




Naive Prediction of Protein Backbone Phi and Psi Dihedral Angles Using Deep Learning



Abstract
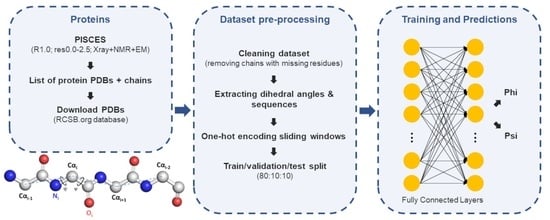
:
1. Introduction
2. Results
2.1. Mean Absolute Prediction Error
2.2. Measured vs. Predicted Values
2.3. Dihedral Angle Predictability in Amino Acids
2.4. Three-State Secondary Structure Prediction
3. Materials and Methods
3.1. Dataset Preparation
3.2. Neural Network
3.3. Loss Function
3.4. Optimization
3.5. Predicted Outputs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| 7PCP | 7 physicochemical properties |
| AMD | Advanced Micro Devices |
| ASA | accessible surface area |
| B | isolated β-bridge |
| BAP | protein backbone angle predictions |
| BLAST | Basic Local Alignment Search Tool |
| BRNN | bidirectional recurrent neural network |
| C | carbon atom ooil |
| CASP | Critical Assessment of protein Structure Prediction |
| CNN | convolutional neural network |
| CoDNas | Conformational Diversity of Native State |
| Cα | alpha carbon atom |
| DNN | deep neural network |
| E | parallel/anti-parallel β sheet conformation |
| E | sheet |
| FCNN | fully connected neural network |
| FM | free modeling |
| G | 310 helix |
| H | α-helix |
| HHBlits | HMM-HMM-based lightning-fast iterative sequence search |
| HMM | hidden Markov model |
| I | π-helix |
| LSTM | long short-term memory |
| LSTM-BRNNs | long short-term memory and bidirectional recurrent neural networks |
| MAE | mean absolute error |
| MD | molecular dynamics |
| MTS | mitoargetingtargetting sequence |
| MnSOD | manganese superoxide dismutase |
| N | nitrogen atom |
| PDB | Protein Data Bank |
| PISCES | Protein sequence culling server |
| PSP | protein structure prediction |
| PSSM | position-specific scoring matrix |
| PSSP | protein secondary structure prediction |
| Q3 | three-state model |
| Q8 | eight-state model |
| R-free | Free R-value |
| RMSE | root mean squared error |
| ReLU | Rectified Linear Unit |
| ResNets | Residual Networks |
| ResNets | residual networks |
| S | bend |
| SAP | structure analysis and prediction |
| SNP | single nucleotide polymorphism |
| SS | secondary structure |
| SSPro | Secondary Structure Prediction |
| SVM | support vector machine |
| T | turn |
| Å | Angstrom |
| τ | tao |
| ψ | psi dihedral angle |
| ω | omega dihedral angle |
| ϕ | phi dihedral angle |
References
- Cutello, V.; Narzisi, G.; Nicosia, G. A multi-objective evolutionary approach to the protein structure prediction problem. J. R. Soc. Interface 2005, 3, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- AlQuraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)-round XIII. Proteins 2019, 87, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.; Simpkin, A.J.; Hartmann, M.D.; Rigden, D.J.; Keegan, R.M.; Lupas, A.N. High-accuracy protein structure prediction in CASP14. Proteins 2021, 89, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-T.; Ellrott, K.; Xu, Y. A historical perspective of template-based protein structure prediction. In Protein Structure Prediction; Humana: Totowa, NJ, USA, 2008; pp. 3–42. [Google Scholar]
- Zhou, Y.; Duan, Y.; Yang, Y.; Faraggi, E.; Lei, H. Trends in template/fragment-free protein structure prediction. Theor. Chem. Acc. 2011, 128, 3–16. [Google Scholar] [CrossRef]
- Maurice, K.J. SSThread: Template-free protein structure prediction by threading pairs of contacting secondary structures followed by assembly of overlapping pairs. J. Comput. Chem. 2014, 35, 644–656. [Google Scholar] [CrossRef]
- Rost, B. Protein secondary structure prediction continues to rise. J. Struct. Biol. 2001, 134, 204–218. [Google Scholar] [CrossRef]
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Oostenbrink, C. Dihedral-based segment identification and classification of biopolymers I: Proteins. J. Chem. Inf. Model. 2014, 54, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Rost, B.; Sander, C. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 1993, 232, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Dor, O.; Zhou, Y. Achieving 80% ten-fold cross-validated accuracy for secondary structure prediction by large-scale training. Proteins 2007, 66, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Faraggi, E.; Zhang, T.; Yang, Y.; Kurgan, L.; Zhou, Y. SPINE X: Improving protein secondary structure prediction by multistep learning coupled with prediction of solvent accessible surface area and backbone torsion angles. J. Comput. Chem. 2012, 33, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Bettella, F.; Rasinski, D.; Knapp, E.W. Protein secondary structure prediction with SPARROW. J. Chem. Inf. Model. 2012, 52, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, C.; Pollastri, G. Porter, PaleAle 4.0: High-accuracy prediction of protein secondary structure and relative solvent accessibility. Bioinformatics 2013, 29, 2056–2058. [Google Scholar] [CrossRef]
- Yaseen, A.; Li, Y. Context-based features enhance protein secondary structure prediction accuracy. J. Chem. Inf. Model. 2014, 54, 992–1002. [Google Scholar] [CrossRef]
- Heffernan, R.; Paliwal, K.; Lyons, J.; Dehzangi, A.; Sharma, A.; Wang, J.; Sattar, A.; Yang, Y.; Zhou, Y. Improving prediction of secondary structure, local backbone angles, and solvent accessibility with a single neural network. Sci. Rep. 2015, 83, 1201–1214. [Google Scholar]
- Cuff, J.A.; Barton, G.J. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 2000, 40, 502–511. [Google Scholar] [CrossRef]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Peng, J.; Ma, J.; Xu, J. Protein secondary structure prediction using deep convolutional neural fields. Sci. Rep. 2016, 6, 18962. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, R.; Yang, Y.; Paliwal, K.; Zhou, Y. Capturing non-local interactions by long short-term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers, and solvent accessibility. Bioinformatics 2017, 33, 2842–2849. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Shang, Y.; Xu, D. MUFOLD-SS: New deep inception-inside-inception networks for protein secondary structure prediction. Proteins 2018, 86, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sønderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins Struct. Funct. Bioinform. 2019, 87, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, J.; Lü, Q. Prediction of 8-state protein secondary structures by a novel deep learning architecture. BMC Bioinform. 2018, 19, 293. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Wang, Q.; Ma, J. OPUS-TASS: A protein backbone torsion angles and secondary structure predictor based on ensemble neural networks. Bioinformatics 2020, 36, 5021–5026. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Hou, J.; Cheng, J. DNSS2: Improved ab initio protein secondary structure prediction using advanced deep learning architectures. Proteins 2021, 89, 207–217. [Google Scholar] [CrossRef]
- Pollastri, G.; Przybylski, D.; Rost, B.; Baldi, P. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins 2002, 47, 228–235. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, F.; Peng, J.; Xu, J. Protein 8-class secondary structure prediction using conditional neural fields. Proteomics 2011, 11, 3786–3792. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, A.; Li, Y. Template-based C8-SCORPION: A protein 8-state secondary structure prediction method using structural information and context-based features. Bioinformatics 2014, 15, S3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Deep supervised and convolutional generative stochastic network for protein secondary structure prediction. In Proceedings of the 31st International Conference on International Conference on Machine Learning, Beijing, China, 21–26 June 2014. [Google Scholar]
- Simons, K.T.; Kooperberg, C.; Huang, E.; Baker, D.; Petrey, D. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins Struct. Funct. Bioinform. 1999, 34, 82–95. [Google Scholar] [CrossRef]
- Faraggi, E.; Yang, Y.; Zhang, S.; Zhou, Y. Predicting continuous local structure and the effect of its substitution for secondary structure in fragment-free protein structure prediction. Structure 2009, 17, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.; Dehzangi, A.; Heffernan, R.; Sharma, A.; Paliwal, K.; Sattar, A.; Zhou, Y.; Yang, Y. Predicting backbone cα angles and dihedrals from protein sequences by stacked sparse auto-encoder deep neural network. J. Comput. Chem. 2014, 35, 2040–2046. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.; Paliwal, K.; Litfin, T.; Yang, Y.; Zhou, Y. Improving prediction of protein secondary structure, backbone angles, solvent accessibility and contact numbers by using predicted contact maps and an ensemble of recurrent and residual convolutional neural networks. Bioinformatics 2018, 35, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Fang, C. Applications of Deep Neural Networks to Protein Structure Prediction. Ph.D. Thesis, University of Missouri, Columbia, MO, USA, 2018. [Google Scholar]
- Wu, S.; Zhang, Y. Anglor: A composite machine-learning algorithm for protein backbone torsion angle prediction. PLoS ONE 2008, 3, e3400. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by hmm-hmm alignment. Nat. Methods 2012, 9, 173. [Google Scholar] [CrossRef]
- Heffernan, R.; Paliwal, K.; Lyons, J.; Singh, J.; Yang, Y.; Zhou, Y. Single-sequence-based prediction of protein secondary structures and solvent accessibility by deep whole-sequence learning. J. Comput. Chem. 2018, 39, 2210–2216. [Google Scholar] [CrossRef]
- Hanson, J.; Paliwal, K.; Litfin, T.; Yang, Y.; Zhou, Y. Accurate prediction of protein contact maps by coupling residual two-dimensional bidirectional long short-term memory with convolutional neural networks. Bioinformatics 2018, 34, 4039–4045. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, S.; Deng, M.; Xu, J. Raptorx-angle: Real-value prediction of protein backbone dihedral angles through a hybrid method of clustering and deep learning. Bioinformatics 2018, 19, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Mataeimoghadam, F.; Hakim Newton, M.A.; Dehzangi, A.; Karim, A.; Jayaram, B.; Ranganathan, S.; Sattar, A. Enhancing protein backbone angle prediction by using simpler models of deep neural networks. Sci. Rep. 2020, 10, 5016. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.A.H.; Mataeimoghadam, F.; Zaman, R.; Sattar, A. Secondary structure specific simpler prediction models for protein backbone angles. Bioinformatics 2022, 23, 6. [Google Scholar] [CrossRef] [PubMed]
- McKinney, W. Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; Volume 445, pp. 51–56. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Chen, K.; Kurgan, L.; Ruan, J. Optimization of the Sliding Window Size for Protein Structure Prediction. In Proceedings of the IEEE Symposium on Computational Intelligence and Bioinformatics and Computational Biology, Toronto, ON, Canada, 28–29 September 2006; pp. 1–7. [Google Scholar]
- Balasco, N.; Esposito, L.; De Simone, A.; Vitagliano, L. Local Backbone Geometry Plays a Critical Role in Determining Conformational Preferences of Amino Acid Residues in Proteins. Biomolecules 2022, 12, 1184. [Google Scholar] [CrossRef] [PubMed]
- Swindells, M.B.; MacArthur, M.W.; Thornton, J.M. Intrinsic φ,ψ propensities of amino acids, derived from the coil regions of known structures. Nat. Struct. Mol. Biol. 1995, 2, 596–603. [Google Scholar] [CrossRef]
- Heinig, M.; Frishman, D. STRIDE: A Web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Teeter, M.M. Water structure of a hydrophobic protein at atomic resolution: Pentagon rings of water molecules in crystals of crambin. Proc. Natl. Acad. Sci. USA 1984, 81, 6014–6018. [Google Scholar] [CrossRef]
- Groll, M.; Huber, R.; Potts, B.C. Crystal structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J. Am. Chem. Soc. 2006, 128, 5136–5141. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Srajer, V.; Knapp, J.E.; Royer, W.E., Jr. Cooperative macromolecular device revealed by meta-analysis of static and time-resolved structures. Proc. Natl. Acad. Sci. USA 2012, 109, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Brvar, M.; Perdih, A.; Renko, M.; Anderluh, G.; Turk, D.; Solmajer, T. Structure-based discovery of subst’tuted 4,5’-bithiazoles as novel DNA gyrase inhibitors. J. Med. Chem. 2012, 55, 6413–6426. [Google Scholar] [CrossRef] [PubMed]
- Ken-ichi, A.; Shigeyuki, M. Solution Structure of IFN alpha8. Available online: https://www.rcsb.org/structure/6jhd (accessed on 3 October 2023).
- Nakazawa, H.; Onodera-Sugano, T.; Sugiyama, A.; Tanaka, Y.; Hattori, T.; Niide, T.; Ogata, H.; Asano, R.; Kumagai, I.; Umetsu, M. Association behavior and control of the quality of cancer therapeutic bispecific diabodies expressed in Escherichia coli. Biochem. Eng. J. 2020, 160, 107636. [Google Scholar] [CrossRef]
- Guan, H.; Wang, Y.; Yu, T.; Huang, Y.; Li, M.; Saeed, A.F.U.H.; Perčulija, V.; Li, D.; Xiao, J.; Wang, D.; et al. Cryo-EM structures of the human PA200 and PA200-20S complex reveal regulation of proteasome gate opening and two PA200 apertures. PLoS Biol. 2020, 18, e3000654. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; An, Q.; Shen, X.F.; Sui, M.; Li, C.; Jia, D.; Luo, Y.; Sun, Q. Structure-Guided Design of the First Noncovalent Small-Molecule Inhibitor of CRM1. J. Med. Chem. 2021, 64, 6596–6607. [Google Scholar] [CrossRef] [PubMed]
- Aljedani, S.S.; Liban, T.J.; Tran, K.; Phad, G.; Singh, S.; Dubrovskaya, V.; Pushparaj, P.; Martinez-Murillo, P.; Rodarte, J.; Mileant, A.; et al. Structurally related but genetically unrelated antibody lineages converge on an immunodominant HIV-1 Env neutralizing determinant following trimer immunization. PLoS Pathog. 2021, 17, e1009543. [Google Scholar] [CrossRef] [PubMed]
- Azadmanesh, J.; Trickel, S.R.; Borgstahl, G.E.O. Substrate-analog binding and electrostatic surfaces of human manganese superoxide dismutase. J. Struct. Biol. 2017, 199, 68–75. [Google Scholar] [CrossRef]
- Broz, M.; Furlan, V.; Lešnik, S.; Jukič, M.; Bren, U. The Effect of the Ala16Val Mutation on the Secondary Structure of the Manganese Superoxide Dismutase Mitochondrial Targeting Sequence. Antioxidants 2022, 11, 2348. [Google Scholar] [CrossRef]
- Wang, G.; Dunbrack, R.L., Jr. PISCES: A protein sequence culling server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Cock, P.J.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A system for large-scale machine learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Chollet, F.; Keras. GitHub. Available online: https://github.com/fchollet/keras (accessed on 12 February 2022).
- Zeiler, M.D. ADADELTA: An Adaptive Learning Rate Method. arXiv 2012, arXiv:1212.5701. [Google Scholar]
- Močkus, J. Bayesian Approach to Global Optimization. Mathematics and Its Applications, 37th ed.; Kluwer Academic Publishers Group: Dordrecht, The Netherlands, 1989; ISBN 978-94-009-0909-0. [Google Scholar]
- Takuya, A.; Shotaro, S.; Yanase, T.; Ohta, T. Optuna: A Next-generation Hyperparameter Optimization Framework. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, New York, NY, USA, 4–8 August 2009; pp. 2623–2631. [Google Scholar]
- Bergstra, J.; Bardenet, R.; Bengio, Y.; Kégl, B. Algorithms for Hyper-Parameter Optimization. In Proceedings of the 24th International Conference on Neural Information Processing Systems, Granada, Spain, 12–14 December 2011; pp. 2546–2554. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SS Label | Helix (Predicted) | Sheet (Predicted) | Undesignated (Predicted) |
|---|---|---|---|
| Helix (real) | 73.2% (141,380) | 16.0% (30,837) | 10.8% (20,908) |
| Sheet (real) | 14.9% (33,535) | 73.9% (166,673) | 11.2% (25,300) |
| Undesignated (real) | 27.1% (23,151) | 26.8% (22,905) | 46.1% (39,335) |
| Sliding Window | Phi MAE [deg] | Psi MAE [deg] | Epoch Duration [s] |
|---|---|---|---|
| 3 | 28.37 | 64.09 | 57 |
| 7 | 25.67 | 53.36 | 102 |
| 11 | 24.51 | 48.98 | 130 |
| 15 | 23.96 | 46.74 | 155 |
| 21 | 23.53 | 44.14 | 210 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broz, M.; Jukič, M.; Bren, U. Naive Prediction of Protein Backbone Phi and Psi Dihedral Angles Using Deep Learning. Molecules 2023, 28, 7046. https://doi.org/10.3390/molecules28207046
Broz M, Jukič M, Bren U. Naive Prediction of Protein Backbone Phi and Psi Dihedral Angles Using Deep Learning. Molecules. 2023; 28(20):7046. https://doi.org/10.3390/molecules28207046
Chicago/Turabian StyleBroz, Matic, Marko Jukič, and Urban Bren. 2023. "Naive Prediction of Protein Backbone Phi and Psi Dihedral Angles Using Deep Learning" Molecules 28, no. 20: 7046. https://doi.org/10.3390/molecules28207046
APA StyleBroz, M., Jukič, M., & Bren, U. (2023). Naive Prediction of Protein Backbone Phi and Psi Dihedral Angles Using Deep Learning. Molecules, 28(20), 7046. https://doi.org/10.3390/molecules28207046