10.3390/molecules28020474