

Synthesis of D-π-A′-π-A Chromophores with Quinoxaline Core as Auxiliary Acceptor and Effect of Various Silicon-Substituted Donor Moieties on Thermal and Nonlinear Optical Properties at Molecular and Material Level

 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
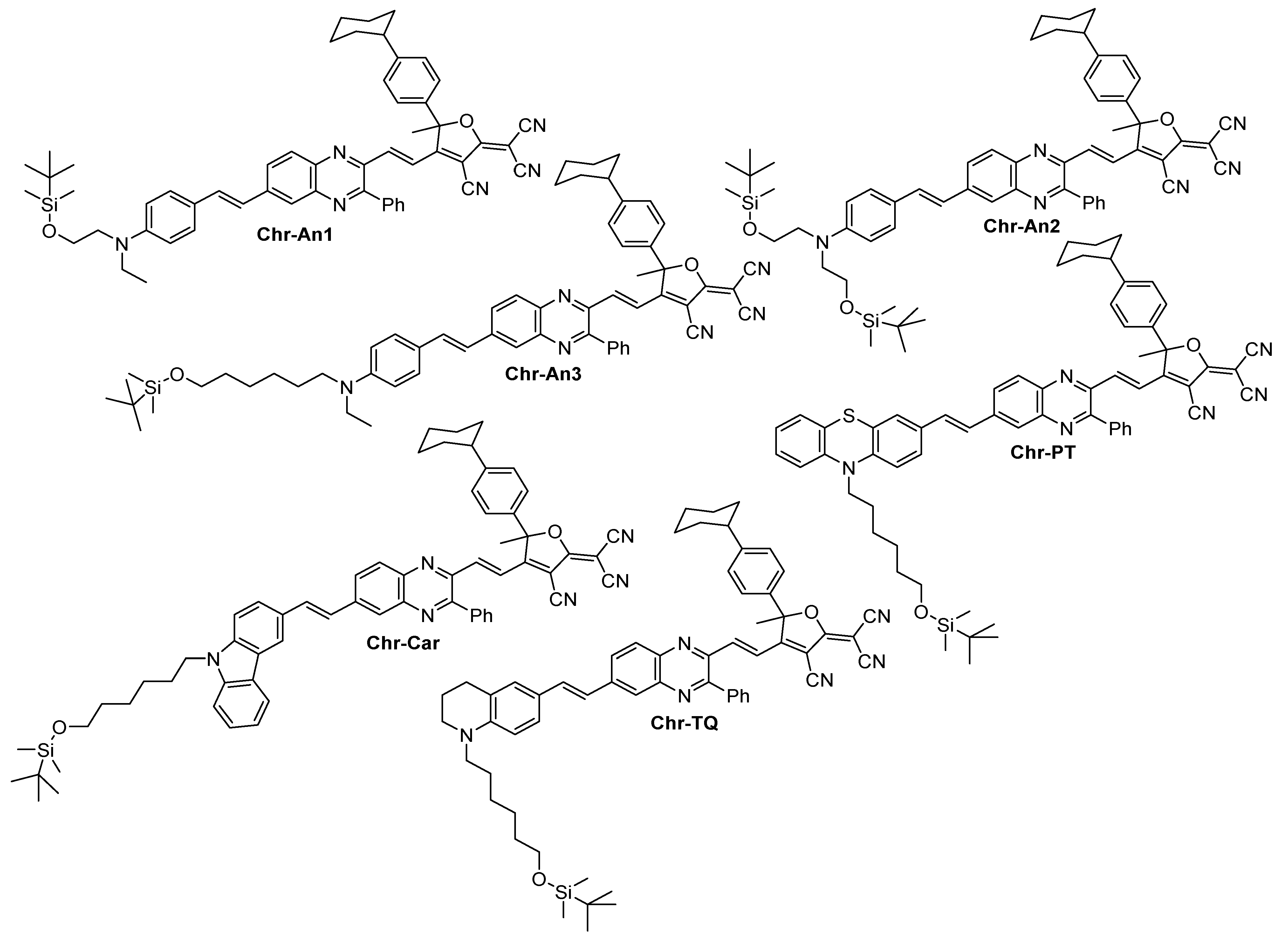
2.1. Synthesis

2.2. Linear Optical Properties
2.3. Thermal Properties
2.4. Quantum-Chemical Calculations and Molecular Modeling
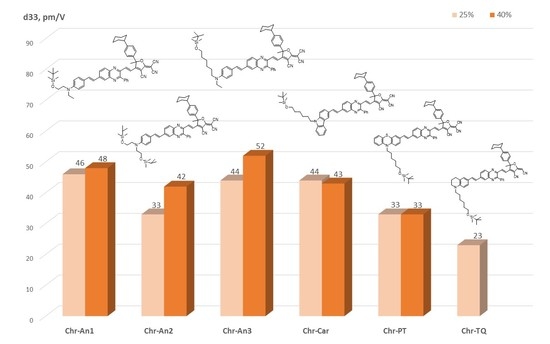
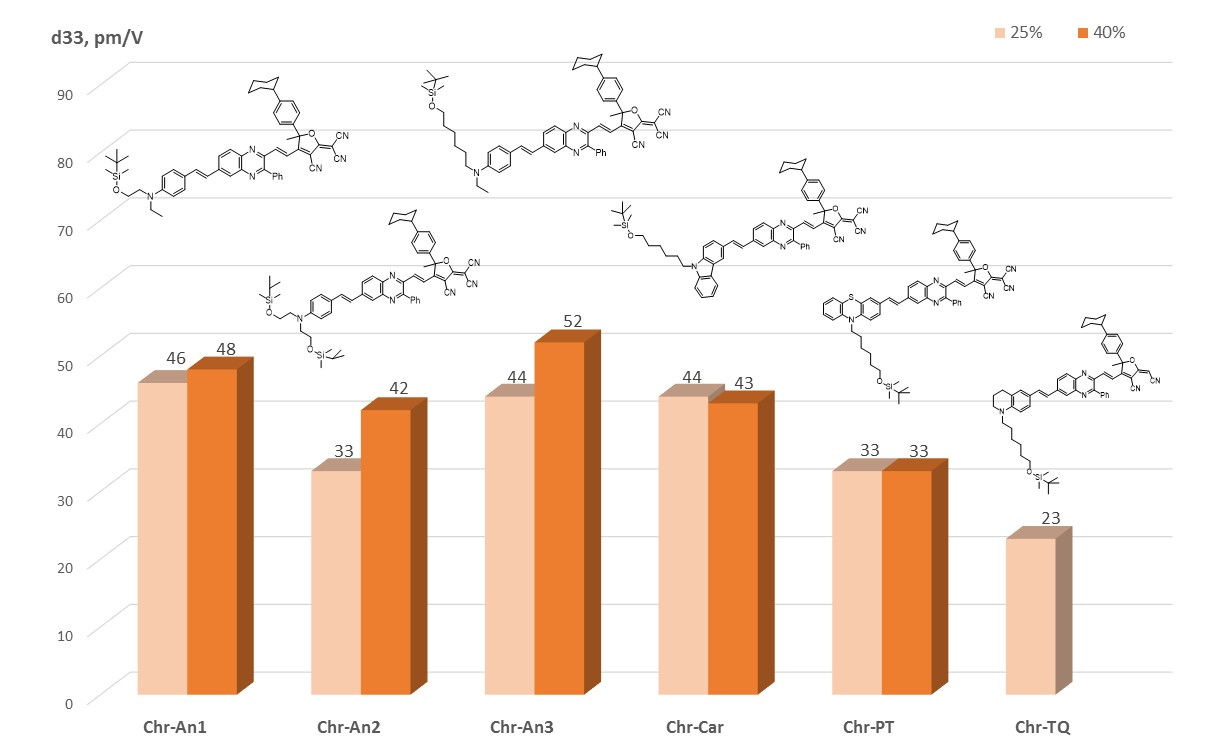
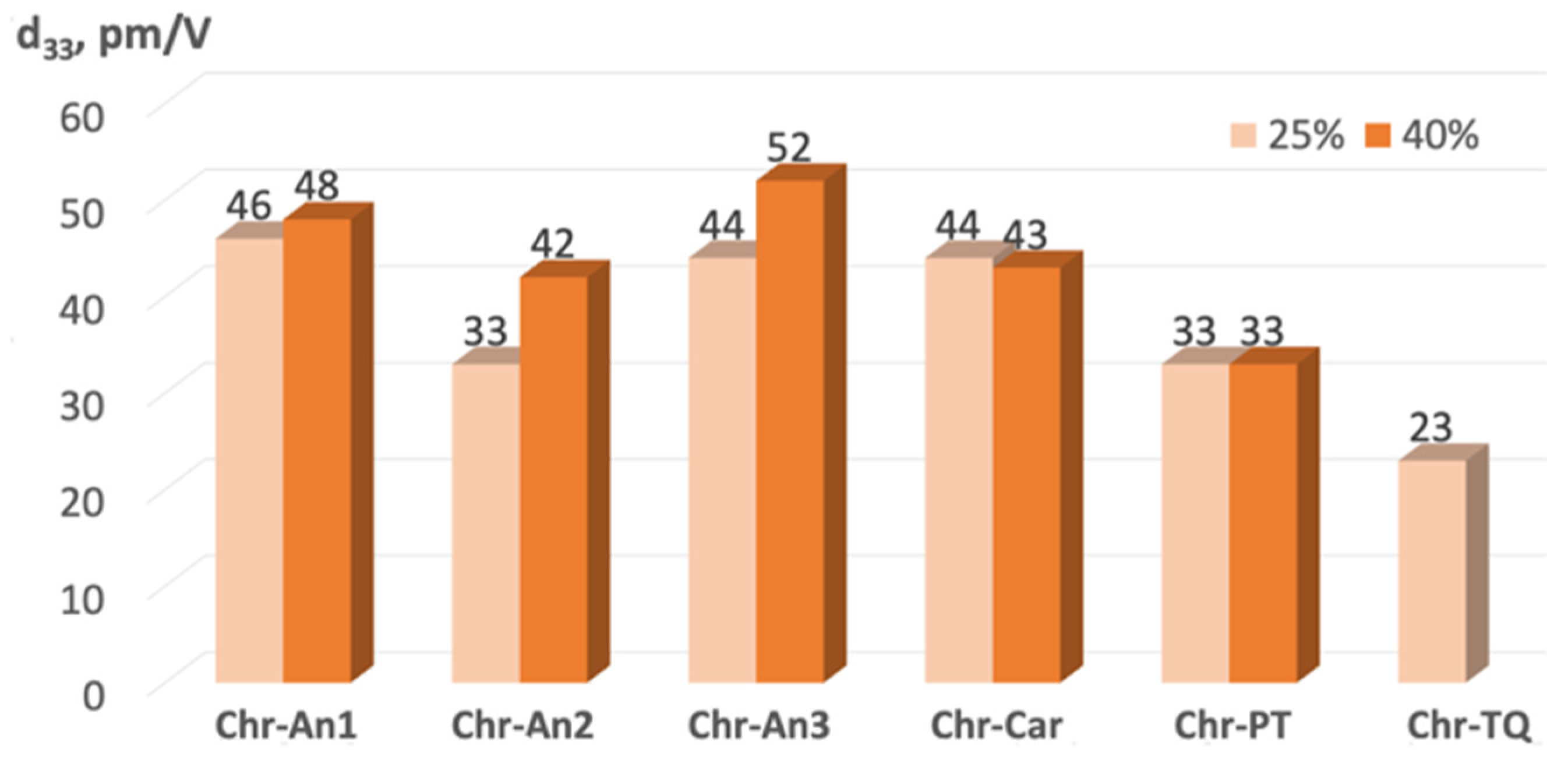
2.5. Experimental NLO Activity of Composite Polymer Materials Doped by Chromophores
3. Materials and Methods
3.1. General
3.2. 6-(10H-Phenothiazin-10-yl)hexan-1-ol
3.3. 6-(10H-Phenothiazin-10-yl)hexyl Acetate
3.4. 6-(3-Formyl-10H-phenothiazin-10-yl)hexyl Acetate (1e)
3.5. General Procedure for Synthesis of Compounds 2
3.5.1. ((4-Vinylphenyl)azanediyl)bis(ethane-2,1-diyl) Diacetate (2b)
3.5.2. 6-(Ethyl(4-vinylphenyl)amino)hexyl Acetate (2c)
3.5.3. 6-(3-Vinyl-10H-phenothiazin-10-yl)hexan-1-ol (2e′)
3.5.4. 1-(6-((tert-Butyldimethylsilyl)oxy)hexyl)-6-vinyl-1,2,3,4-tetrahydroquinoline (2f′)
3.6. General Procedure for Synthesis of 4a–d, 5e′ and 7f
3.6.1. (E)-((4-(2-(2-Methyl-3-phenylquinoxalin-6-yl)vinyl)phenyl)azanediyl)bis(ethane-2,1-diyl) diacetate (4b)
3.6.2. (E)-6-(Ethyl(4-(2-(2-methyl-3-phenylquinoxalin-6-yl)vinyl)phenyl)amino)hexyl Acetate (4c)
3.6.3. (E)-6-(3-(2-(2-Methyl-3-phenylquinoxalin-6-yl)vinyl)-10H-phenothiazin-10-yl)hexan-1-ol (5e)
3.6.4. (E)-6-(2-(1-(6-((tert-butyldimethylsilyl)oxy)hexyl)-1,2,3,4-tetrahydroquinolin-6-yl)vinyl)-3-phenylquinoxaline-2-carbaldehyde (7f)
3.7. General Procedure for Synthesis of 5a–d
3.7.1. (E)-2,2′-((4-(2-(2-Methyl-3-phenylquinoxalin-6-yl)vinyl)phenyl)azanediyl)bis(ethan-1-ol) (5b)
3.7.2. (E)-6-(Ethyl(4-(2-(2-methyl-3-phenylquinoxalin-6-yl)vinyl)phenyl)amino)hexan-1-ol (5c)
3.8. General Procedure for Synthesis of 6a–e and 1f′
3.8.1. (E)-N,N-Bis(2-((tert-butyldimethylsilyl)oxy)ethyl)-4-(2-(2-methyl-3-phenylquinoxalin-6-yl)vinyl)aniline (6b)
3.8.2. (E)-N-(6-((tert-Butyldimethylsilyl)oxy)hexyl)-N-ethyl-4-(2-(2-methyl-3-phenylquinoxalin-6-yl)vinyl)aniline (6c)
3.8.3. (E)-10-(6-((tert-Butyldimethylsilyl)oxy)hexyl)-3-(2-(2-methyl-3-phenylquinoxalin-6-yl)vinyl)-10H-phenothiazine (6e)
3.8.4. 1-(6-((tert-Butyldimethylsilyl)oxy)hexyl)-1,2,3,4-tetrahydroquinoline-6-carbaldehyde (1f’)
3.9. General Procedure for Synthesis of 7a–e and 3′
3.9.1. (E)-6-(4-(Bis(2-((tert-butyldimethylsilyl)oxy)ethyl)amino)styryl)-3-phenylquinoxaline-2-carbaldehyde (7b)
3.9.2. (E)-N-(6-((tert-butyldimethylsilyl)oxy)hexyl)-N-ethyl)amino)styryl)-3-phenylquinoxaline-2-carbaldehyde (7c)
3.9.3. (E)-6-(2-(10-(6-((tert-Butyldimethylsilyl)oxy)hexyl)-10H-phenothiazin-3-yl)vinyl)-3-phenylquinoxaline-2-carbaldehyde (7e)
3.9.4. 6-Bromo-3-phenylquinoxline-2-carbaldehyde (3′)
3.10. General Procedure for Chromophore Synthesis
3.10.1. 2-(4-((E)-2-(6-((E)-4-(bis(2-((tert-butyldimethylsilyl)oxy)ethyl)amino)styryl)-3-phenylquinoxline-2-yl)vinyl)-3-cyano-5-(4-cyclohexylphenyl)-5-methylfuran-2(5H)-ylidene)malononitrile (Chr-An2)
3.10.2. 2-(4-((E)-2-(6-((E)-4-((6-((tert-butyldimethylsilyl)oxy)hexyl)(ethyl)amino)styryl)-3phenylquinoxalin-2-yl)vinyl)-3-cyano-5-(4-cyclohexylphenyl)-5-methylfuran-2(5H)ylidene) malononitrile (Chr-An3)
3.10.3. 2-(4-((E)-2-(6-((E)-2-(10-(6-((tert-butyldimethylsilyl)oxy)hexyl)-10H-phenothiazin-3-yl)vinyl)-3-phenylquinoxalin-2-yl)vinyl)-3-cyano-5-(4-cyclohexylphenyl)-5-methylfuran-2(5H)-ylidene)malononitrile (Chr-PT)
3.10.4. 2-(4-((E)-2-(6-((E)-2-(1-(6-((tert-Butyldimethylsilyl)oxy)hexyl)-1,2,3,4-tetrahydroquinolin-6-yl)vinyl)-3-phenylquinoxalin-2-yl)vinyl)-3-cyano-5-(4-cyclohexylphenyl)-5methylfuran-2(5H)-ylidene)malononitrile (Chr-TQ)
3.11. Computational Details
3.12. Film Fabrication, Poling and NLO Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Negwer, M.; Scharnow, H.G. Organic Chemical Drugs and Their Synonyms: (An International Survey); Akademie: Berlin, Germany, 1987. [Google Scholar]
- Montana, M.; Mathias, F.; Terme, T.; Vanelle, P. Antitumoral activity of quinoxaline derivatives: A systematic review. Eur. J. Med. Chem. 2019, 163, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Soleymani, M.; Chegeni, M. The Chemistry and Applications of the Quinoxaline Compounds. Curr. Org. Chem. 2019, 23, 1789–1827. [Google Scholar] [CrossRef]
- Mamedov, V.A.; Kalinin, A.A. Advances in the synthesis of imidazo[1,5-a]-and imidazo[1,2-a]quinoxalines. Russ. Chem. Rev. 2014, 83, 820–847. [Google Scholar] [CrossRef]
- Achelle, S.; Baudequin, C.; Plé, N. Luminescent materials incorporating pyrazine or quinoxaline moieties. Dyes Pigment. 2013, 98, 575–600. [Google Scholar] [CrossRef] [Green Version]
- Nosova, E.V.; Achelle, S.; Lipunova, G.N.; Charushin, V.N.; Chupakhin, O.N. Functionalized benzazines as luminescent materials and components for optoelectronics. Russ. Chem. Rev. 2019, 88, 1128–1178. [Google Scholar] [CrossRef]
- Gedefaw, D.; Prosa, M.; Bolognesi, M.; Seri, M.; Andersson, M.R. Recent Development of Quinoxaline Based Polymers/Small Molecules for Organic Photovoltaics. Adv. Energy Mater. 2017, 7, 1700575. [Google Scholar] [CrossRef]
- Liu, M.; Gao, Y.; Zhang, Y.; Liu, Z.; Zhao, L. Quinoxaline-based conjugated polymers for polymer solar cells. Polym. Chem. 2017, 8, 4613–4636. [Google Scholar] [CrossRef]
- Yuan, J.; Ouyang, J.; Cimrová, V.; Leclerc, M.; Najari, A.; Zou, Y. Development of quinoxaline based polymers for photovoltaic applications. J. Mater. Chem. C 2017, 5, 1858–1879. [Google Scholar] [CrossRef]
- Black, C.B.; Andrioletti, B.; Try, A.C.; Ruiperez, C.; Sessler, J.L. Dipyrrolylquinoxalines: Efficient Sensors for Fluoride Anion in Organic Solution. J. Am. Chem. Soc. 1999, 121, 10438–10439. [Google Scholar] [CrossRef]
- Sessler, J.L.; Maeda, H.; Mizuno, T.; Lynch, V.M.; Furuta, H. Quinoxaline-Bridged Porphyrinoids. J. Am. Chem. Soc. 2002, 124, 13474–13479. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, X.-J.; Wong, W.-Y.; Guo, J.-P.; Wong, W.K.; Li, Z.-Y. Dipyrrolylquinoxaline-bridged Schiff bases: A new class of fluorescent sensors for mercury(ii). Dalton Trans. 2005, 19, 3235–3240. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Liu, Y.R.; Dong, C.; Han, X.-E. Novel ratio fluorescence probes for selectively detecting zinc ion based on Y-type quinoxaline framework. J. Lumin. 2017, 183, 513–518. [Google Scholar] [CrossRef]
- Mamedov, V.A.; Kalinin, A.A.; Gubaidullin, A.T.; Katsuba, S.A.; Syakaev, V.V.; Rizvanov, I.K.; Latypov, S.K. Efficient synthesis and structure peculiarity of macrocycles with bi-indolizinylquinoxalinone moieties. Tetrahedron 2013, 69, 10675–10687. [Google Scholar] [CrossRef]
- Mamedov, V.A.; Kalinin, A.A.; Samigullina, A.I.; Mironova, E.V.; Krivolapov, D.B.; Gubaidullin, A.T.; Il’dar, K.R. Iodine–sodium acetate (I2–NaOAc) mediated oxidative dimerization of indolizines: An efficient method for the synthesis of biindolizines. Tetrahedron Lett. 2013, 54, 3348–3352. [Google Scholar] [CrossRef]
- Yanilkin, V.V.; Nastapova, N.V.; Stepanov, A.S.; Kalinin, A.A.; Mamedov, V.A. Cation binding by 21,31-diphenyl-l 2,42-dioxo-7,10,13-trioxa-1,4(3,1)-diquinoxalina-2(2,3),3(3,2)-diindolizinacyclopentadecaphane and its acyclic analog. Russ. Chem. Bull. 2009, 58, 89–94. [Google Scholar] [CrossRef]
- Mamedov, V.A.; Kalinin, A.A.; Yanilkin, V.V.; Gubaidullin, A.T.; Latypov, S.K.; Balandina, A.A.; Isaikina, O.G.; Toropchina, A.V.; Nastapova, N.V.; Iglamova, N.A.; et al. 3-Indolizin-2-ylquinoxalines and the derived monopodands. Russ. Chem. Bull. 2005, 54, 2616–2625. [Google Scholar] [CrossRef]
- Reddy, M.R.; Han, S.H.; Lee, J.Y.; Seo, S. Synthesis and characterization of quinoxaline derivative for high performance phosphorescent organic light-emitting diodes. Dyes Pigment. 2018, 153, 132–136. [Google Scholar] [CrossRef]
- Gupta, S.; Milton, M.D. Y-shaped novel AIEE active push-pull quinoxaline derivatives displaying acidochromism and use towards white light emission by controlled protonation. Dyes Pigment. 2021, 195, 109690. [Google Scholar] [CrossRef]
- Yin, X.; Sun, H.; Zeng, W.; Xiang, Y.; Zhou, T.; Ma, D.; Yang, C. Manipulating the LUMO distribution of quinoxaline-containing architectures to design electron transport materials: Efficient blue phosphorescent organic light-emitting diodes. Org. Electron. 2016, 37, 439–447. [Google Scholar] [CrossRef]
- Gerasimova, T.P.; Burganov, T.I.; Katsyuba, S.A.; Kalinin, A.A.; Islamova, L.N.; Fazleeva, G.M.; Ahmadeev, B.S.; Mustafina, A.R.; Monari, A.; Assfeld, X.; et al. Halochromic luminescent quinoxalinones as a basis for pH-sensing in organic and aqueous solutions. Dyes Pigment. 2020, 186, 108958. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, J.; Simalou, O.; Wang, H.; Peng, J.; Zhai, L.; Xue, P.; Lu, R. Multi-stimuli-responsive fluorescent aminostyrylquinoxalines: Synthesis, solvatochromism, mechanofluorochromism and acidochromism. Dyes Pigment. 2018, 151, 296–302. [Google Scholar] [CrossRef]
- Zhan, Y.; Hu, H. Modulation of the emission behavior and mechanofluorochromism by electron-donating moiety of D-π-A type quinoxaline derivatives. Dyes Pigment. 2019, 167, 127–134. [Google Scholar] [CrossRef]
- Li, K.; Xue, P.; Shen, Y.; Liu, J. Ultrasound-and protonation-induced gelation of a carbazole-substituted divinylquinoxaline derivative with short alkyl chain. Dyes Pigment. 2018, 151, 279–286. [Google Scholar] [CrossRef]
- Burganov, T.I.; Katsyuba, S.A.; Sharipova, S.M.; Kalinin, A.A.; Monari, A.; Assfeld, X. Novel quinoxalinone-based push–pull chromophores with highly sensitive emission and absorption properties towards small structural modifications. Phys. Chem. Chem. Phys. 2018, 20, 21515–21527. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Jung, C.Y.; Wang, Y.; Choi, H.D.; Park, S.J.; Ou, P.; Jang, W.-D.; Jaung, J.Y. Effect of regioisomeric substitution patterns on the performance of quinoxaline-based dye-sensitized solar cells. Electrochim. Acta 2019, 298, 650–662. [Google Scholar] [CrossRef]
- Levitskaya, A.I.; Kalinin, A.A.; Fominykh, O.D.; Balakina, M.Y. The effect of rotational isomerism on the first hyperpolarizability of chromophores with divinyl quinoxaline conjugated bridge. Chem. Phys. Lett. 2017, 681, 16–21. [Google Scholar] [CrossRef]
- Fominykh, O.D.; Kalinin, A.A.; Sharipova, A.V.; Levitskaya, A.I.; Balakina, M.Y. The effect of various substituents in donor moiety on the aggregation of nonlinear-optical quinoxaline-based chromophores in composite polymer materials. Comput. Mater. Sci. 2020, 183, 109900. [Google Scholar] [CrossRef]
- Kalinin, A.A.; Sharipova, S.M.; Burganov, T.I.; Levitskaya, A.I.; Fominykh, O.D.; Vakhonina, T.A.; Ivanova, N.V.; Khamatgalimov, A.R.; Katsyuba, S.A.; Balakina, M.Y. Large nonlinear optical activity of chromophores with divinylquinoxaline conjugated π-bridge. J. Photochem. Photobiol. A Chem. 2019, 370, 58–66. [Google Scholar] [CrossRef]
- Kalinin, A.A.; Islamova, L.N.; Shmelev, A.G.; Fazleeva, G.M.; Fominykh, O.D.; Dudkina, Y.B.; Vakhonina, T.A.; Levitskaya, A.I.; Sharipova, A.V.; Mukhtarov, A.S.; et al. D-π-A chromophores with a quinoxaline core in the π-bridge and bulky aryl groups in the acceptor: Synthesis, properties, and femtosecond nonlinear optical activity of the chromophore/PMMA guest-host materials. Dyes Pigment. 2021, 184, 108801. [Google Scholar] [CrossRef]
- Xu, H.; Elder, D.L.; Johnson, L.E.; Heni, W.; de Coene, Y.; De Leo, E.; Destraz, M.; Meier, N.; Ghinst, W.V.; Hammond, S.R.; et al. Design and synthesis of chromophores with enhanced electro-optic activities in both bulk and plasmonic–organic hybrid devices. Mater. Horiz. 2022, 9, 261–270. [Google Scholar] [CrossRef]
- Jin, W.; Johnston, P.V.; Elder, D.L.; Manner, K.T.; Garrett, K.E.; Kaminsky, W.; Xu, R.; Robinson, B.H.; Dalton, L.R. Structure–function relationship exploration for enhanced thermal stability and electro-optic activity in monolithic organic NLO chromophores. J. Mater. Chem. C 2016, 4, 3119–3124. [Google Scholar] [CrossRef]
- Elder, D.L.; Haffner, C.; Heni, W.; Fedoryshyn, Y.; Garrett, K.E.; Johnson, L.E.; Campbell, R.A.; Avila, J.D.; Robinson, B.H.; Leuthold, J.; et al. Effect of Rigid Bridge-Protection Units, Quadrupolar Interactions, and Blending in Organic Electro-Optic Chromophores. Chem. Mater. 2017, 29, 6457–6471. [Google Scholar] [CrossRef]
- Xu, H.; Liu, F.; Elder, D.L.; Johnson, L.E.; de Coene, Y.; Clays, K.; Robinson, B.H.; Dalton, L.R. Ultrahigh Electro-Optic Coefficients, High Index of Refraction, and Long-Term Stability from Diels–Alder Cross-Linkable Binary Molecular Glasses. Chem. Mater. 2020, 32, 1408–1421. [Google Scholar] [CrossRef]
- Xu, H.; Johnson, L.E.; de Coene, Y.; Elder, D.L.; Hammond, S.R.; Clays, K.; Dalton, L.R.; Robinson, B.H. Bis(4-dialkylaminophenyl)heteroarylamino donor chromophores exhibiting exceptional hyperpolarizabilities. J. Mater. Chem. C 2021, 9, 2721–2728. [Google Scholar] [CrossRef]
- Tillack, A.F.; Robinson, B.H. Toward optimal EO response from ONLO chromophores: A statistical mechanics study of optimizing shape. JOSA B 2016, 33, E121–E129. [Google Scholar] [CrossRef]
- Xu, H.; Elder, D.L.; Johnson, L.E.; de Coene, Y.; Hammond, S.R.; Ghinst, W.V.; Clays, K.; Dalton, L.R.; Robinson, B.H. Electro—Optic Activity in Excess of 1000 pm V−1 Achieved via Theory—Guided Organic Chromophore Design. Adv. Mater. 2021, 33, 2104174. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, J.; Xiao, H.; Zhen, Z.; Bo, S. The important role of the isolation group (TBDPS) in designing efficient organic nonlinear optical FTC type chromophores. Dyes Pigment. 2017, 139, 239–246. [Google Scholar] [CrossRef]
- Sharipova, S.M.; Gilmutdinova, A.A.; Krivolapov, D.B.; Khisametdinova, Z.R.; Kataeva, O.N.; Kalinin, A.A. Synthesis of isomeric (E)-[4-(dimethylamino)phenyl]-vinylquinoxalines–precursors for a new class of nonlinear optical chromophores. Chem. Heterocycl. Compd. 2017, 53, 504–510. [Google Scholar] [CrossRef]
- Kalinin, A.A.; Isaikina, O.G.; Mamedov, V.A. Quinoxaline-benzimidazole rearrangements in the reactions of 3-alkanoylquinoxalin-2-ones with 1,2-phenylenediamines. Chem. Heterocycl. Compd. 2007, 43, 1307–1314. [Google Scholar] [CrossRef]
- Mamedov, V.A.; Kalinin, A.A.; Gubaidullin, A.T.; Isaikina, O.G.; Litvinov, I.A. Synthesis and Functionalization of 3-Ethylquinoxalin-2(1H)-one. Russ. J. Org. Chem. 2005, 41, 599–606. [Google Scholar] [CrossRef]
- Fazleeva, G.M.; Islamova, L.N.; Shaikhutdinova, G.R.; Kalinin, A.A. Synthesis of E,E-4-(6-(N-hydroxyethyl(N-ethyl)-aminostyrylquinoxalin-2-yl)vinyl)-2-dicyanomethylene-3-cyano-2,5-dihydrofurans. Synth. Commun. 2019, 49, 3528–3535. [Google Scholar] [CrossRef]
- Kalinin, A.A.; Sharipova, S.M.; Islamova, L.N.; Fazleeva, G.M.; Busyurova, D.N.; Sharipova, A.V.; Fominykh, O.D.; Balakina, M.Y. Chromophores with quinoxaline core in π-bridge and aniline or carbazole donor moiety: Synthesis and comparison of their linear and nonlinear optical properties. Russ. Chem. Bull. 2022, 71, 1009–1018. [Google Scholar] [CrossRef]
- Coe, B.J.; Harris, J.A.; Hall, J.J.; Brunschwig, B.S.; Hung, S.-T.; Libaers, W.; Clays, K.; Coles, S.J.; Horton, P.N.; Light, M.E.; et al. Syntheses and Quadratic Nonlinear Optical Properties of Salts Containing Benzothiazolium Electron-Acceptor Groups. Chem. Mater. 2006, 18, 5907–5918. [Google Scholar] [CrossRef] [Green Version]
- Kalinin, A.A.; Sharipova, S.M.; Levitskaya, A.I.; Dudkina, Y.B.; Burganov, T.I.; Fominykh, O.D.; Katsyuba, S.A.; Budnikova, Y.H.; Balakina, M.Y. D-π-A’-π-A chromophores with quinoxaline core in the π-electron bridge and charged heterocyclic acceptor moiety: Synthesis, DFT calculations, photophysical and electro-chemical properties. J. Photochem. Photobiol. A Chem. 2021, 407, 113042. [Google Scholar] [CrossRef]
- Dalton, L.R.; Xu, C.; Harper, A.W.; Ghosn, R.; Wu, B.; Liang, Z.; Montgomery, R.; Jen, A.K.-Y. Development and application of organic electro-optic modulators. Mol. Cryst. Liq. Cryst. Sci. Technol. Sect. B Nonlinear Opt. 1995, 10, 383–407. [Google Scholar]
- Zhu, Z.; Li, Z.; Tan, Y.; Li, Z.; Li, Q.; Zeng, Q.; Ye, C.; Qin, J. New hyperbranched polymers containing second-order nonlinear optical chromophores: Synthesis and nonlinear optical characterization. Polymer 2006, 47, 7881–7888. [Google Scholar] [CrossRef]
- Yuquan, S.; Yuxia, Z.; Zao, L.; Jianghong, W.; Ling, Q.; Shixiong, L.; Jianfeng, Z.; Jiayun, Z. The synthesis of highly active thiophene ring-containing chromophore components for photonic polymers based on a newly designed route. J. Chem. Soc. Perkin Trans. 1 1999, 3691–3695. [Google Scholar] [CrossRef]
- He, M.; Twieg, R.J.; Gubler, U.; Wright, D.; Moerner, W.E. Synthesis and Photorefractive Properties of Multifunctional Glasses. Chem. Mater. 2003, 15, 1156–1164. [Google Scholar] [CrossRef]
- Allen, D.E.; Demartino, R.N.; Yoon, H.-N. Acrylic Polymers Exhibiting Nonlinear Optical Response. Patent WO 91/03504, 21 March 1991. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Schrodinger Release 2022-1: Materials Science Suite; Schrödinger: New York, NY, USA, 2022.
- Suponitsky, K.Y.; Tafur, S.; Masunov, A. Applicability of hybrid density functional theory methods to calculation of molecular hyperpolarizability. J. Chem. Phys. 2008, 129, 044109. [Google Scholar] [CrossRef]
- Johnson, L.E.; Dalton, L.R.; Robinson, B.H. Optimizing Calculations of Electronic Excitations and Relative Hyperpolarizabilities of Electrooptic Chromophores. Acc. Chem. Res. 2014, 47, 3258–3265. [Google Scholar] [CrossRef] [PubMed]
- Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef] [PubMed]
- Levitskaya, A.I.; Kalinin, A.A.; Fominykh, O.D.; Balakina, M.Y. Theoretical predictions of nonlinear optical characteristics of novel chromophores with quinoxalinone moieties. Comput. Theor. Chem. 2015, 1074, 91–100. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Schrödinger Release 2022–1: Desmond Molecular Dynamics System, DE Shaw Research, New York, NY, 2022 Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2022.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λmax, nm/eV (ε, 103·M−1·cm−1) | Δλmax a, Nm | Δλmax b, nm | ||||
|---|---|---|---|---|---|---|
| Chromophore | 1,4-dioxane | CHCl3 | CH2Cl2 | CH3CN | ||
| Chr-An1 [43] | 585/2.12 (36.5) | 630/1.97 (36.0) | 619/2.00 (29.4) | 585/2.12 (31.3) | 45/0.15 | 45/0.15 |
| Chr-An2 | 585/2.12 (31.3) | 627/1.98 (31.1) | 616/2.01 (30.8) | 582/2.13 (21.8) | 42/0.14 | 45/0.15 |
| Chr-An3 | 595/2.08 (35.6) | 642/1.93 (31.9) | 629/1.97 (30.1) | 592/2.09 (31.2) | 47/0.15 | 50/0.16 |
| Chr-Car [43] | 536/2.31 (35.0) | 568/2.18 (32.6) | 555/2.23 (36.5) | 528/2.35 (31.0) | 32/0.13 | 40/0.17 |
| Chr-PT | 542 (29.4)/2.28 447 (30.2) 377 (27.4) | 575/2.15 (23.8) 455 (29.5) 382 (24.0) | 562/2.20 (21.6) 454 (25.6) 381 (21.3) | 527/2.35 (27.2) 448 (28.2) 373 (25.2) | 33/0.13 | 48/0.20 |
| Chr-TQ | 613/2.02 (32.2) | 665/1.86 (31.0) | 653/1.90 (31.8) | 612/2.03 (27.1) | 52/0.16 | 53/0.17 |
| Chromophore | Chr-An1 | Chr-An2 | Chr-An3 | Chr-Car | Chr-PT | Chr-TQ |
|---|---|---|---|---|---|---|
| Td a, °C | 262 | 275 | 334 | 271 | 268 | 259 |
| Td b, °C | 239 | 270 | 226 | 266 | 264 | 171 |
| mp, °C | 219 | 262 | 192 | 212 | 208 | 156 |
| Chr-An1 [43] | Chr-An2 | Chr-An3 | Chr-Car [43] | Chr-PT | Chr-TQ | |
|---|---|---|---|---|---|---|
| μ, D | 19.5 | 20.2 | 18.7 | 16.7 | 17.3 | 18.6 |
| α(av), 10−24 esu | 142.3 | 160.0 | 149.1 | 150.6 | 155.3 | 154.9 |
| β(x), 10−30 esu | −46.9 | 289.2 | 706.5 | −455.5 | −456.5 | −413.5 |
| β(y), 10−30 esu | 605.7 | 844.9 | −363.5 | −121.3 | 95.5 | 841.3 |
| β(z), 10−30 esu | 515.8 | 188.5 | 73.3 | 157.8 | 86.5 | −14.3 |
| βtot, 10−30 esu | 797.0 | 912.7 | 797.8 | 497.0 | 474.3 | 937.4 |
| DBA-VQV-TCFCyPh | Chr-An1 | Chr-An2 | Chr-An3 | Chr-Car | Chr-PT | Chr-TQ | |
|---|---|---|---|---|---|---|---|
| ϕ1 a | 4.2 | 21.7 | 24.3 | 20.6 | 29.4 | 13.0 | 23.6 |
| ϕ2 b | 1.1 | 39.4 | |||||
| ϕ3 c | 3.7 | 1.6 | 2.4 | 2.2 | 1.9 | 2.6 | 3.0 |
| ϕ4 d | 24.2 | 12.2 | 3.5 | 9.3 | 7.9 | 13.0 | 10.1 |
| ϕ5 e | 26.4 | 32.8 | 28.2 | 19.5 | 35.7 | 22.6 | 28.2 |
| Chromophore | Chromophore Content, % | Number of Chromophores in a Cell | Number of π–π Bonds | Number of Noncovalently Bound Chromophores | Portion of Noncovalently Bound Chromophores, % | Maximal Size of the Aggregate |
|---|---|---|---|---|---|---|
| DBA-VQV-TCFCyPh | 25 | 25 | 10 | 40 | 4 | |
| 30 | 33 | 16 | 48 | 3 | ||
| Chr-An1 | 25 | 23 | 6 | 8 | 35 | 3 |
| 40 | 47 | 20 | 24 | 51 | 4 | |
| Chr-An2 | 25 | 20 | 7 | 8 | 40 | 2 |
| 40 | 43 | 12 | 15 | 35 | 3 | |
| Chr-An3 | 25 | 23 | 12 | 7 | 30 | 2 |
| 40 | 44 | 18 | 14 | 32 | 2 | |
| Chr-Car | 25 | 22 | 13 | 11 | 50 | 3 |
| 40 | 43 | 45 | 23 | 53 | 3 | |
| Chr-PT | 25 | 20 | 7 | 8 | 40 | 2 |
| 40 | 40 | 22 | 23 | 57 | 5 | |
| Chr-TQ | 25 | 22 | 9 | 5 | 23 | 3 |
| 40 | 44 | 28 | 24 | 54 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalinin, A.A.; Islamova, L.N.; Sharipova, S.M.; Fazleeva, G.M.; Shustikov, A.A.; Gaysin, A.I.; Shmelev, A.G.; Sharipova, A.V.; Vakhonina, T.A.; Fominykh, O.D.; et al. Synthesis of D-π-A′-π-A Chromophores with Quinoxaline Core as Auxiliary Acceptor and Effect of Various Silicon-Substituted Donor Moieties on Thermal and Nonlinear Optical Properties at Molecular and Material Level. Molecules 2023, 28, 531. https://doi.org/10.3390/molecules28020531
Kalinin AA, Islamova LN, Sharipova SM, Fazleeva GM, Shustikov AA, Gaysin AI, Shmelev AG, Sharipova AV, Vakhonina TA, Fominykh OD, et al. Synthesis of D-π-A′-π-A Chromophores with Quinoxaline Core as Auxiliary Acceptor and Effect of Various Silicon-Substituted Donor Moieties on Thermal and Nonlinear Optical Properties at Molecular and Material Level. Molecules. 2023; 28(2):531. https://doi.org/10.3390/molecules28020531
Chicago/Turabian StyleKalinin, Alexey A., Liliya N. Islamova, Sirina M. Sharipova, Guzel M. Fazleeva, Alexey A. Shustikov, Adel I. Gaysin, Artemiy G. Shmelev, Anastasiya V. Sharipova, Tatyana A. Vakhonina, Olga D. Fominykh, and et al. 2023. "Synthesis of D-π-A′-π-A Chromophores with Quinoxaline Core as Auxiliary Acceptor and Effect of Various Silicon-Substituted Donor Moieties on Thermal and Nonlinear Optical Properties at Molecular and Material Level" Molecules 28, no. 2: 531. https://doi.org/10.3390/molecules28020531
APA StyleKalinin, A. A., Islamova, L. N., Sharipova, S. M., Fazleeva, G. M., Shustikov, A. A., Gaysin, A. I., Shmelev, A. G., Sharipova, A. V., Vakhonina, T. A., Fominykh, O. D., Babaeva, O. B., Khamatgalimov, A. R., & Balakina, M. Y. (2023). Synthesis of D-π-A′-π-A Chromophores with Quinoxaline Core as Auxiliary Acceptor and Effect of Various Silicon-Substituted Donor Moieties on Thermal and Nonlinear Optical Properties at Molecular and Material Level. Molecules, 28(2), 531. https://doi.org/10.3390/molecules28020531